


Abstract
The ability of modafinil to affect human hepatic cytochrome P450 (CYP) activities was examined in vitro. The potential for inhibition of CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, and CYP4A9/11 by modafinil (5–250 μM) was evaluated with pooled human liver microsomes. Modafinil exhibited minimal capacity to inhibit any CYP enzyme, except CYP2C19. Modafinil inhibited the 4′-hydroxylation ofS-mephenytoin, a marker substrate for CYP2C19, reversibly and competitively with a Ki value of 39 μM, which approximates the steady-stateCmax value of modafinil in human plasma at a dosage of 400 mg/day. No irreversible inhibition of any CYP enzyme was observed, and there was no evidence of metabolism-dependent inhibition. The potential for induction of CYP activity was evaluated by exposing primary cultures of human hepatocytes to modafinil (10–300 μM). Microsomes were then prepared and assayed for CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5 activities. The mean activities of microsomal CYP1A2, CYP2B6, and CYP3A4/5 from modafinil-treated hepatocytes were higher (up to 2-fold) than those in the solvent-treated controls but were less than those produced by reference inducers of these enzymes. At high concentrations of modafinil (≥100 μM), the mean activity of CYP2C9 was decreased (up to 60%) relative to that in the solvent controls. Overall, modafinil was shown to have effects on human hepatic CYP1A2, CYP2B6, CYP2C9, CYP2C19, and CYP3A4/5 activities in vitro. Although effects obtained in vitro are not always predictive of effects in vivo, such results provide a rational basis for understanding drug-drug interactions that are observed clinically and for planning subsequent investigations.
Modafinil (dl-2-[(diphenylmethyl)sulfinyl]acetamide; Fig.1), a nonamphetamine-like wakefulness-promoting agent, has recently been approved in the United States, United Kingdom, and Ireland (under the tradename Provigil) for the treatment of excessive daytime sleepiness associated with narcolepsy. The compound, which was discovered by Laboratoire L. Lafon (Boivin et al., 1993), is already on the market in France (under the tradename Modiodal).

Structure of modafinil.
In clinical use, modafinil is likely to be administered in combination with other medications, and an understanding of the potential for drug-drug interactions is therefore very important. Such interactions could be pharmacokinetic or pharmacodynamic in nature, or both. The present study focused on potential pharmacokinetic interactions arising from inhibition or induction of cytochrome P450 drug-metabolizing enzymes by modafinil.
In a previous in vitro study in primary human hepatocytes (Moachon et al., 1996), modafinil was evaluated for its ability to induce the activities of cytochrome P450 enzymes, including ethoxyresorufinO-deethylase, pentoxyresorufin O-dealkylase,S-mephenytoin 4′-hydroxylase, dextromethorphanO-demethylase, nifedipine oxidase, and lauric acid hydroxylase. At concentrations (10 and/or 100 μM) approximating those achieved clinically, modafinil was found to induce ethoxyresorufinO-deethylase and nifedipine oxidase activities in human hepatocytes, although the extent of induction observed was less than that obtained in mice, rats, or dogs and less than those produced by known inducers of these cytochrome P450 enzymes. At the highest concentration tested (1000 μM), more pronounced changes were observed in the activities of ethoxyresorufin O-deethylase (increased), dextromethorphan O-demethylase (increased), and nifedipine oxidase (decreased) relative to those in the solvent-treated controls. The activity of S-mephenytoin 4′-hydroxylase was decreased at all concentrations of modafinil. The conclusion from this study was that ethoxyresorufin O-deethylase [cytochrome P450 (CYP)21A] and nifedipine oxidase (CYP3A) activities were slightly induced by modafinil, which also increased the activity of dextromethorphan demethylase (CYP2D6) at the highest concentration tested (1000 μM).
Drawing definitive conclusions from these results was complicated by the substantial degree of intersubject variability that was observed and by the fact that modafinil was probably still present in the hepatocytes during the assay of enzymatic activities. In addition, the highest concentration tested (1000 μM) was substantially higher than the aqueous solubility of modafinil and was well above the concentration range obtained clinically (Wong et al., 1999).
A second in vitro induction study in human hepatocytes was therefore conducted to test whether the earlier results could be confirmed in a different laboratory, using a somewhat different and extended experimental design. In addition, the ability of modafinil to inhibit cytochrome P450 enzymes was studied in vitro in human liver microsomes (HLMs). The results of these latter two in vitro studies form the basis for the present communication.
Materials and Methods
Chemicals, Enzymes, and Antibodies.
Modafinil was supplied by Cephalon, Inc. (West Chester, PA). Rifampin, α-naphthoflavone, nicotine, quinidine, 4-methylpyrazole, ketoconazole, baccatin, and 8-methoxypsoralen were purchased from Sigma Chemical Co. (St. Louis, MO). 7-Ethoxy-4-trifluoromethylcoumarin (EFC) was obtained from Molecular Probes (Junction City, OR). Paclitaxel and 6α-hydroxypaclitaxel were obtained from Hauser Chemical Co. (Boulder, CO) and Gentest Corp. (Woburn, MA), respectively.S-Mephenytoin, (±)-4′-hydroxymephenytoin, and hydroxymethyltolbutamide were purchased from Ultrafine Chemicals (Manchester, England). Furafylline was obtained from Research Biochemicals Inc. (Natick, MA). Hexobarbital was purchased from Sterling-Winthrop (Rensselaer, NY). Sulfaphenazole was obtained from Ciba-Geigy Ltd. (Basel, Switzerland). Troleandomycin was obtained from Pfizer, Inc. (Brooklyn, NY). Sources of other chemicals, including culture media components, were as specified by Pearce et al. (1996a)and Madan et al. (1999).
HLMs (individual livers), previously assayed for their activities of cytochrome P450 enzymes, were provided by XenoTech, L.L.C. (Kansas City, KS). cDNA-expressed enzymes were purchased from Gentest Corp. (Woburn, MA).
Purified polyclonal antibodies (rabbit-derived) for CYP1A1/CYP1A2, for CYP2B6, and for CYP3A4/CYP3A5 and monoclonal antibodies (mouse-derived) for CYP2A6/CYP2C8/CYP2C19, used in Western immunoblotting analysis, were commercial products provided by XenoTech, L.L.C.
Human Hepatocytes.
Hepatocytes were isolated from human liver tissue obtained as surgical waste or from rejected donor livers via a modification of the two-step collagenase digestion method (Seglen et al., 1980; Quistorff et al., 1989; LeCluyse et al., 1996). Briefly, human liver tissue was perfused with pH 7.4 buffer containing 118 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 25 mM NaHCO3, 5.5 mM glucose, and 0.5 mM EGTA, followed by the same buffer lacking EGTA but containing 1.5 mM CaCl2 and 0.2–0.5 mg/ml collagenase. Viability (trypan blue exclusion) was ≥70% for all preparations used.
Enzyme Inhibition.
Direct inhibition.
Modafinil was incubated with HLMs (pool of seven subjects) at concentrations up to 250 μM. Significantly higher concentrations could not be tested due to the limited aqueous solubility of the compound. Modafinil was added in DMSO (final concentration, 0.1%), except for assay of CYP2E1, which is strongly inhibited by DMSO. For CYP2E1, modafinil was dissolved directly in the buffer mixture.
The substrates used for each enzyme and the concentrations tested (representing Km/2,Km, and 4Km) are presented in Table 1. A reference inhibitor for each enzyme, when available, was also included as a positive control.
Summary of the cytochrome P450 enzyme assay conditions
Incubations without DMSO solvent served as additional negative controls. The effects of the DMSO were minimal, except on 6β-hydroxylation of testosterone (CYP3A4/5), whose rates were decreased by up to 19% in the DMSO control. This extent of inhibition was not considered sufficient to compromise the results with modafinil.
Metabolism-dependent (mechanism-based) inhibition.
To test for reversible inhibition, HLMs (two individual samples and a pool of seven) were preincubated with modafinil plus NADPH for 15 min before the addition of the substrate to start the assay. The concentrations of modafinil and of the substrates investigated are summarized in Table 1. Solvent controls, containing all components except modafinil, were also examined.
To test for irreversible inhibition, modafinil was incubated for 15 min with HLMs as was done for reversible inhibition but at 10- to 20-fold higher protein concentrations. Before assay, each microsomal mixture was diluted 10- to 20-fold to reduce any effects from reversible inhibition by modafinil or its metabolites.
Enzyme Induction.
Freshly isolated human hepatocytes were cultured according to the method described by LeCluyse et al. (1994, 1996). Approximately 3 × 106 cells were added to 60-mm culture dishes coated with collagen and allowed to attach for 2 to 3 h. Unattached cells were aspirated, and serum-free modified Chee's medium containing 0.1 μM dexamethasone, 1% insulin-transferrin-selenium premix, and 0.25 mg/ml Matrigel was added. The cells were then maintained in culture for ∼3 days, with daily changes of medium, before the initiation of experiments. Only preparations that contained morphologically normal hepatocytes, as evaluated by phase contrast light microscopy, without significant contamination from other cell types were treated with modafinil or reference inducers.
Hepatocytes were treated with modafinil at varying concentrations or with β-naphthoflavone (33 μM), phenobarbital (250 μM), or rifampicin (50 μM) to serve as positive controls. The final concentration of the solvent (DMSO) in the medium was 0.1%. Treatment was continued for 3 days, with daily renewal of the medium plus the test drug or reference compound.
Before harvest, hepatocytes were photographed to document the status of the cells after treatment. Approximately 24 h after the final treatment, the hepatocytes were rinsed and collected, and microsomes were prepared (Madan et al., 1999). Resuspension was in 0.25 M sucrose at a protein concentration of 1 to 10 mg/ml (BCA Protein Assay Kit; Pearce Chemical Co., Rockford, IL), with storage at −80°C, to preserve the activity of the cytochrome P450 enzymes (Pearce et al., 1996a).
Assay Procedures.
Assays were performed as described in detail by Pearce et al. (1996a)or as described later. In each assay described later, the reaction mixture volume was 1 ml, containing 50 μg of microsomal protein, and the reactions were carried out at 37°C.
The equipment used for HPLC analysis in the paclitaxel and diclofenac assays included a Shimadzu LC-6A or LC-10A binary gradient HPLC system with an SIL-6A or SIL-10A autosampler and an SPD-6A or SPD-10A variable-wavelength UV detector. The column for both assays was a Supelcosil LC-18 reverse phase octyldecylsilane column (5 μm particle size; 4.6 mm i.d. × 15 cm) preceded by a Supelcosil LC-18 guard column (40 μm particle size; 4.6 mm i.d. × 2 cm) (Supelco, Bellefonte, PA). Column temperature was maintained at 30 ± 1°C with a CH-30 column heater controlled with a TC-50 temperature controller (Eppendorf, Inc., Madison, WI).
The O-dealkylation of EFC was measured using a modification of the fluorophotometric method of Buters et al. (1993). EFC (25 μM) was added in 5 μl of DMSO. Reactions proceeded for 5.0 min and were stopped by the addition of 2.0 ml of ice-cold acetone. Precipitated protein was removed by centrifugation. Concentrations of 7-hydroxy-4-trifluoromethylcoumarin in the supernatant were determined with a Shimadzu RF-540 spectrofluorometer (λex= 410 nm; λem = 510 nm). Zero time incubations served as blanks, and blanks spiked with 20 to 1000 pmol of 7-hydroxy-4-trifluoromethylcoumarin served as standards.
The oxidation of paclitaxel was monitored by reverse phase HPLC, based on the method described by Richheimer et al. (1992) and Cresteil et al. (1994), with slight modifications. Paclitaxel (10 μM) was added in 10 μl of acidic methanol. Reactions were started by the addition of an NADPH-generating solution and were stopped after 60 min by the addition of 5.9 ml of dichloromethane. Zero time incubations served as blanks, and blanks spiked with 120 to 1200 pmol of 6α-hydroxypaclitaxel (added in 40 μl of methanol), as standards. Each sample was spiked with 3 nmol of the internal standard, baccatin (in 100 μl of dichloromethane), and vigorously mixed on a batch vortexer. After the two phases were separated by low-speed centrifugation, the aqueous (upper) phase was aspirated and discarded. An aliquot (4 ml) of the organic phase was transferred to a culture tube and evaporated in a Speed-Vac concentrator (Savant Instruments, Farmingdale, NY). The residue was redissolved in 200 μl of mobile phase, and a 50-μl aliquot was analyzed by HPLC. The isocratic mobile phase was water/acetonitrile 60:40 (v/v), at a total flow rate of 1.5 ml/min. Eluates were monitored at 235 nm. Total analysis time was 20 min/run, and the retention times for baccatin, 6α-hydroxypaclitaxel, and paclitaxel were ∼3.4, ∼8.6, and ∼15.0 min, respectively. Paclitaxel and 6α-hydroxypaclitaxel were quantified by peak area compared with authentic standards, with correction for variation in extraction efficiency based on recovery of the internal standard.
The 4′-hydroxylation of diclofenac (100 μM) was measured by reverse phase HPLC, based on the method described by Leemann et al. (1993). Reactions were started by the addition of an NADPH-generating system and were stopped after 30 min by addition of 1.0 ml of methanol. Precipitated protein was removed by low-speed centrifugation, and a 400-μl aliquot of the supernatant fraction was analyzed by HPLC. Zero time incubations served as blanks, and blanks spiked with 50 to 1000 pmol of 4′-hydroxydiclofenac (added in 20 μl of methanol) served as standards. The isocratic mobile phase was 20 mM potassium phosphate buffer (pH 7.0)/acetonitrile 75:25 (v/v), at a total flow rate of 1.0 ml/min. Total analysis time was 15 min/run, and the retention times of 4′-hydroxydiclofenac and diclofenac were ∼3.7 and ∼11.0 min, respectively. Eluates were monitored at 282 nm. 4′-Hydroxydiclofenac was quantified using peak area compared with authentic standards.
Western Immunoblotting Procedures.
CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, and CYP3A4 in the hepatocyte-derived microsomes were analyzed via Western immunoblotting using essentially the procedures described by Madan et al. (1999).
Data Analysis.
All incubations for enzyme activity assays were in duplicate; the data reported are averages of those duplicate determinations.
The results of the inhibition study were analyzed by Dixon and Eadie-Hofstee plots to determine the type of inhibition and the value of the inhibitory constant (Ki). If an enzyme was not inhibited at the highest concentration of modafinil tested, an estimated minimum value of Kiwas calculated using the following equation for competitive inhibition (Todhunter, 1979):
To determine significant differences between group mean values in the induction study, data were first determined to be parametrically distributed; then, a one-way ANOVA test for repeated measures was carried out. When significance (P < .05) was observed, a Dunnett's post hoc test was used to identify the group mean values that were significantly different from the controls (P< .05).
Results
Inhibition of Human Cytochromes P450.
The rates of metabolism of marker substrates for nine cytochrome P450 enzymes (Table 1) were determined in HLMs (pool of seven) in the presence and absence of modafinil (5–250 μM). The presence of modafinil had minimal effect on any reaction except the 4′-hydroxylation of S-mephenytoin (CYP2C19), which was reversibly inhibited with a Ki value of ∼39 μM (Fig. 2). This concentration approximates the steady-state Cmax value of modafinil in human plasma at a dosage of 400 mg/day (Wong et al., 1999). The sample-to-sample variation in the inhibition of CYP2C19 was determined with 10 samples of individual HLMs at substrate concentrations equal to Km and modafinil concentrations equal to ∼2Ki. As expected, modafinil produced substantial inhibition (∼50%) of CYP2C19 in all microsomal samples (Fig.3), consistent with its ability to function as a competitive inhibitor.

Evaluation of modafinil as a reversible inhibitor of S-mephenytoin 4′-hydroxylase (CYP2C19) activity in pooled HLMs.
Dixon and Eadie-Hofstee plots of the rates of S-mephenytoin 4′-hydroxylation by pooled HLMs. The microsomes were incubated withS-mephenytoin for 30 min at concentrations of the substrate that were approximately equal to Km/2,Km, and 4Km and in the absence (solvent control; 0.1% DMSO) or presence of modafinil at concentrations of 5, 25, 100, or 250 μM.

Sample-to-sample variation in the inhibition of S-mephenytoin 4′-hydroxylase (CYP2C19) activity in HLMs by modafinil.
The rates of S-mephenytoin 4′-hydroxylation were measured in the presence and absence (solvent control; 0.1% DMSO) of modafinil in individual 30-min incubations of microsomes prepared from nine different human livers. The substrate concentration was equal toKm, and the concentration of modafinil, when present, was 75 μM.
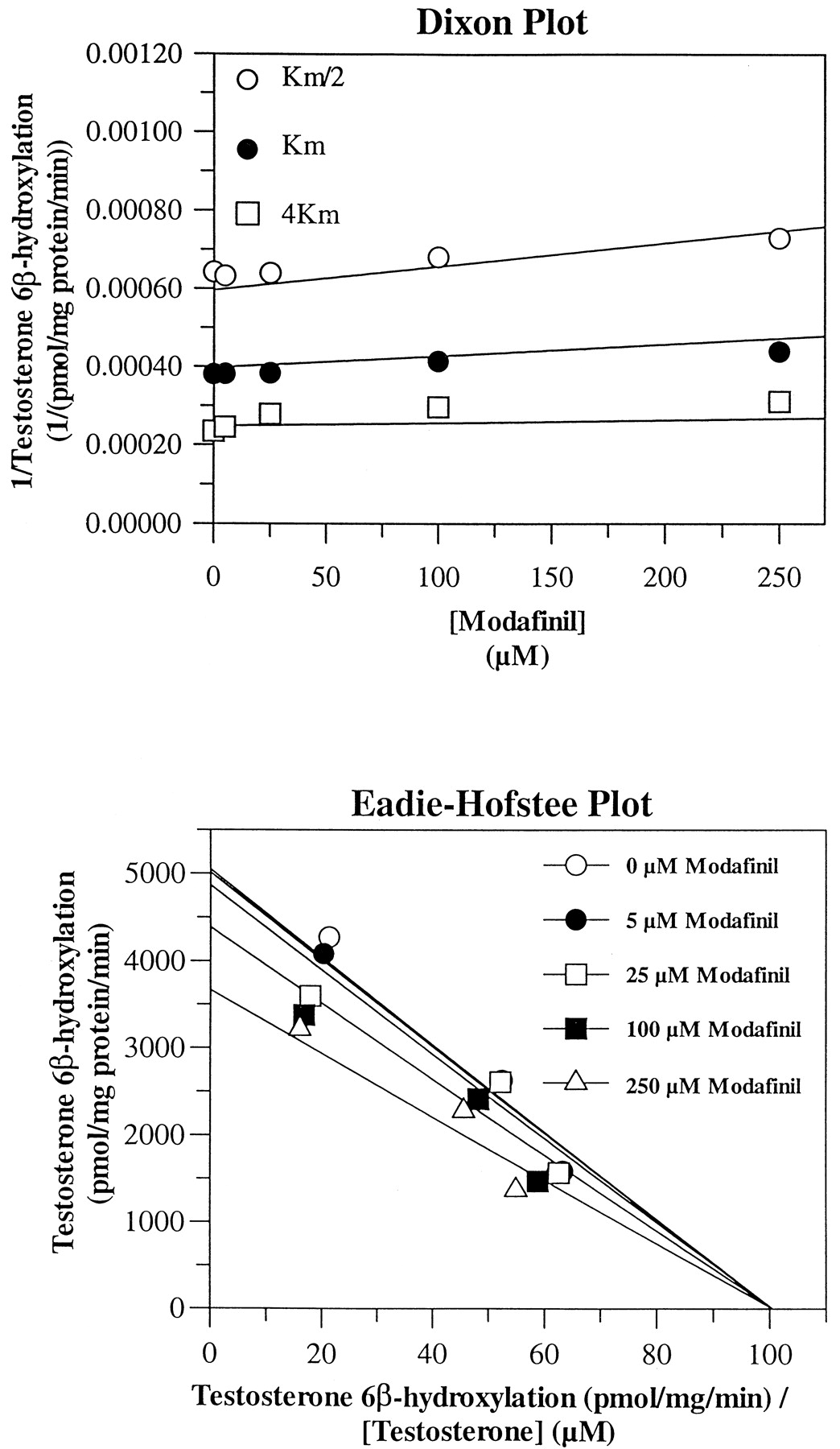
In addition, CYP3A4/5 and CYP2B6 were weakly inhibited. 6β-Hydroxylation of testosterone (CYP3A4/5) was uncompetitively inhibited with a Ki value of ∼632 μM (Fig. 4), and O-dealkylation of EFC (CYP2B6) was noncompetitively inhibited with aKi value of ∼1200 μM (data not shown). However, these Ki values are both much higher than the concentrations of modafinil attained clinically (Wong et al., 1999). The other cytochrome P450 activities appeared to be unaffected (Ki > 1500 μM) by the presence of modafinil at the concentrations tested.

Evaluation of modafinil as a reversible inhibitor of testosterone 6β-hydroxylase (CYP3A4/5) activity in pooled HLMs.
Dixon and Eadie-Hofstee plots of testosterone 6β-hydroxylation by pooled HLMs. The microsomes were incubated with testosterone for 8 min at concentrations of the substrate that were approximately equal to Km/2, Km, and 4Km and in the absence (solvent control; 0.1% DMSO) or presence of modafinil at concentrations of 5, 25, 100, or 250 μM.
The ability of metabolites of modafinil to affect the activities of hepatic drug-metabolizing enzymes was examined by preincubating the microsomes and cofactors with modafinil before the addition of each marker substrate and then comparing the rates of metabolism obtained under those conditions with the rates obtained when the substrate was introduced at the same time as modafinil. No reversible or irreversible metabolism-dependent inhibition was observed. A representative data set displaying the effect of modafinil on testosterone 6β-hydroxylase activity (CYP3A4/5) is shown in Fig. 5.

Evaluation of modafinil as a metabolism-dependent (mechanism-based) inhibitor of testosterone 6β-hydroxylase (CYP3A4/5) activity in HLMs.
HLMs (two individual samples and a pool of seven) were incubated with modafinil (250 μM) or with the solvent only (DMSO; final concentration, 0.1%) for 15 min under the assay conditions before starting the assay reaction by introduction of the marker substrate, testosterone (50 μM). After 8 min, the reaction was quenched, and the rate of formation of 6β-hydroxytestosterone was calculated. The rates without preincubation were then compared with those after incubation for both the modafinil-treated microsomes and the solvent controls. To test for reversible inhibition, the substrate was added to undiluted microsomal mixtures. To test for irreversible inhibition, the preincubation was conducted at microsomal protein concentrations that were 10- to 20-fold higher than those normally used in the CYP3A4/5 assay. Immediately before the addition of the substrate, these microsomal mixtures were diluted 10- to 20-fold to reduce any effect from reversible inhibition.
Induction of Human Cytochromes P450.
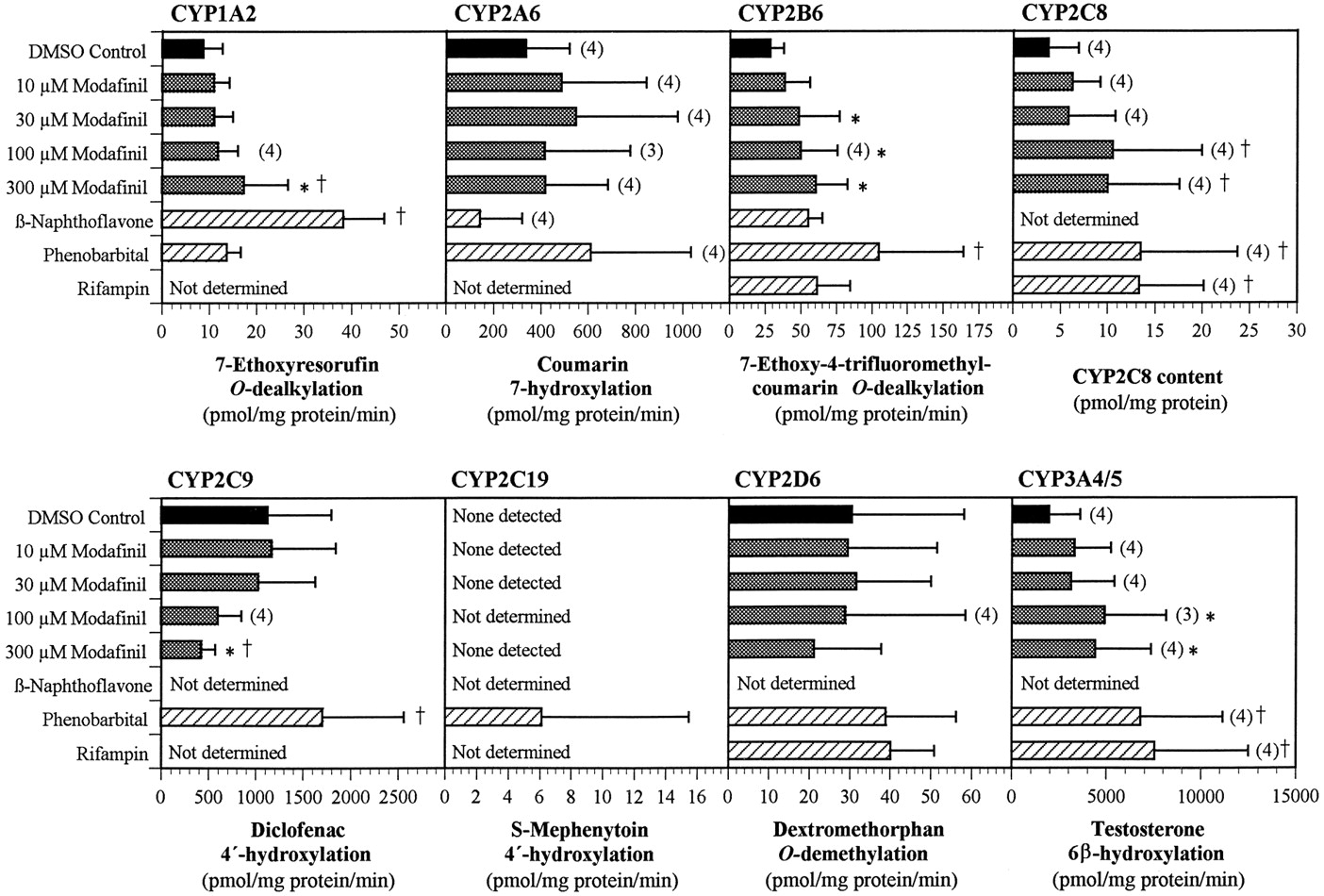
The ability of modafinil to induce cytochrome P450 activities was examined in vitro in primary cultures of human hepatocytes. After ∼3 days in culture, the hepatocytes were exposed to modafinil at concentrations of 10 to 300 μM for 3 additional days. The hepatocytes were then harvested, and microsomes were prepared and assayed for the activities of eight cytochrome P450 enzymes. The mean results are presented in Fig. 6. [Note: Due to low hepatocyte yield from one human liver (HL-5), the 100 μM treatment group was omitted for that liver to ensure that the other treatment conditions could be effectively evaluated.]
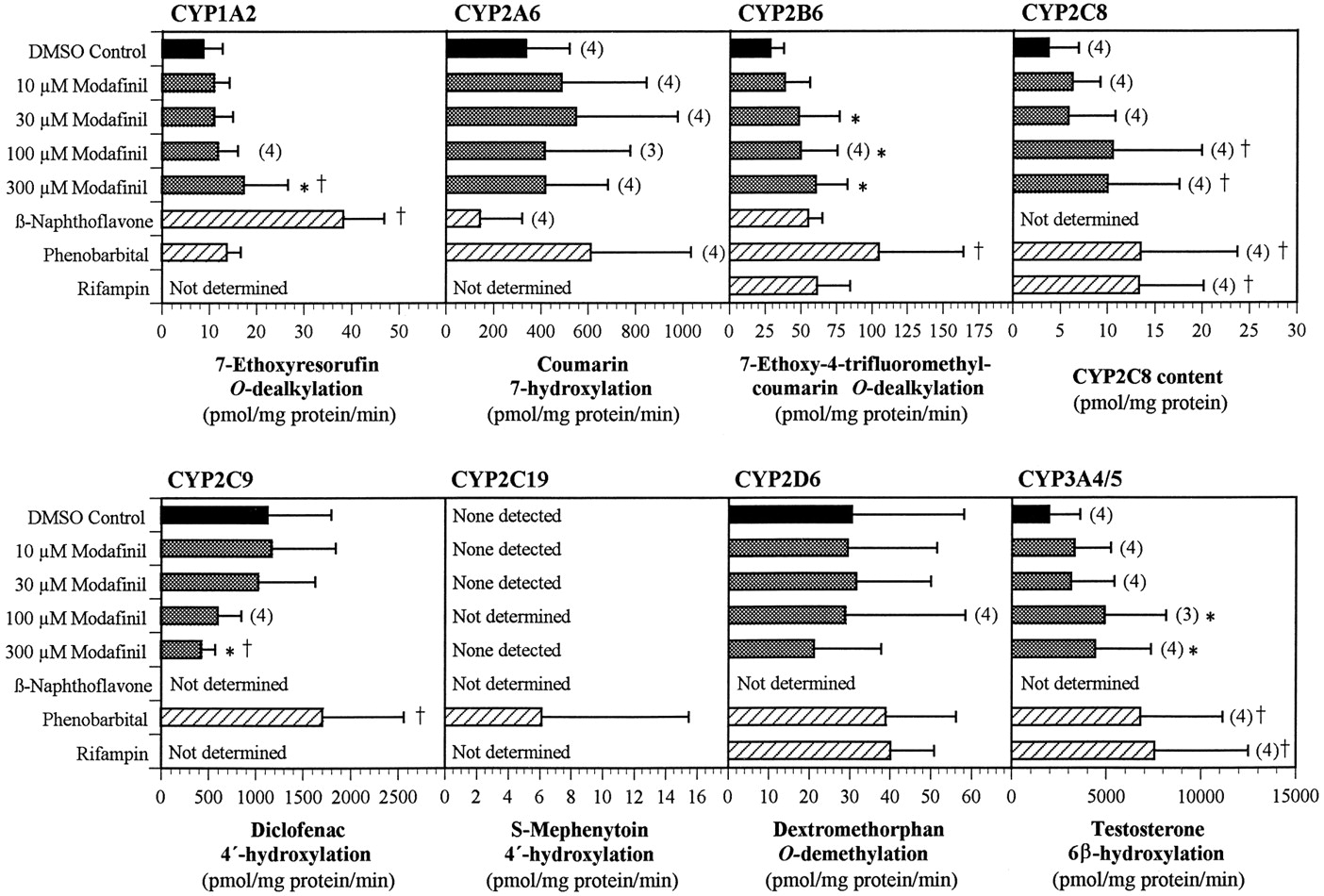
Effect of treating primary human hepatocyte cultures with modafinil and prototypical cytochrome P450 enzyme inducers on the activities of CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2D6, and CYP3A4/5 and on the protein level of CYP2C8.
Human hepatocytes from five donors (HL-1 to HL-5) were placed in culture for 3 days before the initiation of treatment with modafinil at 0 (solvent control), 10, 30, 100, or 300 μM for 3 days, with renewal of the medium and test article every 24 h. Additional culture dishes were similarly exposed to β-naphthoflavone (33 μM), phenobarbital (250 μM), or rifampicin (50 μM) and served as positive controls. The final concentration of the solvent (DMSO) in the medium was 0.1%. Microsomes were prepared for each human liver from each of the treatment groups. The enzymatic activity of each of the CYP enzymes was then determined for each liver and treatment group using marker substrates. In the case of CYP2C8, the rate of 6α-hydroxylation of paclitaxel, the marker substrate, was too low to be measured; hence, the levels of CYP2C8 protein (Western immunoblots) were used to assess induction. For each assay, there were five determinations per treatment group, unless otherwise noted above the bar. Values represent mean ± S.D. *P < .05 in the comparison of modafinil-treated samples with the corresponding DMSO controls. †P < .05 in the comparison of all treated samples with the corresponding DMSO controls.
No significant changes were observed in the activities of CYP2A6, CYP2C8, CYP2C19, or CYP2D6 (Fig. 6). However, the activities of CYP2C19 in the microsomes from modafinil-treated and solvent control hepatocytes were below the limits of detection of the assay; only the positive controls produced detectable levels of enzyme activity. Hence, an induction of CYP2C19 to levels less than those in the positive controls might not have been detected.
The enzymatic activity of CYP2C8 was also below the limit of detection in all microsomal samples, except those from one phenobarbital-treated control. However, CYP2C8 protein was detected in all of the microsomal preparations by Western immunoblotting. The levels of the protein were low relative to those in microsomes prepared directly from liver and were not consistent within or between the hepatocyte preparations and treatment groups. An apparent, slight increase in mean CYP2C8 protein concentration was observed at the two highest concentrations of modafinil, but the differences between the solvent control and modafinil-treated preparations were not statistically significant (P > .05). Although a weak induction of CYP2C8 by high concentrations of modafinil cannot be ruled out on the basis of these data, the likelihood of a substantive effect appears to be low.
The enzymatic activities associated with CYP3A4/5 and CYP1A2 were increased slightly, in a generally concentration-related manner, over those in the solvent-treated controls (Fig. 6). The activity of EFCO-deethylase, a marker for CYP2B6, was also increased, but this compound is also a substrate for human CYP1A2. Therefore, at least part of the increase in EFC O-deethylase activity reflected the induction of CYP1A2.
The activity of CYP2C9 (which was not examined in the previous study reported by Moachon et al., 1996) was decreased up to 60% in modafinil-treated hepatocytes relative to the activity in solvent-only treated cells (Fig. 6). The decrease was concentration-dependent but did not represent an appreciable change in activity except at concentrations of modafinil of ≥100 μM.
To determine whether the changes in enzyme activity were due to changes in enzyme concentration, Western immunoblotting was carried out with antibodies to CYP1A2, CYP2A6/2C8/2C19, CYP2B6, and CYP3A4/3A5. The results for CYP1A2 and CYP3A4 were consistent with those obtained by determination of enzymatic activity. A picture of two representative Western immunoblots of CYP3A4 protein, representing three of the hepatocyte preparations, is shown in Fig.7. In addition, the immunoblots showed modest induction of CYP2B6, whose level of induction could not be determined solely from the enzymatic assay.

Effect of treating primary cultures of human hepatocytes with modafinil on the level of CYP3A4 expression (Western immunoblotting).
Human hepatocytes from five donors (HL-1 to HL-5) were placed in culture for 3 days before initiation of treatment with modafinil at 0 (solvent control), 10, 30, 100, or 300 μM for 3 days, with renewal of the medium and test article every 24 h. Additional culture dishes were similarly exposed to β-naphthoflavone (33 μM), phenobarbital (250 μM), or rifampicin (50 μM) and served as positive controls. The final concentration of the solvent (DMSO) in the medium was 0.1%. Microsomes were prepared for each human liver from each of the treatment groups, and a portion of each was subjected to Western immunoblotting to measure the levels of immunoreactive CYP3A4 using a polyclonal antibody against rat CYP3A1. For comparison, HLMs prepared directly from the livers of two individual subjects (HLM15 and HLM16), plus a pool of microsomes, were also evaluated, in addition to known concentrations of cDNA-derived CYP3A4.
Discussion
In the present study, the ability of modafinil to inhibit or to induce cytochrome P450 enzyme activities was studied in human liver preparations. Such information is important for the design of effective treatment programs that will include administration of modafinil, because such enzymatic interactions can either enhance or diminish the effectiveness and/or safety of concomitant medications.
Cytochrome P450 inhibition by modafinil appears to be limited to CYP2C19, whose substrates and inhibitors have been extensively reviewed (e.g., Flockhart, 1995; Parkinson, 1996; Rendic and Di Carlo, 1997). Modafinil does not itself appear to be a substrate for CYP2C19, and there are a relatively small number of marketed pharmaceutical products that are predominantly or even largely metabolized by the enzyme. Examples are S-mephenytoin (Goldstein et al., 1994), omeprazole (Ko et al., 1997), lansoprazole (Pearce et al., 1996b), proguanil (Wright et al., 1995), diazepam (Jung et al., 1997), and propranolol (Ward et al., 1989). (Omeprazole and lansoprazole are also potent inhibitors of CYP2C19.) Caution should be exercised when initiating cotherapy with modafinil in patients receiving these medications, but with the possible exception of diazepam or other sedative benzodiazepines, they generally are not drugs with which modafinil is likely to be a frequent comedication.
However, as demonstrated by a recent report (Grözinger et al., 1998) of an apparent metabolic drug-drug interaction of modafinil with clomipramine, inhibition of CYP2C19 could, in special cases, also be important for compounds that are not normally considered to be significant substrates for the enzyme. In the case reported, the plasma concentrations of clomipramine were found to have increased, along with those of its pharmacologically active des-methyl metabolite, after the addition of modafinil as a comedication. Because clomipramine and des-methylclomipramine normally are largely eliminated through metabolism by CYP2D6 (Nielsen et al., 1992, 1996), a significant effect by an inhibitor of CYP2C19 was unexpected. However, the patient was subsequently determined to be CYP2D6-deficient (Grözinger et al., 1998), belonging to a subset of the human population who have no functional CYP2D6 enzyme (i.e., 7–10% of whites and equal or smaller portions of other ethnic groups;Eichelbaum, 1984; Setiabudy et al., 1994). In these “poor metabolizers” of CYP2D6 substrates, such as dextromethorphan, debrisoquine, and sparteine, the fractional contributions of alternative metabolic pathways for clomipramine anddes-methylclomipramine through CYP2C19 and other cytochrome P450 enzymes could assume substantially more important roles than would be the case in individuals with normal CYP2D6 activity.
The inhibition of CYP2C19 by modafinil would likely have minimal therapeutic consequences for patients at steady state for modafinil and for whom a tricyclic antidepressant would be prescribed as cotherapy, because the dosage of the antidepressant would generally be titrated to identify a safe and effective dose. However, these results would suggest that in patients at steady state for clomipramine or similar tricyclic antidepressants, the addition of modafinil as cotherapy may require a dosage reduction for the antidepressant, particularly in CYP2D6-deficient individuals.
Modafinil also caused an induction of cytochrome P450 activities in vitro in human hepatocytes. Three enzymes appeared to be induced by modafinil: CYP1A2, CYP3A4, and CYP2B6. [The results for CYP1A2 and CYP3A4 were generally consistent with results obtained previously (Moachon et al., 1996); CYP2B6 was not previously examined.] The extent of induction of each enzyme, although statistically significant at one or more concentrations of modafinil, was modest, especially in comparison with those produced by reference inducers, when available, and in comparison with interindividual variability.
No significant effect of modafinil treatment (10–300 μM) was observed in the activities of CYP2A6, CYP2C8, CYP2C19, or CYP2D6. The apparent suppression of CYP2C19 reported previously (Moachon et al., 1996) was likely due to inhibition of the enzyme by residual modafinil that remained in the hepatocyte preparations during the enzymatic assays. The lack of induction of CYP2D6 in the present study at concentrations of up to 300 μM is consistent with the previous results for all except the highest concentration tested in that study (i.e., 1000 μM). The reason for the increased enzymatic activity at that concentration is not known, but the concentration is far beyond those that have any clinical relevance.
Of the three cytochrome P450 enzymes apparently inducible by modafinil, CYP1A2 and CYP2B6 do not appear to be of major concern. CYP1A2 provides the primary metabolic pathway for relatively few pharmacologically important substrates, and these do not have narrow therapeutic indices (Tassaneeyakul et al., 1993; Brøsen, 1995; Bertz and Granneman, 1997). In addition, the extent of induction observed in the present study, or in the previous one (Moachon et al., 1996), was small relative to the ∼40-fold interindividual variability observed for this enzyme. In the case of CYP2B6, the activity of the enzyme is extremely low in human livers (Shimada et al., 1994), and it appears to contribute minimally to the metabolism of pharmaceutical products.
In contrast, CYP3A4 represents the largest single portion of the cytochrome P450 protein and activity in the human liver and plays a substantial role in the metabolism of a vast array of pharmaceutical products (Guengerich, 1995; Parkinson, 1996; Bertz and Granneman, 1997;Rendic and Di Carlo, 1997). Of particular concern are compounds that are predominantly or exclusively metabolized by CYP3A4 and also have a narrow therapeutic margin (e.g., cyclosporine A and steroidal contraceptives containing ethinyl estradiol).
As with the inhibition of CYP2C19, the apparently low degree of induction of CYP3A4 by modafinil would be most likely to produce clinical effects if modafinil were added as cotherapy to a patient already at steady state for a narrow-margin CYP3A4 substrate. A single case of apparent interaction has been reported in a patient in whom the effectiveness of treatment with cyclosporine A decreased after the addition of modafinil as a cotherapy (Le Cacheux et al., 1997). However, insufficient information is available to establish the cause of the effect.
Finally, the apparent suppression of CYP2C9 activity in human hepatocytes by treatment with modafinil is potentially important due primarily to one compound, warfarin (Coumadin), which has a narrow therapeutic index and whose more active enantiomer (S-warfarin) is primarily a substrate for CYP2C9 (Rettie et al., 1992). The origin of the suppressive effect obtained in vitro and its relevance to the clinical situation are not known, but the finding suggests caution in the initiation of treatment with modafinil in patients who are at steady state on warfarin.
In summary, modafinil has been demonstrated in vitro to be a moderately potent, reversible inhibitor of CYP2C19 in HLMs and a modest inducer of CYP1A2, CYP3A4, and CYP2B6 in vitro in human hepatocytes. In addition, CYP2C9 appeared to be suppressed in vitro in human hepatocytes after treatment with modafinil. Overall, these results suggest that there is potential for metabolic drug-drug interactions between modafinil and certain possible concomitant medications.
Due to the relatively low degree of alteration of the enzyme activities in vitro and to the concentrations of modafinil required to obtain appreciable effects, a high incidence of clinically significant interactions would not be expected. However, these in vitro results are being used in evaluation of clinical reports of apparent drug-drug interactions and in the design of subsequent studies targeted at further elucidation of the clinical relevance, if any, of these in vitro findings.
Acknowledgments
We gratefully acknowledge the contributions of Dr. David Stong, Kathy Carroll, Dan Mudra, Rick Graham, Jason Latham, Kevin Smith, and Alayne Burton to the success of this project.
Footnotes
-
Send reprint requests to: Dr Philmore Robertson, Jr., Department of Drug Safety and Disposition, Cephalon, Inc., 145 Brandywine Parkway, West Chester, PA 19380-4245. E-mail:proberts{at}cephalon.com
-
↵1 Current address: Department of Pharmacology and Physiology, University of Rochester, Rochester, NY 14642.
-
A preliminary report of this study was presented as a poster at the 12th International Symposium on Microsomes and Drug Oxidations, July 20–24, 1998, in Montpellier, France, Abstract 314.
- Abbreviations used are::
- CYP
- cytochrome P450
- EFC
- 7-ethoxy-4-trifluorocoumarin
- 7-ethoxyresorufin
- 7-ethoxyphenoxazone
- HLM
- human liver microsomes
- 6β-hydroxytestosterone
- 4-androsten-6β,17β-diol-3-one
- resorufin
- 7-hydroxyphenoxazone
- testosterone
- 4-androsten-17β-ol-3-one
- Received September 21, 1999.
- Accepted March 3, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}