


Abstract
P-glycoprotein (Pgp)-mediated drug efflux is a major factor contributing to the variance of absorption and distribution of many drugs (Hall et al., 1999). A simple and reliable in vitro method to identify inhibitors of Pgp helps to prevent the potential of drug interactions. Using daunorubicin as a fluorescent marker and vanadate as a positive control compound, a functional flow cytometry method for assessing the ability of a drug to inhibit Pgp-mediated drug efflux from CR1R12 multidrug-resistant cells has been evaluated. Quantitation of the relative fluorescence was used to compare potency of individual inhibitors. Known Pgp inhibitors, such as cyclosporin A, nicardipine, verapamil, quinidine, terfenadine, tamoxifen, and vinblastine were demonstrated to inhibit the Pgp-mediated efflux of daunorubicin. Cyclosporin A and terfenadine were the most potent inhibitors among the compounds tested. Tetraphenylphosphonium and α-tocopherol had little inhibitory effect. Progesterone produced significant inhibition at relatively high concentrations. This study demonstrated that this in vitro flow cytometry method is a simple, sensitive, and quantitative tool to assess the capacity of a drug to inhibit Pgp transporters, and is useful for screening or identifying inhibitors of Pgp as well as evaluation of potential for drug interactions.
One of the main obstacles to successful cancer chemotherapy is the development of multiple drug resistance (MDR)1to a variety of structurally unrelated cytotoxic drugs. Overexpression of P-glycoprotein (Pgp; 170–180 kDa), the product of theMDR1 gene, is the most commonly observed characteristic of multidrug-resistant cells grown in vitro (Gottesman and Pastan, 1993;Ling, 1995; Schinkel et al., 1995) and in a number of tumors (Redmond et al., 1991; Decker et al., 1995). Pgp, an ATP-dependent multidrug pump belonging to the ATP-binding cassette (ABC) superfamily of proteins (Hyde et al., 1990), protects cells from cytotoxic compounds by transporting them out of the cells and reducing the intracellular levels below their effective concentrations. Physiologically, Pgp is widely expressed in the epithelial cells of intestine, liver and kidney, and in the endothelial cells of brain and placenta. The broad substrate specificity and distinctive expression locations suggest that Pgp may have a direct role related to the absorption and disposition of drugs or xenobiotics, and is quickly becoming recognized as a critical factor in the disposition of many drugs and xenobiotics. Consequently, the inhibition of this membrane transporter could result in far reaching implications, including drug interactions.
A large number of compounds that interact with the Pgp efflux pump have been identified, and some are under development as drugs. These compounds have no common chemical structural features except for hydrophobicity. Some of them are positively charged at physiological pH (Chin et al., 1993). The early generation of modulators of Pgp, such as cyclosporin A (Sonneveld et al., 1992), verapamil (Watanabe et al., 1995), and quinidine (Wishart et al., 1992), failed to show clinical significance due to limited efficacy and their own dose-related toxicity or profound alterations in pharmacokinetics when used in combination with anticancer drugs. The more recently developed modulators possess a higher affinity for Pgp; however, their efficacy is still under clinical evaluation. Examples of this class include the cyclosporin A analog PSC 833 (Keller et al., 1992), the acridonecarboxamide GF120918 (Hyafil et al., 1993), LY335979 (Slate et al., 1995), the triazinoaminopiperidine derivative S9788 (Merlin et al., 1995), a yohimbine analog, trimethoxybenzoylyohimbine (Pearce et al., 1989), and other compounds, including MS-073 (Sato et al., 1991) and the R-isomer of verapamil (Toffoli et al., 1995).
To assess the capacity of a drug to inhibit Pgp-mediated drug efflux from multidrug-resistant cells, a sensitive and reliable method must be developed. Previously, functional flow cytometry assays using a fluorescent marker (rhodamine) have been used to evaluate the inhibitory potential of Pgp modulators in cells expressing Pgp. It is recognized that test compounds affect the active Pgp-mediated transport of rhodamine 123 and daunorubicin (DNR) differently as there are multiple binding sites on Pgp for the substrates with one selective for rhodamine 123 (Shapiro and Ling, 1997). In this study, DNR was used as a fluorescent marker and vanadate as a positive control agent. Use of the positive control (inactivator) made it possible to quantitatively assess the potency and efficacy of known Pgp inhibitors and unknown test drugs to inhibit the Pgp-mediated DNR efflux. Therefore, this method provides a more rigorous means to evaluate the potential and efficiency of drugs as inhibitors of the Pgp transporter as well as offer quantitation.
Materials and Methods
Chemicals.
DNR, verapamil, vinblastine, colchicine, quinidine, progesterone, gramicidin, nicardipine, α-tocopherol, EGTA, EDTA, TRIZMA base, HEPES, and cyclosporin A were purchased from Sigma Chemical Co. (St. Louis, MO). Hanks' balanced salt solution, α-minimum essential medium (α-MEM), Dulbecco's modified Eagle's medium, penicillin/streptomycin, fetal bovine serum (FBS), and trypsin-EDTA were obtained from Life Technologies, Inc. (Rockville, MD). Sodium orthovanadate was purchased from Pfaltz & Bauer Inc. (Waterbury, CT). Tetraphenylphosphonium (TPP) was obtained from Aldrich Chemical Co. (Milwaukee, WI). Microplates (Costar 96-well), plastic tubes, and cell culture flasks (75 cm2) were purchased from Corning Inc. (Corning, NY). All other reagents were of the highest grade commercially available.
Tumor Cells.
CR1R12 cell line, provided by Dr. Alan Senior (University of Rochester, Rochester, NY), was maintained in complete α-MEM supplemented with 10% FBS, penicillin/streptomycin 50 U/50 μg/ml in a 5% CO2/95% air atmosphere at 37°C. Colchicine (0.5 μg/ml) was added to the culture medium. Cells were grown to 80 to 90% confluency and treated with trypsin-EDTA before subculturing.
Fluorescence-Activated Cell Sorter (FACS) Flow Cytometry.
Fluorescence measurements of individual cells were performed with a Becton-Dickinson FACScalibur (San Jose, CA) equipped with an ultraviolet argon laser (excitation at 488 nm, emission at 530/30 and 570/30 nm band-pass filters). Analysis was gated to include single cells on the basis of forward and side light-scatter and was based on acquisition of data from 10,000 cells. Log fluorescence was collected and displayed as single parameter histograms.
A direct functional assay for the Pgp efflux pump in CR1R12 cells was performed with the flow cytometer. When DNR, a fluorescent substrate, is diffused into the cell, Pgp actively pumps out the fluorochrome. If another compound that is a substrate and/or inhibitor is presented in the same manner along with the fluorescence marker, the fluorescent marker accumulates in the cell, resulting in a higher intensity of fluorescence. Compounds that impede the efflux of the fluorescent substrate markers can thus be quantitatively analyzed and compared with respect to their potency to inhibit the Pgp efflux pump.
Cell Incubation Assays.
On the day of the experiment, the cell media was replaced with fresh 10% FBS α-MEM containing no Pgp inducers 60 min before cell detachment with trypsin-EDTA. Cells with 80 to 90% confluency were routinely used for the transport studies. After the media was removed, trypsin-EDTA was used to detach the cells from the plates. The cells were centrifuged (50g for 3 min) at room temperature and resuspended in 10% FBS α-MEM at a concentration of 6,000,000 cells/ml. Aliquots (50 μl) of the cell suspension were transferred to plastic tubes containing 1.95 ml of incubation media containing DNR. In the accumulation phase, cells were incubated in the presence or absence of test drugs in 10% FBS α-MEM with 0.1 to 5 μM DNR for 30 min at 37°C. After centrifugation (50g for 3 min at 4°C) and removal of supernatant, the cells were reincubated in 10% FBS α-MEM in the presence or absence of modulators without DNR for an additional 30 min at 37°C (the efflux phase). After final centrifugation (50g for 3 min at 4°C), the supernatant was removed. Cold Hanks' buffer was then added to each tube, and the cell suspension was transferred to FACS tubes, which were placed on ice (less than 1 h) until analysis.
Cell Viability Test.
Cell viability was tested with 0.4% trypan blue and then propidium iodide staining. Dead cells in which propidium iodide was bound to double strands of DNA or RNA were detected in certain regions of cytometry dot plots and were not included in the final data calculations.
Calculation of Relative Fluorescence Channel Numbers.
The DNR fluorescence intensities of individual cells are recorded as histograms. The mean fluorescence intensity of 10,000 cells is used for comparison among different conditions. Vanadate was selected as a positive control and standard compound because it can maximally inactivate the Pgp efflux pump. Figure 3 shows vanadate (5 mM) reaching maximal geometric mean fluorescence for DNR retention. This concentration was therefore used in each set of experiments. Relative fluorescence was used for quantitation and comparison between different compounds. The relative fluorescence (percent inactivation) represents a ratio obtained through the following formula: the geometric mean (because the data is acquired as a logarithm) fluorescence of a discrete sample divided by the geometric mean fluorescence in the presence of 5 mM vanadate, times 100 or expressed as:

Effect of vanadate on intracellular retention of DNR in CR1R12 cells.
Graph represents fluorescent intensity expressed as relative fluorescence (% inhibition) versus concentrations of vanadate.
Results
DNR Dose-Dependent Fluorescence Intensity in CR1R12 Cells.
Three concentrations (0.1, 1, and 5 μM) of DNR were tested in viable CR1R12 cells for their fluorescence intensity at the end of a 30-min accumulation phase, at the end of a 30-min efflux phase, and at the end of a 30-min efflux phase with 1 μM verapamil. When a DNR concentration of 0.1 μM was incubated with the CR1R12 cells in media (Fig. 1a), the difference of fluorescent intensities among accumulation and efflux incubations were not significant; only in the presence of inhibitor (1 μM verapamil) was there a significant difference. When the DNR concentration was increased to 1 μM (Fig. 1b), there was a marked difference between the accumulation and efflux phases. The geometric means of fluorescent intensity for efflux, accumulation, and verapamil are 59, 109, and 219 in 1b, respectively. The separation was even greater at 5 μM DNR (Fig. 1c). These results indicated that a concentration of between 1 and 5 μM DNR provides reasonable fluorescence to distinguish differences, thereby making this range suitable for additional inhibition studies. Additionally, this indicates that as much as 5 μM was not cytotoxic over the duration of the experiment based on the ability of the cell to retain DNR. However, to avoid potential cytotoxic effects, a concentration of 2 μM was considered optimal. This is very conservative as the ED50 for DNR is 26.5 μM in this MDR cell line (Al-Shawi and Senior, 1993; The ED50 is the concentration of drug required to reduce colony formation to 50% of that observed in the absence of the drug.).
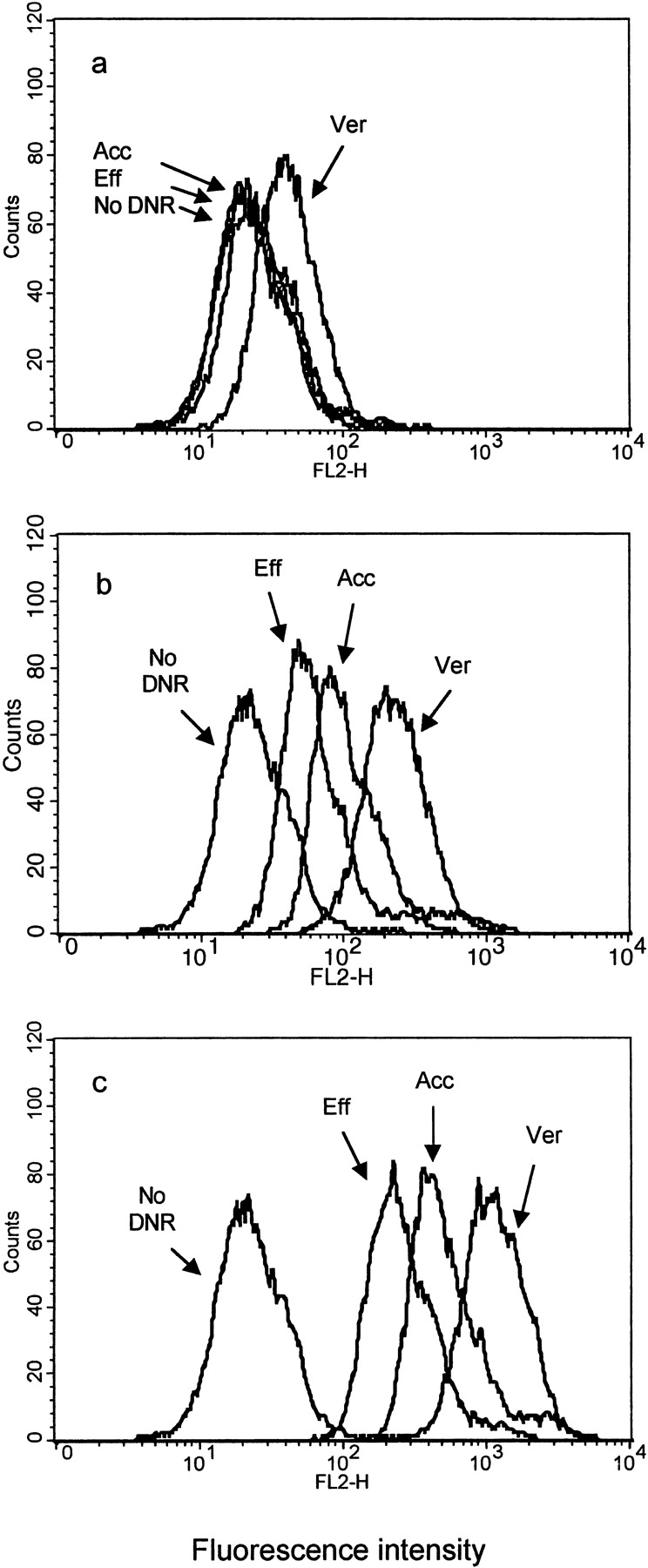
Intracellular retention of DNR from multidrug-resistant CR1R12 cells in the presence of 0.1 μM DNR (a), 1 μM DNR (b), and 5 μM DNR (c).
Cells were treated as described in Materials and Methods before the flow cytometry analysis. Histograms represent the cell counter numbers versus fluorescent intensity expressed as log relative fluorescence. In each figure, four overlaid histograms represent, from left to right, the peak from autofluorescent control cells without DNR (No DNR), the peak from 30-min efflux cells (Eff), the peak from 30-min accumulation cells (Acc), and the peak from cells coincubated with 1 μM verapamil (Ver). The geometric means of fluorescent intensity for No DNR, Eff, Acc, and Ver are 22.54, 23.52, 27.11, and 40.88, respectively, in 1a; 22.54, 59.44, 108.98, and 218.74, respectively, in 1b; and 22.54, 251.00, 513.15, and 1104.05, respectively, in 1c.
Comparison of Effects of Drug Influx and Efflux.
When DNR, a Pgp substrate, was coincubated with a Pgp inhibitor or test compound in the cell culture, the test compound might interact with Pgp and affect transport of DNR. During the accumulation phase, the concentration of DNR is higher outside the cell. Because the passive diffusion of DNR into cells and the Pgp-mediated efflux are in opposite directions, the inhibition of the Pgp pump caused a relative increase in DNR levels within the cells. In the efflux phase, the concentration of DNR is lower outside the cell, and thus the direction of the passive diffusion and the Pgp-mediated efflux is the same. When the Pgp pump is inhibited in the efflux phase, DNR exits the cell primarily by passive diffusion. As a result, DNR retention in the efflux phase (Fig. 2b) was lower compared with that in the accumulation phase (Fig. 2a) at given concentrations of verapamil. Increasing concentrations of verapamil caused a concentration-dependent increase in fluorescent intensity in both the accumulation and efflux phases with slightly greater separation during the efflux phase. Although DNR and verapamil are known to be substrates of the Pgp efflux enzyme, a nonPgp substrate fluorescent indicator was used to further test for a nonspecific effect on DNR retention by verapamil and the other test compounds. The presence of saturating inhibition concentrations (see below) of cyclosporin A (50 μM), quinidine (100 μM), verapamil (100 μM), ketoconazole (100 μM), terfenadine (25 μM), progesterone (500 μM), had no detectable effect on the retention of resorufin fluorescence, which has similar fluorescence qualities as DNR (data not shown).
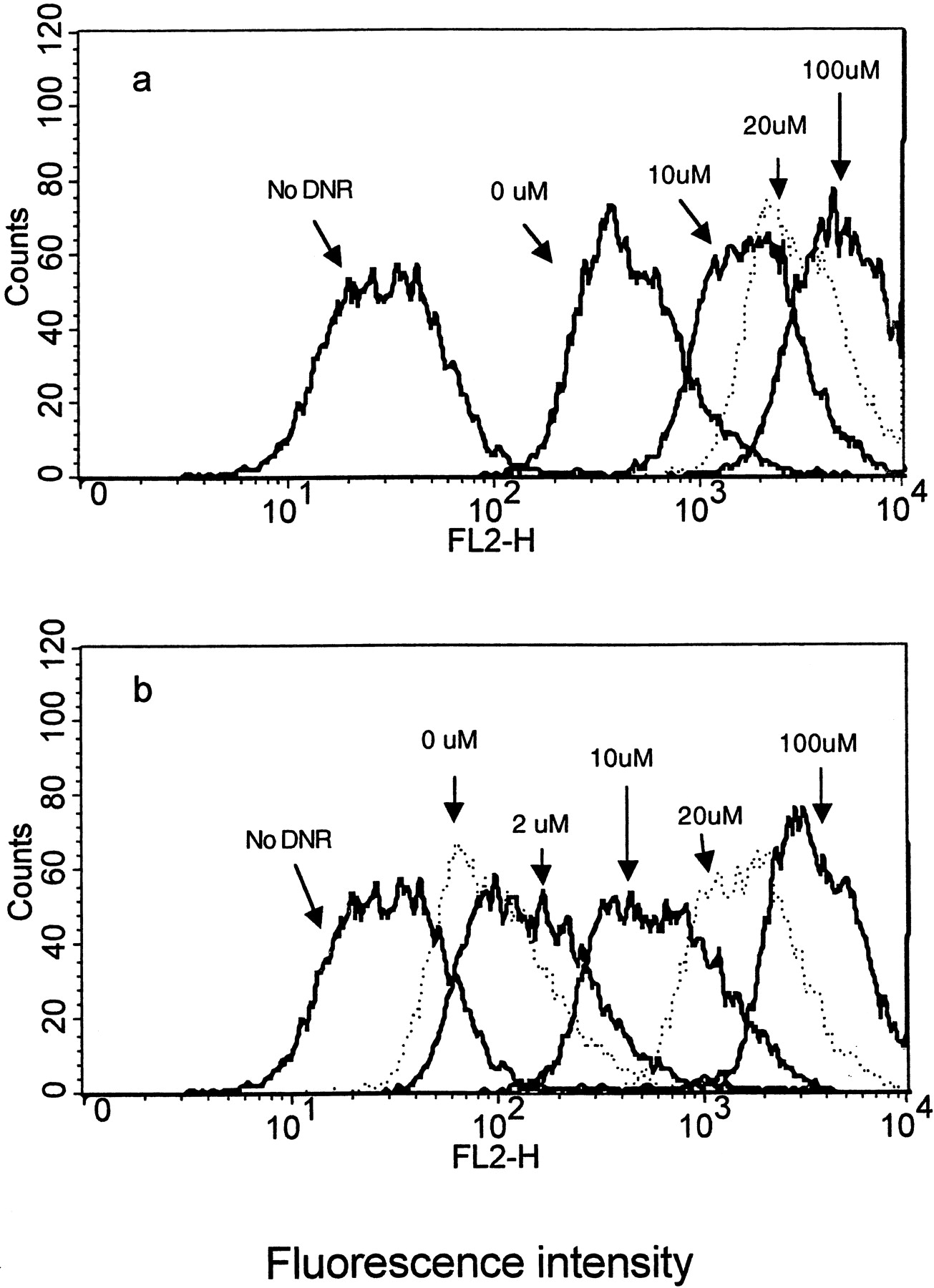
Effect of verapamil (0–100 μM) on intracellular retention of DNR in CR1R12 cells either in the accumulation phase (a) and/or in the efflux phase (b).
Histograms represent the cell counter numbers versus fluorescent intensity expressed as log relative fluorescence. The geometric means of fluorescent intensity for No DNR, 0, 10, 20, and 100 μM verapamil are 29.22, 459.91, 1738.99, 2986.73 and 4692.86, respectively, in 2a; for No DNR, 0, 2, 10, 20, and 100 μM are 29.22, 98.19, 144.83, 586.59, 1602.54, and 3324.83, respectively, in 2b.
Effects of Vanadate on Accumulation of DNR in CR1R12 Cells.
Vanadate is a competitive inhibitor of the Pgp-ATPase at the ATP binding site and, therefore, is distinct from other MDR1 transport site inhibitors. As shown in Fig. 3, vanadate caused a concentration-dependent increase in DNR retention in CR1R12 cells. Vanadate concentrations less than 100 μM had little effect on DNR retention. However, vanadate concentrations between 100 and 12,000 μM produced a sigmoidal DNR retention response curve, with maximal accumulation at ∼5 mM.
Time Dependence of DNR Influx and Efflux in CR1R12 Cells.
DNR diffused into CR1R12 cells very rapidly. Within 2 to 5 min, the accumulation of DNR reached a plateau as shown in Fig.4a. To assure maximal retention of DNR, subsequent experiments were allowed to incubate for 30 min. During the efflux phase (Fig. 4b), the time required to reach steady state with respect to DNR was between 10 and 15 min. For most of the assays, 30 min were allowed for DNR efflux.

Intracellular retention of DNR in CR1R12 cells in the accumulation phase (a) and in the efflux phase (b).
Potency and Efficacy of Known Pgp Inhibitors on DNR Efflux.
To examine the inhibition of the efflux of DNR via Pgp, a number of potential inhibitors were tested (Fig. 5, a–l). The efflux inhibition potency of each compound was compared with that of vanadate. In general, all compounds tested demonstrated only partial inhibition of efflux. Increasing the concentration of most inhibitors increased the intracellular retention of DNR. After reaching a point of maximal inhibition, the inhibitory effect decreased dramatically for most compounds at higher concentrations. For a limited number of compounds, a stable plateau was achieved at higher concentrations (verapamil, for example). Cyclosporin A and progesterone (Fig. 5, f and h) produced a significant inhibition, which was approximately 75 and 60% of that observed with vanadate, respectively. Verapamil and terfenadine achieved 40 to 50% inhibition relative to vanadate (Fig. 5, e and c). Vinblastine, quinidine, tamoxifen, and nicardipine produced 30 to 40% relative inhibition (Fig. 5, a, b, d, and i). Gramicidin (Fig. 5j) had a slight inhibitory effect (14%), and TPP and α-tocopherol (Fig. 5, k and l) had no significant inhibitory effect on DNR efflux.


Effect of drugs or chemicals on intracellular retention of DNR in CR1R12 cells.
Graphs showing fluorescent intensity expressed as relative fluorescence (% inhibition) in the presence of different concentrations of inhibitors.
Discussion
To be suitable for a functional flow cytometry assay, a Pgp marker must satisfy several criteria: 1) it should be primarily transported by the Pgp pump; 2) it should be a good fluorescent marker, and 3) its rate of passive diffusion should be slow compared with the rate of Pgp-mediated active transport. In earlier drug efflux functional assays, DNR was used for determination of cellular drug retention (Krishan and Ganapathi, 1980; Nooter et al., 1983; Krishan et al., 1987; Ross et al., 1995; Baggetto et al., 1998) and for evaluation of Pgp phenotypes in clinical samples (Chin-Yee et al., 1994;Maynadie et al., 1994; Gheuens et al., 1997). Rhodamine123 was introduced as a fluorescent marker due to its excellent fluorescent qualities and its seemingly general interaction with specific Pgp substrates (Lampidis et al., 1985). Other fluorochromes, including SY-38, SY-3150 (Frey et al., 1995), and SYTO16 (Broxterman et al., 1997), have also been used for monitoring drug retention or for assessment of Pgp function. Studies thus far have indicated that at least two substrate binding sites are present in the Pgp transporter, including those for rhodamine123 or H33442 (Shapiro and Ling, 1997) and for verapamil or adriamycin (Wang et al., 1998). The affinity of a Pgp modulator for these two sites varies. The DNR binding site may represent a unique site on the Pgp transporter. We have demonstrated that known Pgp inhibitors or substrates such as cyclosporin A, gramicidin, nicardipine, quinidine, progesterone, verapamil, terfenadine, and tamoxifen do affect the efflux of DNR. However, compounds such as TPP and α-tocopherol had little effect on the efflux of DNR, suggesting that these compounds may interact with other binding sites or have no effect on this Pgp transporter. DNR has several advantages as a fluorescence marker. Its cellular retention represents good evidence of the Pgp transporter being effectively blocked, and its fluorescent spectrum makes it suitable for this type of FACS assay. Concentrations of DNR between 1 and 5 μM provide reasonable fluorescence to distinguish differences in potency of inhibition for most Pgp inhibitors. Furthermore, an additional advantage is the intrinsic specificity. Drug candidates that selectively block the efflux of DNR will enhance the chemosensitivity to anthracyclines.
A typical ABC transporter protein consists of four units; two membrane-bound domains and two nucleotide-binding domains, which bind and hydrolyze ATP with similar efficiency (Loo and Clarke, 1995). As a member of the ABC superfamily, the functional activity of Pgp depends on energy released from ATP hydrolysis. Vanadate forms a complex with ADP, which traps both molecules in the ATP-binding site. This results in competitive inhibition of ATP binding. Because both nucleotide-binding domains must be functional for ATP hydrolysis by Pgp, trapping of vanadate at one of the nucleotide-binding domains is sufficient to completely block all ATPase activity (Urbatsch et al., 1995). Under our experimental conditions, 5 mM vanadate was sufficient to block active transport (inhibition reached saturation). The mean fluorescence at this concentration of vanadate represents complete inhibition (inactivation) of the pump and provides a value for comparison with other Pgp inhibitors. The disadvantage of vanadate is its nonspecific effect on the ATPase of other ABC transporters. Because observation in the presence of vanadate is merely a reference point to calculate the relative degree of inhibition for test compounds, potential nonspecific effects of vanadate will not affect the relationship of the normalized values among experiments with test compounds.
The multidrug-resistant CR1R12 cell line was obtained from the CHRC5 cell line, which in turn was developed from the Chinese hamster ovary cell line AUXB1. CR1R12 cells constitutively overexpress Pgp, which can represent up to 32% of plasma membrane protein weight (Al-Shawi and Senior, 1993), and are tolerant to DNR toxicity (ED50 = 26.5 μM). Because no visible 190-kDa protein band, which would indicate the presence of multidrug resistance associated protein (MRP), was found in a Coomassie Blue-stained gel and anti-Pgp immunoblot for the CR1R12 cell line, any minimal contribution by another transporter should not confound conclusions.
Our results indicated that the capacity of a drug to inhibit the Pgp transporter could be evaluated in both the accumulation and efflux phases. As a method to screen a large number of drug candidates, the accumulation assay provides a simple and efficient way to generate information. To quantitatively assess the potency of a drug candidate with respect to inhibition of the Pgp transporter, the combination of the accumulation and efflux assays is desirable.
Use of cell lines expressing different levels of Pgp may result in differences in the potency determined for related inhibitors. Quantitative values such as IC50 from CR1R12 cells could not be directly compared with those from other cell lines. When compounds are tested in the same cell system for the purpose of comparison, it is important to assess the relative potency of the inhibitors. This study demonstrates that most of the known MDR inhibitors are partial inhibitors of DNR efflux. Exposure to quinidine, nicardipine, vinblastine, and tamoxifen resulted in 33% inhibition of DNR efflux activity. Terfenadine and verapamil blocked 42% of DNR efflux. In contrast to previous reports, which indicated that TPP and α-tocopherol were Pgp modulators (Gros et al., 1992; Relling, 1996;Hall et al., 1999), these studies demonstrated that these compounds do not significantly affect the efflux of DNR. Among compounds tested to date, only cyclosporin A and progesterone achieved greater than 50% inhibition. Most of the inhibitors did not reach a level of 50% inhibition, and the effective concentrations were in the micromolar range. This may explain the limited clinical significance when these inhibitors are used in combination chemotherapy.
In conclusion, this study provides an improved method for assessing the potency of a candidate drug to inhibit the Pgp-mediated efflux pump. With DNR as a fluorescent marker and vanadate as a positive control compound, most known Pgp inhibitors were classified as partial, low-potency antagonists. A significant improvement in chemotherapy may need to include a search for more potent Pgp inhibitors.
Footnotes
-
Send reprint requests to: William W. Johnson, Ph.D., 144 Route 94, P.O. Box 32, Schering-Plough Research Institute, Lafayette, NJ 07848-0032. E-mail: William.W.Johnson{at}spcorp.com
- Abbreviations used are::
- MDR
- multiple drug resistance
- DNR
- daunorubicin
- Pgp
- P-glycoprotein
- ABC
- ATP-binding cassette
- TPP
- tetraphenylphosphonium
- α-MEM
- α-minimum essential medium
- FBS
- fetal bovine serum
- Received September 8, 1999.
- Accepted January 28, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}