


Abstract
Mibefradil, a calcium T- and L-channel blocker developed for use in hypertension, was recently removed from the market after reports of severe drug-drug interactions. Mibefradil is known to inhibit various cytochrome P450 enzymes involved in drug metabolism, particularly CYP3A. However, the extent and the severity of the observed drug interactions in humans suggest that inhibition of additional systems important to drug disposition, such as the drug transporter P-glycoprotein (P-gp), may also have contributed to the severity of the mibefradil interactions. A polarized epithelial cell line, LLC-PK1, which does not express P-gp, and the derived L-MDR1 cell line, which overexpresses human P-gp, were used to study the effects of mibefradil on drug transport. A markedly greater basal-to-apical versus apical-to-basal transport of [H3]mibefradil was seen in the L-MDR1, but not in the LLC-PK1 cells, suggesting that the drug is a substrate of P-gp. Using a human intestinal cancer-derived cell line Caco-2, which constitutively expresses P-gp, mibefradil was shown to be a potent inhibitor of P-gp-mediated digoxin transport, with an IC50 of 1.6 μM. Additionally, the effect of mibefradil on CYP3A was assessed using human liver microsomes. Mibefradil inhibited CYP3A-mediated nifedipine oxidase activity with an IC50 of 0.8 μM, and a Ki of 0.6 μM. Thus, mibefradil is not only a P-gp substrate, but also a potent inhibitor of both P-gp and CYP3A. These data suggest that the severity of drug interactions seen with mibefradil use is due to the dual inhibition of both P-gp and CYP3A.
When a drug enters widespread use after its approval, previously unexpected severe adverse drug interactions can occur. Severe interactions have been reported with the recently developed calcium channel blocker mibefradil (Posicor, Roche Laboratories), an antihypertensive agent that inhibits both T (transient) channel as well as the L (long) channel (Mullins et al., 1998). In the limited population studied before its release, this drug had appeared to be as efficacious and as safe as other currently available calcium channel blocking agents (Bursztyn et al., 1997; Kobrin et al., 1997). However, mibefradil was withdrawn from the market by its manufacturer after drug interactions resulted in increased plasma concentrations of coadministered drugs, including other calcium channel blockers, beta blockers, digoxin, cyclosporine, simvastatin, and tacrolimus (Siepmann et al., 1995;Krahenbuhl et al., 1998; Mullins et al., 1998; Spoendlin et al., 1998;Prueksaritanont et al., 1999). Although increases in plasma concentrations of some of these drugs can be explained by inhibition of drug metabolism, specifically CYP3A, such an explanation would be unlikely to account for the digoxin interaction as digoxin does not undergo significant metabolic biotransformation in humans. An alternative hypothesis is that mibefradil also inhibits membrane-bound drug transport systems such as the cellular efflux transporter P-glycoprotein (P-gp)1. Inhibition of P-gp produces an increase in intestinal absorption and a decrease in hepatic and renal excretion of its substrates, resulting in elevated plasma and organ drug levels, particularly in the brain (Schinkel et al., 1994, 1996). Therefore, the purpose of this study was to test the hypothesis that the severity of drug-drug interactions with mibefradil resulted from its dual inhibition of both drug metabolism (CYP3A) and drug transport.
Experimental Procedures
Materials.
[3H]Digoxin (15 Ci/mmol) was supplied by DuPont-New England Nuclear (Boston, MA), and [3H]mibefradil (specific activity, 15 Ci/mmol, >98% purity by HPLC) was a gift from Hoffmann-La Roche (Basel, Switzerland). L-MDR1 and LLC-PK1 cells were provided by Dr. A.H. Schinkel (Netherlands Cancer Institute, Amsterdam, the Netherlands) and Dr. E.G. Schuetz (St. Jude's Children's Research Hospital, Memphis, TN), and Caco-2 cells were obtained from Dr. R.J. Coffey (Vanderbilt University, Nashville, TN). All other chemicals and reagents, unless stated otherwise, were obtained from Sigma-Aldrich (St. Louis, MO) and were of the highest quality available.
Transport in Cultured LLC-PK1, L-MDR1, and Caco-2 Cells.
LLC-PK1 and L-MDR1 cells were cultured in M199 medium (Life Technologies, Gaithersburg, MD) supplemented with 50 μg streptomycin/ml and 10% v/v fetal calf serum at 37°C in the presence of 5% CO2; cells were subcultured after trypsinization every 6 to 7 days (Schinkel et al., 1995). Cells were grown as monolayers on polycarbonate membrane filters (Transwell; Costar Corporation, Cambridge, MA) as outlined previously (Kim et al., 1998). Transepithelial resistance was measured in each well using a Millicell ERS ohmmeter (Millipore, Bedford, MA); wells registering a resistance of 200 ohms or greater, after correcting for the resistance obtained in control blank wells, were used in the transport experiments. About 1 to 2 h before the start of the transport experiments, the medium in each compartment was replaced with a serum-free medium (Optimem; Life Technologies). At the beginning of each experiment, the medium in each compartment was replaced with 700 μl of serum-free medium (Optimem), with or without drug (radiolabeled or unlabeled). The amount of the drug appearing in the opposite compartment after 1, 2, 3, and 4 h was measured in 25-μl aliquots taken from each compartment.
Inhibition of P-gp-mediated transport by Caco-2 cells was determined in a similar manner after the addition of the putative inhibitor to both the apical and basal compartments, and using radiolabeled digoxin as a P-gp substrate (Delannoy and Silverman, 1992) as described previously (Wandel et al., 1999). Complete inhibition of P-gp-mediated transport would be expected to result in the loss of the basal-to-apical (B→A) versus apical-to-basal (A→B) transport difference for digoxin. Accordingly, percent inhibition was estimated by eq. 1:
CYP3A Activity Measured Using Human Liver Microsomes.
Human liver microsomes were prepared as previously described (Guengerich, 1994), and the total cytochrome P-450 content was measured using a standard method (Omura and Sato, 1964). CYP3A catalytic activity was determined by incubating the microsomes in a total volume of 500 μl of 0.1 M phosphate buffer (pH 7.4) containing 100 pmol of cytochrome P450 and 1.5 mM NADPH, with varying concentrations of nifedipine (15, 20, 30, 60, and 200 μM), in the presence and absence of varying concentrations of inhibitors. All samples including controls contained 2% dimethyl sulfoxide because 2% dimethyl sulfoxide itself inhibited CYP3A activity by 25% in the microsomes used. Incubations were performed at 37°C and stopped after 10 min by the addition of 1 ml of CH2Cl2. Nifedipine metabolite concentrations were determined using an HPLC-based procedure (Guengerich and Bocker, 1988; Guengerich, 1994). The Michaelis-Menten kinetics was calculated using a nonlinear regression program (kcat; BioMetallics, Princeton, NJ). Inhibition type and constant (Ki) were derived using a Lineweaver-Burk plot, and IC50 values using the Hill equation.
Results
Identification of Mibefradil as a P-gp Substrate Using L-MDR1 and LLC-PK1 Cells.
Studies with LLC-PK1 cells, a cell line lacking P-gp, and the derived cell line L-MDR1, which overexpresses human MDR1-encoded P-gp, revealed that mibefradil cellular translocation was markedly polarized (Fig.1); transport is more extensive and significantly greater (P < .01) in the basolateral to apical direction than the reverse. On the other hand, in the LLC-PK1 cell line (lacking P-gp), this vectorial transport difference was not observed (Fig. 1), thus demonstrating that mibefradil is a substrate of P-gp. As shown in Fig. 1, the differential cellular translocation rates seen with mibefradil were similar to those of digoxin, demonstrating that mibefradil is a substrate of P-gp with similar affinity to digoxin.
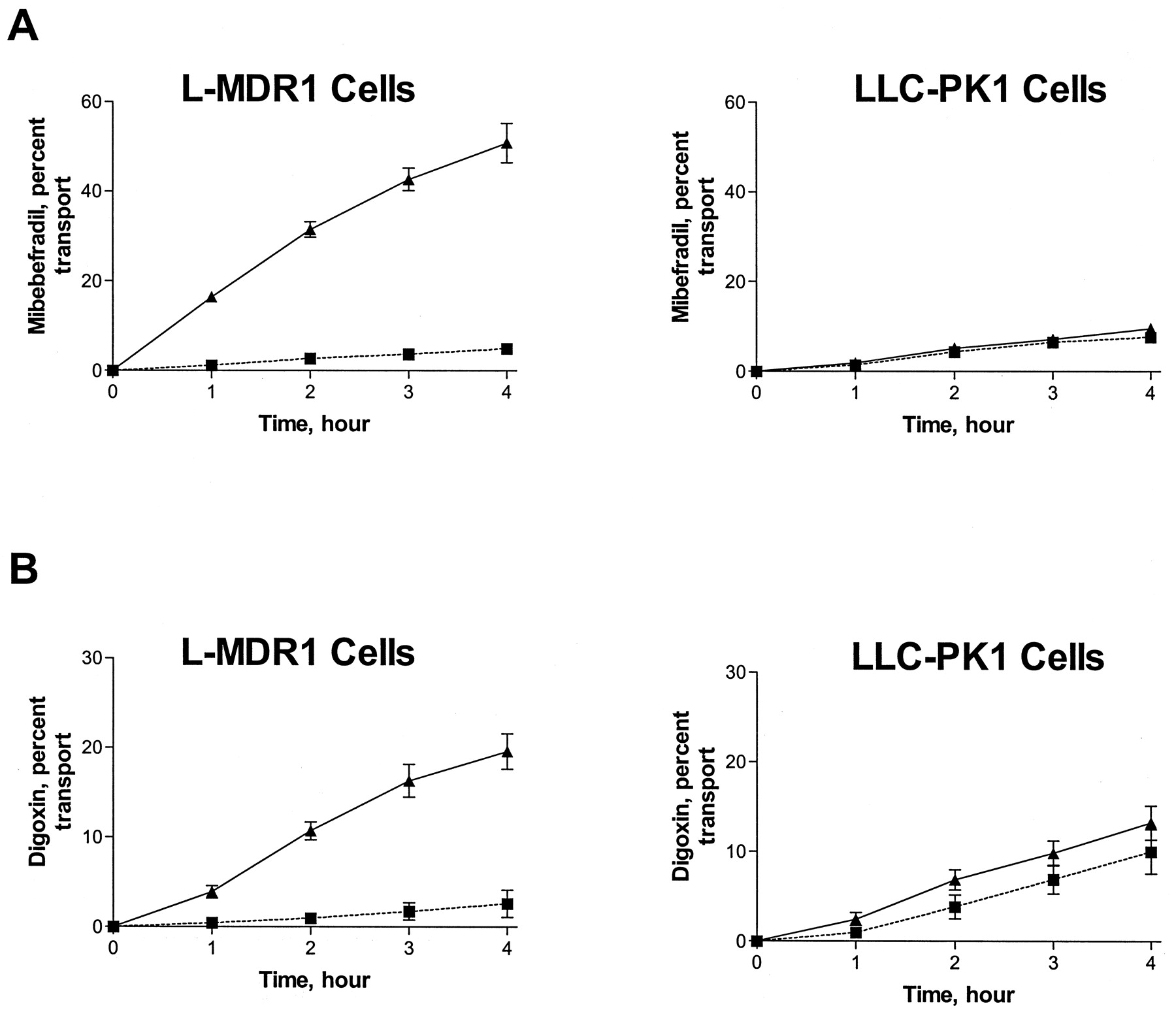
Transepithelial transport across L-MDR1 and LLC-PK1 cell culture monolayers.
A, 5 μM [3H]mibefradil; B, 5 μM [3H]digoxin. Translocation from basal to apical compartments (▴ and solid line); translocation from apical to basal compartments, (▪ and dotted line). Data are mean ± S.E. from three or more experiments.
Inhibition Studies Using Caco-2 Cells.
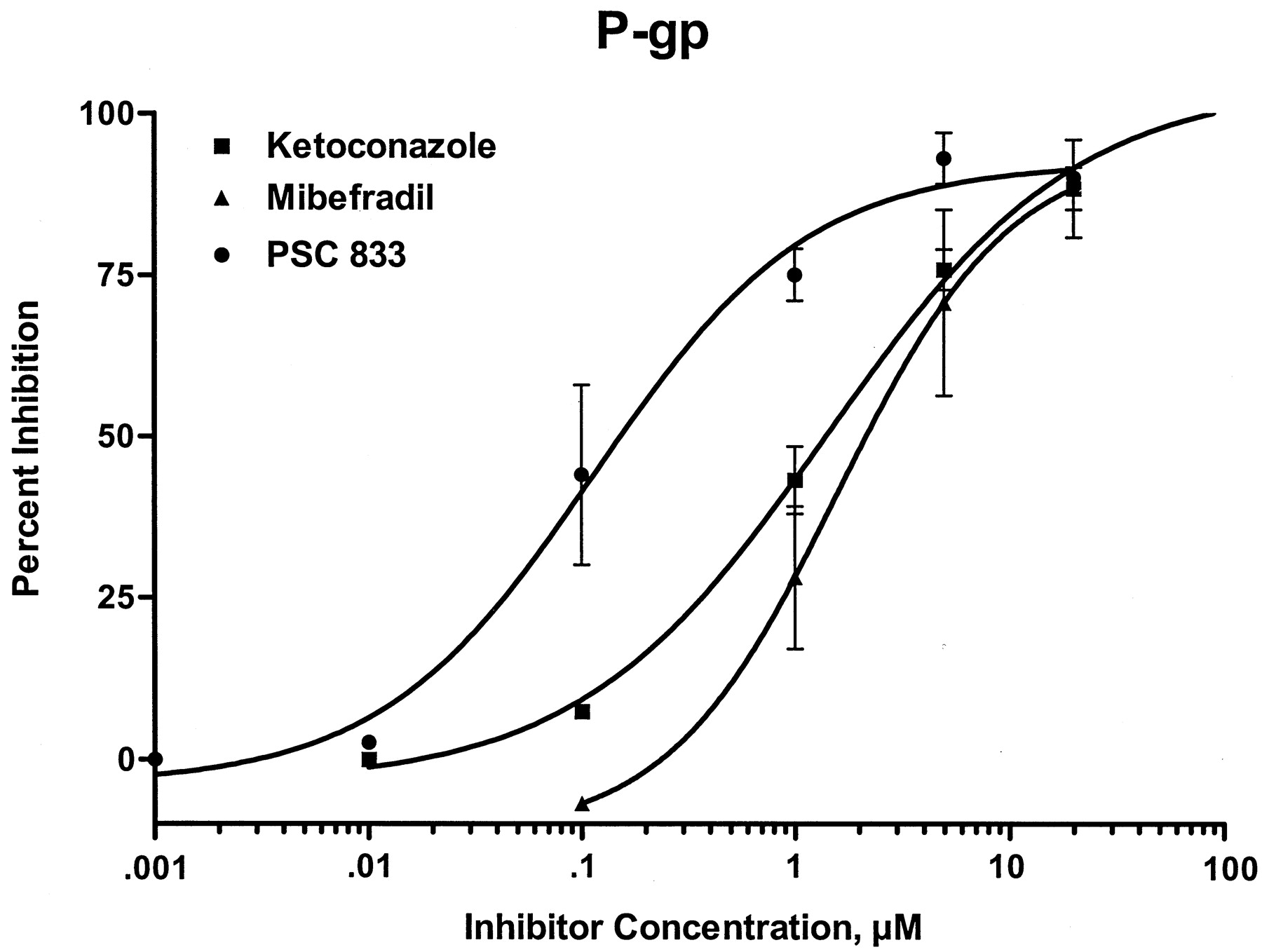
The ability of mibefradil, ketoconazole, and PSC-833 to inhibit P-gp was validated in Caco-2 cells, a human intestinal cancer-derived cell line that constitutively expresses P-gp. Inhibition of 5 μM digoxin transport in the presence of varying concentrations of inhibitors was used to define the IC50. This revealed that mibefradil (Table 1) was a relatively potent P-gp inhibitor with an IC50 value of 1.6 μM, similar to that of ketoconazole, but was not as potent as the specific P-gp inhibitor PSC-833, which had an IC50 of 0.11 μM. The Hill plot of percent inhibition versus inhibitor concentration revealed a relatively steep curve showing P-gp inhibition of about 30% at a concentration of 1 μM mibefradil (Fig. 2).
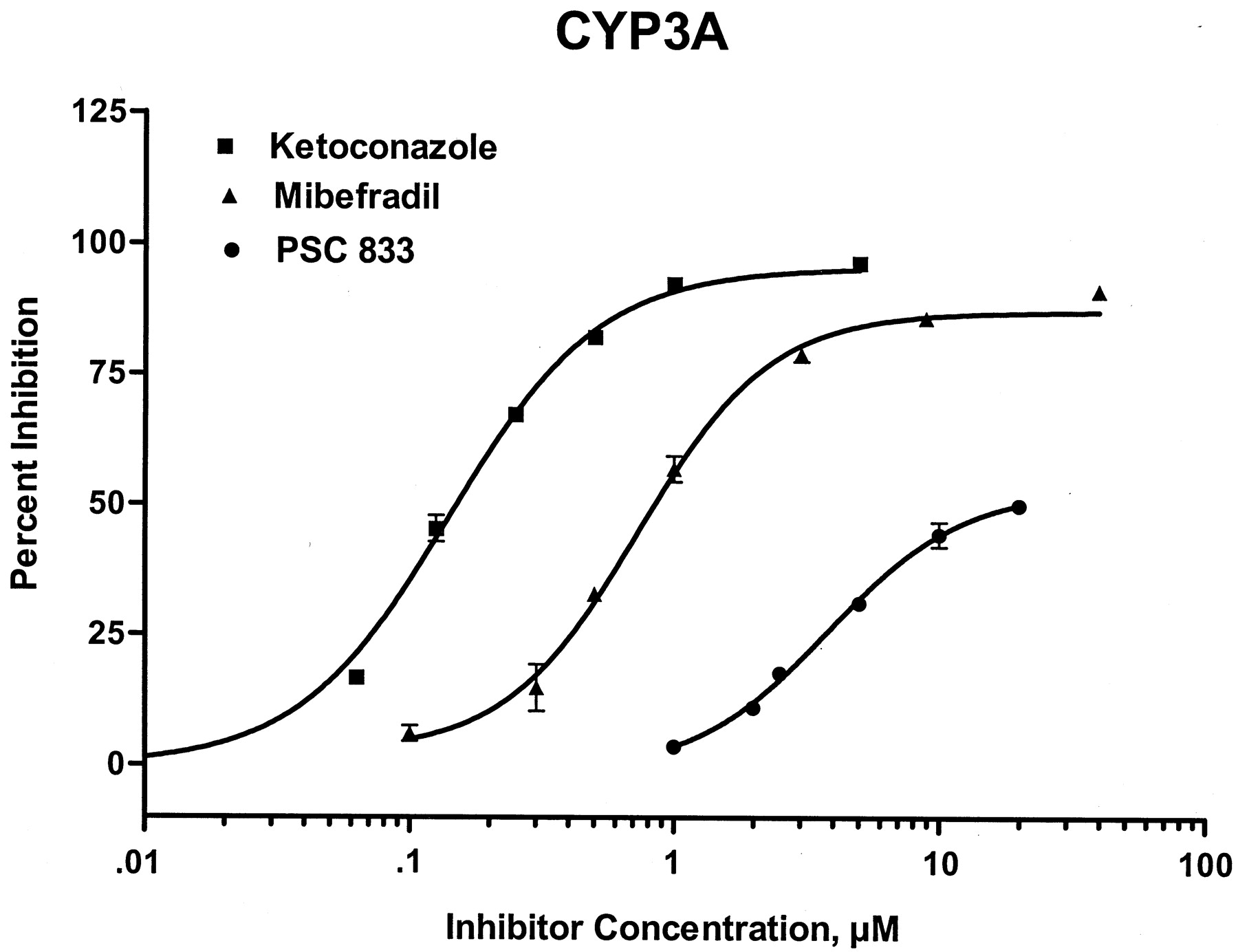
Effect of mibefradil, ketoconazole, and PSC833 on P-gp determined using digoxin transport in Caco-2 cells, and CYP3A-mediated nifedipine oxidase activity in human liver microsomes

Inhibition of P-gp by PSC833 (●),mibefradil (▴), and ketoconazole (▪)measured using 5 μM digoxin transport in Caco-2 cells.
Data are mean ± S.E. from two or more experiments.
Inhibition Studies Using Human Liver Microsomes.
Nifedipine oxidation in human liver microsomes was characterized by an apparent Km of 30 μM, and aVmax of 7 nmol metabolite/nmol of cytochrome P450/min. Mibefradil competitively inhibited oxidation of nifedipine (CYP3A) with an apparent Ki of 0.6 μM. The IC50 value was 1 μM at a nifedipine concentration of 20 μM. The comparative effects of ketoconazole and PSC833 on microsomal nifedipine oxidation are shown in Table 1 and Fig. 3.

Inhibition of CYP3A by PSC833 (●),mibefradil (▴), and ketoconazole (▪)measured using human liver microsomal nifedipine oxidase activity.
Discussion
It has been estimated that nearly 100,000 patients each year die of adverse drug reactions in the United States (Lazarou et al., 1998). Some of these are due to drug-drug interactions. The voluntary withdrawal of mibefradil by its manufacturer demonstrated that interactions were detected only after the drug was released for general use. There are a number of potent CYP3A inhibitors on the market that appear to be used safely. Therefore why did mibefradil appear to be so toxic? We proposed that this was due to the multiplicity of pathways inhibited by mibefradil, including metabolism by such key cytochrome P450 enzymes as CYP2D6 and CYP3A4 (Abernethy, 1997), the enzymes responsible for the metabolism of a variety of drugs (Guengerich, 1995;Thummel and Wilkinson, 1998); in addition, drug transport by the drug efflux transporter, P-gp was inhibited. This membrane-bound efflux transporter is expressed not only in cancer cells, but also in normal tissues, including the intestine, liver, and kidney (Thiebaut et al., 1989; Pastan and Gottesman, 1991), where P-gp contributes to the elimination of drug substrates into the gut, bile, and urine, respectively. In addition, P-gp also determines drug entry into the central nervous system (Kim et al., 1998). Known drug substrates of P-gp include various calcium channel blockers, some beta blockers, immunosuppressive agents such as cyclosporine and tacrolimus, HIV-1 protease inhibitors, and cardiac glycosides including digoxin (Mayer et al., 1996; Schinkel et al., 1996; Terao et al., 1996; Kim et al., 1998). There is considerable overlap in substrates of P-gp and CYP3A, resulting in many drugs being both transported by P-gp and metabolized by CYP3A (Wacher et al., 1995; Wandel et al., 1999). Therefore, the simultaneous inhibition of both systems will result in a substantial increase in plasma drug concentrations with resultant toxicity.
It is important to emphasize the potential for drug interactions to occur due to both CYP3A and P-gp inhibition in the gut. CYP3A and P-gp in enterocytes contribute to reducing systemic availability of drugs administered orally so that inhibition of both systems may result in a substantial increase in plasma drug concentrations and patient exposure to drug.
We have previously shown that the predominant mechanism to account for the clinically significant digoxin-quinidine interaction is the inhibition of P-gp by quinidine, resulting in reduced P-gp-mediated digoxin elimination (Fromm et al., 1998). We therefore hypothesized that the previously described drug-drug interactions involving mibefradil and digoxin (Siepmann et al., 1995) might be explained by similar P-gp inhibition by mibefradil. This study has confirmed that hypothesis, and demonstrated that mibefradil is both a substrate and an inhibitor of P-gp. In addition, we confirmed the potent inhibitory effect of mibefradil on CYP3A. However, it is the likely dual inhibition of both systems that contributed to its notable toxicity in practice. As shown in Table 1, mibefradil inhibits nifedipine oxidation activity with an IC50 andKi values similar to the potent CYP3A inhibitor, ketoconazole. The potency of mibefradil as a P-gp inhibitor, although less than that of drugs specifically developed to inhibit P-gp, such as PSC833, is remarkably high so that inhibition of both P-gp and CYP3A will occur at clinically relevant concentrations. After chronic mibefradil dosing of 100 mg/day in human subjects, the trough levels are approximately 1 μM (Abernethy, 1997). Accordingly, patients receiving mibefradil will have plasma concentrations throughout the dosing interval that may significantly inhibit both P-gp and CYP3A, resulting in both distributional and metabolic drug interactions.
In conclusion, our data demonstrate that mibefradil is both a substrate with similar basal to apical translocation rates to digoxin and a potent inhibitor of the P-gp transporter while also being a potent inhibitor of human CYP3A and is therefore another example of the overlapping substrate specificity of CYP3A and P-gp. Accordingly, the severity of drug-drug interactions observed during clinical use of mibefradil is explained by its ability to inhibit multiple systems important to drug disposition. Studies aimed at determining the role of drug transport systems, and not just drug metabolism at an early stage in drug development, have the potential to better predict drug-drug interactions before widespread general use of a drug and prevent subsequent toxicity.
Acknowledgments
We thank B. Leake, K. Ghebreselasie, and H. Waldrop for expert technical assistance.
Footnotes
-
Send reprint requests to: Alastair J. J. Wood, Division of Clinical Pharmacology, Vanderbilt University School of Medicine, Nashville, TN 37232-6602. E-mail:alastair.wood{at}mcmail.vanderbilt.edu
-
This work was supported in part by U.S. Public Health Service Grants GM 31304, HL 56251, CA 44353, ES 00267, and the Deutsche Forschungsgemeinschaft (C.W.).
- Abbreviation used is::
- P-gp
- P-glycoprotein
- Received November 8, 1999.
- Accepted April 21, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}