


Abstract
Cytochrome P450 inhibition studies are performed in the pharmaceutical industry in the discovery stage to screen candidates that may have the potential for clinical drug-drug interactions. A 96-well microtiter plate assay using recombinant cytochrome P450 (Supersomes) has been used to increase the overall throughput. The IC50 values for the inhibition of CYP3A4 by 52 new chemical entities (NCEs) were determined using the Supersomes assay with resorufin benzyl ether as a substrate, and the data were compared with those obtained in human liver microsomes (HLM) using midazolam as a substrate. Among the 52 compounds tested, 25 showed IC50values within a 5-fold difference in the two assays. For all compounds that showed a >5-fold difference, the IC50 values in the Supersomes assay were lower than those obtained in HLM, except for one compound. Further studies suggested that this discrepancy was not related to difference in protein concentrations between the two assays. In addition, the IC50 values for 16 compounds with a wide range of inhibition potency were determined in HLM using testosterone and dextromethorphan as substrates. The results showed an 80 to 93% match within a 5-fold difference between the three probe substrates. However, for certain compounds including ketoconazole, there were substrate-dependent differences in the inhibition. The results suggest that the difference between the Supersomes and HLM could be partially attributed to differences in the substrate used, and to metabolism by other cytochrome P450s present in the HLM but not in the Supersomes. Furthermore, multiple CYP3A4 substrates should be used to improve the reliability of estimating potential drug-drug interaction of NCEs.
Oxidative metabolism of xenobiotics is primarily carried out by a family of heme-containing enzymes found in the endoplasmic reticulum, known as cytochrome P450 (Gonzalez, 1989). In human liver, the five isoforms that are primarily responsible for the metabolism of most drugs are CYP3A4, CYP2D6, CYP2C9, CYP2C19, and CYP1A2 (Guengerich, 1996). CYP3A4 is the most abundant isoform, and it is believed to be expressed in all human livers (Draper et al., 1998). This isoform accounts for approximately 30% of the total microsomal cytochrome P450 protein in human liver, and it is involved in the metabolism of approximately 50% of all marketed drugs, although the levels vary by as much as 20-fold among individuals (Guengerich, 1992a,b, 1995, 1996; Shimada et al., 1994; Parkinson, 1996). CYP3A4 has been also shown to be expressed in extrahepatic tissues such as intestine, lung, kidney, and brain (Shimada et al., 1989, 1996; De Waziers et al., 1990; Kaminsky and Fasco, 1992; Kolars et al., 1992;Ravindranath and Boyd, 1995). Inhibition of this enzyme by coadministered drugs has been shown to result in adverse clinical drug-drug interactions, including fatalities (Honig et al., 1993). Therefore, it is important to determine the inhibition potential of new chemical entities (NCEs1) for CYP3A4 early in the discovery stage as to predict, anticipate, and ultimately manage potential drug-drug interactions.
A 96-well microtiter plate assay using microsomes from recombinant baculovirus-infected insect cells expressing a single cytochrome P450 (Supersomes) has been used to increase the overall throughput (Crespi et al., 1997). The Supersomes assay is rapid and reproducible, whereas assessment of the inhibition in human liver microsomes (HLM) using conventional HPLC methods is much more time-consuming. Our laboratory has evaluated the reliability of the Supersomes assay for predicting the potential inhibition of CYP2D6 in HLM using the conventional dextromethorphan O-demethylase assay (Palamanda et al., 1998). The IC50 values for the two assays closely matched for 53 of the 62 (85%) compounds evaluated (difference ≤ 5-fold). When the inhibition in the Supersomes assay was determined at 10 min instead of the recommended 45 min, the IC50 values determined in the Supersomes matched more closely those determined in HLM, resulting in 95% match (Favreau et al., 1999).
As part of the evaluation of the Supersomes assay, we have determined the IC50 values for the inhibition of CYP3A4 for 52 NCEs and compared the data with values obtained for the inhibition of midazolam hydroxylation in HLM. Furthermore, the IC50 values for the inhibition of CYP3A4 in HLM were determined for selected compounds using the testosterone-6β-hydroxylase and dextromethorphan-N-demethylase enzyme activity assays. The objectives were to compare the results of the Supersomes assay using recombinant CYP3A4 to those obtained in HLM and to evaluate the effect of substrate on the IC50 in HLM in a drug discovery and selection program.
Materials and Methods
The Supersomes Assay.
The Supersomes assay for CYP3A4 was performed according to the method described by Crespi et al. (1997), with minor modifications. Test articles were synthesized by the Chemical Research Division, Schering-Plough Research Institute (Kenilworth, NJ), as part of the discovery program. All compounds were initially dissolved in methanol. Stock solutions for inhibition assays were made in 20% aqueous methanol, and the final methanol concentration in the assay was 0.5%. A 10 mM resorufin benzyl ether solution was prepared in acetonitrile. After the addition of 5 μl of the test compound, 95 μl of a 200 mM potassium phosphate buffer, pH 7.4, solution containing 3 mM β-NADPH, 0.02% pluronic F-68 (a nonionic surfactant of polyoxyethylene-polyoxypropylene copolymer used to help solubilize the substrate), and 50 μM resorufin benzyl ether was added to each well. The plate was kept at 37°C for 5 min, and then the reaction was initiated by the addition of 100 μl of a 200 mM potassium phosphate buffer, pH 7.4, containing 50 pmol/ml of insect cell-expressed CYP3A4 (Supersomes, GENTEST Corp., Woburn, MA). The plates were incubated for 30 min at 37°C followed by the addition of a 100 μl of a reaction stop solution containing 60% acetonitrile and 40% 0.1 M Tris, pH 9. In some experiments, after the addition of the Supersomes to start the reaction, fluorescence data were acquired every 6 min for 30 min by scanning while the plate remained in the instrument. The fluorescence was determined using a Cytofluor series 4000 Multi-well Plate Reader (PerSeptive Biosystems, Framingham, MA). All analyses were conducted within the linear ranges for cytochrome P450 concentrations and incubation times. All assays were carried out in triplicate.
Data analysis.
Relative CYP3A4 activity was calculated from the fluorescence data using CytoCalc Data Analysis Software (PerSeptive Biosystems). The IC50 values were estimated by comparisons with control values, i.e., enzyme, substrate, NADPH, and 0.5% methanol in the reaction mixture (Palamanda et al., 1998; Favreau et al., 1999).
Human Liver Microsomes Assays.
Human liver microsomes were prepared according to the methods described by Guengerich (1989). The protein content was determined using the Pierce protein assay kit (Rockford, IL) using albumin to construct standard curves. Test articles were prepared as 100× stock solutions in methanol to achieve final concentrations in the microsomal incubations of 0.15, 0.3, 3, and 30 μM, each containing 1% methanol. Ketoconazole and troleandomycin (TAO) were used as prototype CYP3A4 inhibitors. Midazolam, dextromethorphan, and testosterone were used as probe substrates to monitor the changes in CYP3A4 activity during exposure to each test material or known inhibitor. Midazolam, testosterone, and dextromethorphan were prepared to achieve final concentrations in the microsomal incubations of 4, 80, and 400 μM, respectively. The concentrations of the probe substrate in reaction mixtures were chosen to be near the Kmvalue for each substrate for the CYP3A4 activity.
The microsomal incubations were carried out in a 0.1 M Tris-acetate buffer, pH 7.4, containing (final concentrations in 1-ml reaction volume) microsomal protein (0.5 mg/ml), test article (at 0.15, 0.3, 3, and 30 μM), and either midazolam (4 μM), testosterone (80 μM), or dextromethorphan (400 μM). Vehicle controls, containing 1% methanol, and ketoconazole were also included along with each probe substrate. NADPH-regenerating system (NRS) was prepared separately in 2% sodium bicarbonate solution (pH 7.4) to contain (final concentrations in 1-ml reaction volume) β-NADP (0.5 mM), glucose 6-phosphate (7 mM), and glucose-6-phosphate dehydrogenase (1.5 units/ml). Reaction tubes and NRS were kept at 37°C for 5 min before the addition of NRS to initiate the metabolic reactions. Once initiated, the reactions were incubated in a shaking water bath (37°C; 150 rpm) for 10 min (midazolam and testosterone assays) or 15 min (dextromethorphan assay). Reactions were terminated by the addition of 1 ml of methanol to the dextromethorphan and midazolam samples, or 100 μl of perchloric acid to the testosterone samples. Protein was precipitated by placing samples on ice for 15 min before adding internal standards, vortexing, centrifugation, and transferring supernatants to HPLC vials for analysis. Levallorphan and 11β-hydroxytestosterone were used as internal standards for the dextromethorphan and testosterone assays, respectively. No internal standard was added to the midazolam samples. All assays were carried out within the linear range of protein concentrations and reaction time, and they were carried out in triplicate.
HPLC analysis.
Testosterone samples were analyzed using a Waters NovaPak C18 column (4 μm, 15 cm × 3.9 mm), with a gradient elution and UV detection (240 nm) according to the methods described by Sonderfan et al. (1987) and Funae and Imaoka (1987). Samples were quantified by monitoring the ratio of 6β-hydroxytestosterone to the internal standard 11β-hydroxytestosterone. Values were then normalized using the molar absorptivity of each component to calculate the rate of 6β-hydroxytestosterone metabolite formation according to the method described by Reinerink et al. (1991). Hydroxytestosterones were purchased from Steraloids (Newport, RI).
Dextromethorphan samples were analyzed using a Rainin Microsorb MV-phenyl column (4 μm, 30 cm × 4.6 mm) under isocratic conditions with a fluorescence detector set at excitation and emission wavelengths of 270 and 310 nm, respectively. The mobile phase was an aqueous solution containing 30% acetonitrile, 1% acetic acid, and 0.5% triethylamine. Samples were quantified by monitoring the ratio between the 3-methoxymorphinan metabolite and the internal standard levallorphan in each sample and in calibration curves. The 3-methoxymorphinan metabolite of dextromethorphan was purchased from RBI/Sigma (Natick, MA).
Midazolam samples were analyzed using a Waters Symmetry C8 column (5 μm, 15 cm × 3.9 mm) under isocratic conditions and UV detection (220 nm) similar to the methods described by Ring et al. (1995). The mobile phase contained 25% acetonitrile in 20 mM sodium perchlorate buffer, pH 2.5. Samples were quantified against standard curves prepared with 1-hydroxymidazolam. Midazolam and its 1-hydroxy metabolite were obtained as gifts from Hoffman-La Roche (Nutley, NJ).
IC50 determinations.
The IC50 values were determined by plotting the percentage of inhibition of each test article versus log concentration using GraFit 3.0 (Erithacus Software LTD, Horley, Surrey, UK) and determining the concentration of test article that results in 50% inhibition of the probe substrate reaction (IC50) (Palamanda et al., 1998; Favreau et al., 1999). Values from the vehicle controls were used as a basis from which to calculate the percentage of inhibition.
Results
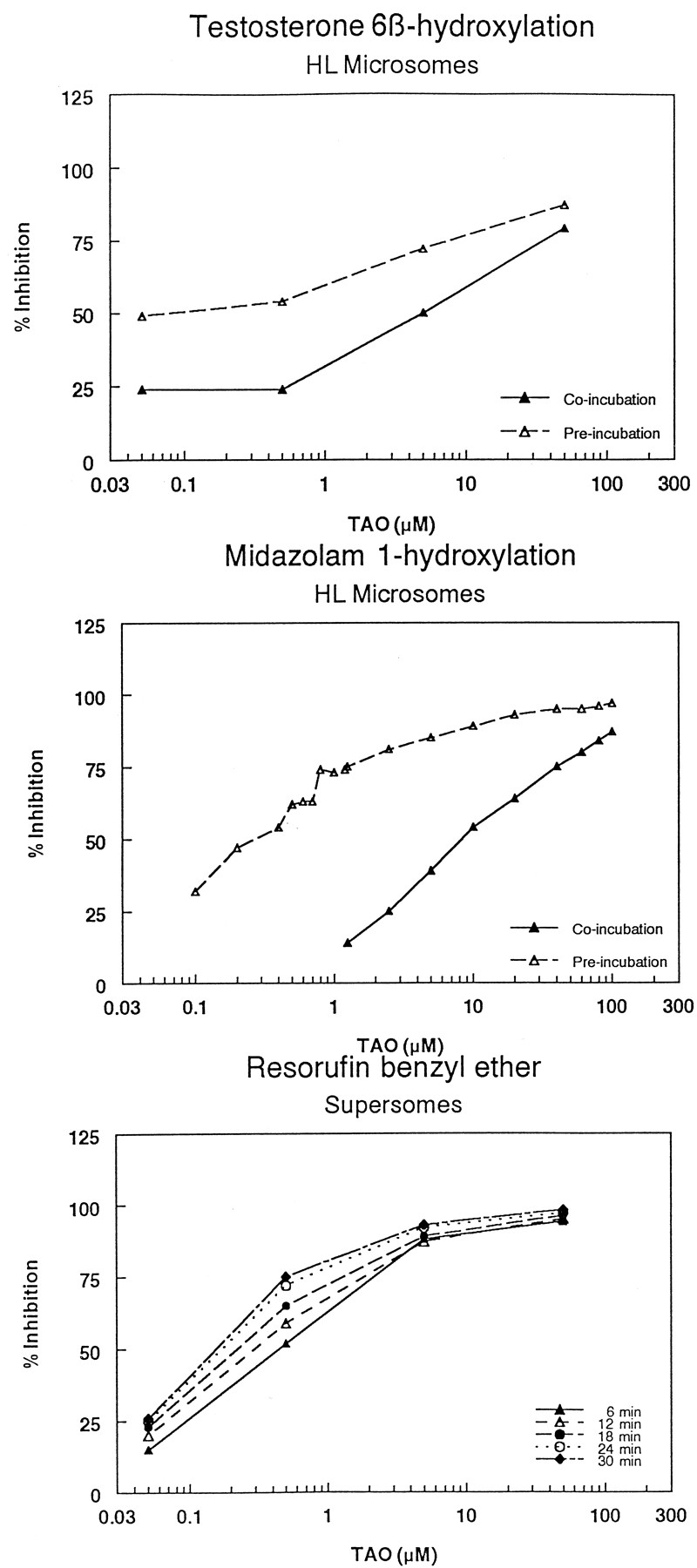
Initially, the IC50 values of the prototype CYP3A4 inhibitors ketoconazole and TAO (a mechanism-based inhibitor of CYP3A4) were determined in the Supersomes assay using resorufin benzyl ether as a substrate, and in HLM using midazolam and testosterone as substrates. The IC50 of ketoconazole in the Supersomes assay was 50 nM, in agreement with 83 nM reported by Crespi et al. (1997). The IC50 values of ketoconazole in human liver microsomes following coincubation using the midazolam and testosterone hydroxylation assays were 70 and 30 nM, respectively, consistent with a Ki of 110 nM (for the midazolam assay) reported by Wrighton and Ring (1994) and in concordance with those determined in the Supersomes assay (Fig.1). Also, when the inhibition profile of ketoconazole was determined in the Supersomes assay at various time intervals (Favreau et al., 1999), the IC50 did not change over time, suggesting that ketoconazole is neither a substrate nor a mechanism-based inhibitor of recombinant CYP3A4 (Fig.1). In HLM, the IC50 values of ketoconazole following a 30-min preincubation (Chu et al., 2000) were shifted to higher IC50 values compared with coincubation (from 70–110 nM for the midazolam assay, and from 30–100 nM for the testosterone assay), suggesting that ketoconazole is subjected to metabolism by enzymes other than CYP3A4. The inhibition profiles of TAO were determined in HLM using both testosterone and midazolam under coincubation and preincubation conditions, and in the Supersomes assay at various times. Both systems showed concrete evidence of mechanism-based inhibition of CYP3A4 as indicated by the shifting of the inhibition profiles toward lower IC50 values following the 30-min preincubation (liver microsomes), or increasing the incubation time for the Supersomes assay (Fig.2). Also, the mechanism of inhibition of two selected NCE compounds was characterized using midazolam as a substrate. The results showed that both compounds reversibly inhibited CYP3A4 with a competitive mechanism (data not shown).
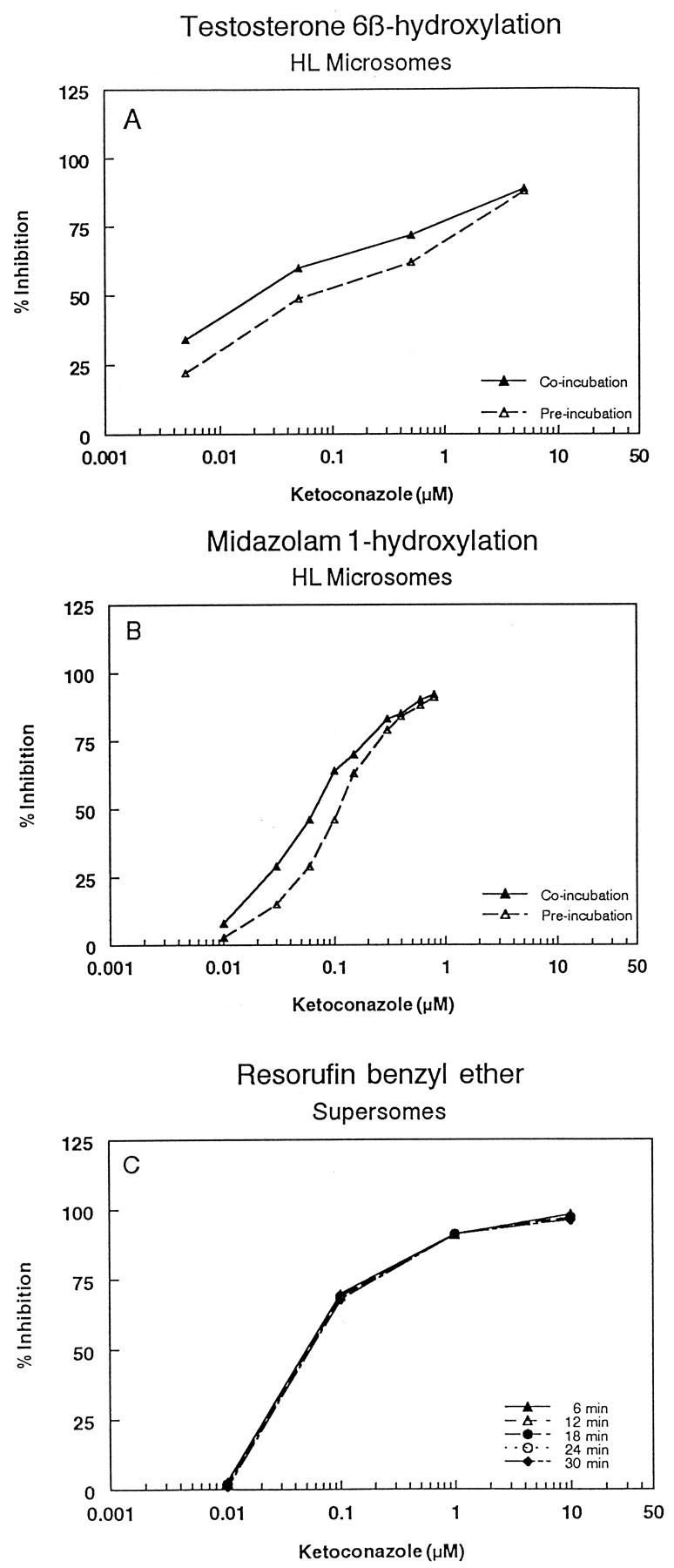
Inhibition profiles of CYP3A4 by ketoconazole in human liver microsomes using testosterone (A) and midazolam (B) as substrates, and in Supersomes using resorufin benzyl ether as a substrate (C).
Coincubation indicates that ketoconazole was present in the reaction mixture that contains microsomes, NADPH, and the substrate. Preincubation indicates that ketoconazole was incubated with the microsomes and NADPH in the absence of the substrate for 30 min, and then the substrate was added to determine the residual activity.

Inhibition profiles of CYP3A4 by TAO in human liver microsomes using testosterone (A) and midazolam (B) as substrates, and in Supersomes using resorufin benzyl ether as a substrate (C).
Coincubation indicates that TAO was present in the reaction mixture that contains microsomes, NADPH, and the substrate. Preincubation indicates that TAO was incubated with the microsomes and NADPH in the absence of the substrate for 30 min, and then the substrate was added to determine the residual activity.
Table 1 shows the IC50 values for the inhibition of CYP3A4 in HLM and in the Supersomes assay for a series of NCEs. The NCEs evaluated are broadly classified into imidazole- and nonimidazole-containing compounds. Most of the NCEs (79%) contained one imidazole ring, and 5% contained two imidazole rings. The remaining 16% contained pyrrolidine, piperidine, or 1-aza cycloheptane ring structures. Among the 52 compounds evaluated, 25 showed IC50 values within a 5-fold difference in the two assays. A difference of 5-fold or less in the IC50 value was considered acceptable because we are comparing data from a single cytochrome P450 isoform (Supersomes) with data from a mixture of cytochrome P450 enzymes (HLM), and because the substrates are different. For all compounds that showed a difference >5-fold, the IC50 values in the Supersomes assay were lower than those for HLM, with the exception of one compound (no. 13).
Inhibition of CYP3A4 in HLM and Supersomes
The difference in the IC50 values between the microsomes and Supersomes was further investigated. Since the protein concentration in the two assays is different (0.036 mg/ml in the Supersomes versus 0.5 mg/ml in the microsomes), the IC50 values for seven selected compounds (with a wide range of IC50 values) were determined in the Supersomes at two additional protein concentrations of 0.10 and 0.20 mg/ml, without changing the amount of CYP3A4 in the reaction mixture. This was achieved by the addition of insect cell microsomes prepared from wild-type baculovirus-infected cells. The results showed that increasing protein concentration did not significantly affect the IC50 in the microtiter plate assay (Table2).
Effect of protein concentration on the inhibition of CYP3A4 in the Supersomes assay
To gain further insight into the difference in the IC50 between the two assays, 16 compounds with a wide range of IC50 values in the Supersomes assay were selected, and their IC50 values were determined in HLM using two other CYP3A4 substrates, testosterone (6β-hydroxylation reaction) and dextromethorphan (N-demethylation reaction) (Table3). In general, there was a good agreement for most compounds; i.e., the IC50values were comparable. However, for some compounds including ketoconazole, there was a discrepancy in the IC50between different substrates. The data were further analyzed by determining the IC50 ratios for each compound in the three assays in HLM, and the ratios were also calculated for each compound with each of the three substrates relative to the IC50 in the Supersomes assay (Table4). In HLM, there were 80 to 93% matches with the three substrates (the IC50 within 5-fold difference). When the IC50 data from HLM were compared with those from the Supersomes, the matches were in the range of 20 to 53%.
Effect of substrate on the IC50 of various compounds for CYP3A4 in HLM
Comparison of the IC50 values
In addition to the substrate-dependent P450 inhibition, other interesting substrate-dependent interactions were observed. Table5 shows the interaction profiles of two selected discovery compounds in HLM using the three different substrates. With SCH A, there was no observed interaction with dextromethorphan as a substrate; there was a partial inhibition with testosterone and a partial activation with midazolam. With SCH B using midazolam as a substrate, there was a significant activation at lower concentrations and a significant inhibition at higher concentrations. With the other two substrates, typical concentration-dependent inhibition profiles of CYP3A4 were observed.
Substrate-dependent interactions with CYP3A4 in HLM
Discussion
Cytochrome P450 inhibition studies are performed routinely in the pharmaceutical industry to uncover candidates with potential clinical drug-drug interaction liabilities (Parkinson, 1996; Lin and Lu, 1997;White, 2000). These inhibition screens previously relied on time-consuming HPLC assays, which were convenient for evaluating small numbers of compounds. Since screening for enzyme inhibition is moving to the discovery stage, these HPLC assays are becoming obsolete. Therefore, higher-throughput assays such as those using the Supersomes described by Crespi et al. (1997) and liquid chromatography/tandem mass spectrometry assays with short run times (Ayrton et al., 1998;Chu et al., 2000) have been developed.
The Supersomes assay that uses microsomes from recombinant baculovirus-infected insect cells expressing a single P450 allows the simultaneous measurement of 96 samples within 10 min, which is much faster than conventional HPLC methods using HLM (Crespi et al., 1997). As part of the evaluation of this approach, we have attempted to explore the use of this assay to evaluate CYP3A4 inhibition in our drug discovery programs.
The results of our studies with CYP3A4 showed that there is approximately only a 50% match between the two assays. This poor correlation was unexpected given the good correlation we obtained earlier with CYP2D6 with the same class of compounds (Palamanda et al., 1998; Favreau et al., 1999). One of the major differences between the Supersomal and the microsomal reactions is the amount of the microsomal protein present (0.036 mg/ml in the Supersomes versus 0.5 mg/ml in the microsomes), which could affect nonspecific binding of the compounds being evaluated. This could have resulted in the presence of greater concentrations of free (unbound) compound in the Supersomes, resulting in a higher potency of inhibition (lower IC50). However, increasing the concentration of the microsomal non-P450 protein in the Supersomes did not significantly affect the IC50 values of seven selected compounds, indicating that protein concentration, within the range evaluated, was not a significant factor in this discrepancy.
Since it has been recently reported that CYP3A4, as well as other P450s, exhibits atypical enzyme kinetics depending on the substrate used (Korzekwa et al., 1998), we have initiated a study in which the IC50 values of 16 compounds were determined in HLM using testosterone and dextromethorphan. The results showed that even though there was 80 to 93% match within 5-fold difference among various substrates (Table 4), there was a substrate-dependent inhibition of certain compounds in HLM. The effect of substrate on the response of human liver microsomal CYP3A4 to certain inhibitors can be demonstrated by the difference in the IC50 values of ketoconazole with the three substrates. The IC50 varied by almost 10-fold between testosterone and dextromethorphan even though the substrate concentrations were at the Km (Tables 3 and4). Similar results were reported by Kenworthy et al. (1999) using microsomes from human β-lymphoblastoid cell line engineered to express a recombinant human CYP3A4 and P450 reductase.
Other substrate-dependent effects have been observed with certain compounds. For example, SCH A showed partial inhibition with testosterone, partial activation with midazolam, and neither inhibition nor activation with dextromethorphan (Table 5). A second compound, SCH B, showed a significant activation at lower concentrations and a significant inhibition at higher concentrations with the same substrate (midazolam), while it showed typical inhibition profiles with testosterone and dextromethorphan (Table 5). These results are in concordance with those recently reported by Wang et al. (2000), who demonstrated that the substrate used to measure the activity influences the results obtained in HLM. Therefore, it is highly likely that the difference in IC50 observed between the microsomes and Supersomes is due, in part, to the difference in substrate effect on the inhibition outcome.
In comparing the results from the Supersomes assay with those obtained in HLM, it is important to also point out that insect cell microsomes containing the expressed enzyme do not contain any other P450 isoforms. Thus, the potential for metabolism by other P450 isoforms in HLM does not exist in the Supersomes. If a compound is metabolized in HLM by one isoform to a less potent inhibitor of a second isoform, it is expected that its IC50 obtained with HLM would be higher than that obtained in the Supersomes assay. Therefore, it is reasonable to speculate that the higher IC50 values obtained in HLM for most compounds might have resulted, in part, from metabolism by or binding to other P450 isoforms. Furthermore, if a compound is metabolized by one isoform to a more potent inhibitor of another isoform, it is expected that the IC50 obtained in HLM would be lower than that obtained in the Supersomes assay. Hence, it is reasonable to speculate that the lower IC50obtained in HLM for compound 13 might have resulted from metabolism to a more potent inhibitor of CYP3A4 by other isoform(s). However, it should be mentioned that using the recombinant CYP3A4 preparation results is a more intrinsically accurate estimate of the IC50 of a particular compound for this isoform with this particular substrate, but may not reflect the apparent IC50 determined in HLM, which is probably more relevant in predicting drug-drug interactions. Furthermore, in the prediction of the in vivo inhibition potential of a specific cytochrome P450, and the consequent clinically relevant drug-drug interaction, plasma levels (better yet, concentrations in the liver) of the compound and the potency of the inhibitor (better expressed asKi) all must be taken into consideration.
In testing P450 inhibition of potential drug candidates, it would be ideal to use the actual drugs that are likely to be coadministered with the particular drug candidate as substrates to avoid differences due to substrate-dependent interactions. In the absence of such an option, it is advisable to use multiple substrates (probably with different reactions, i.e., N-demethylation,O-demethylation, aliphatic hydroxylation, aromatic hydroxylation, etc.) to evaluate potential drug-drug interactions for the selected candidates that would be advanced for development.
In conclusion, the results of this study demonstrate that the difference in inhibition of CYP3A4 between the microsomes and Supersomes could be attributed, in part, to substrate-dependent inhibition and to the fact that other P450 isoforms present in HLM may interact with the compounds being tested. Therefore, it is advisable to use multiple substrates to evaluate the interaction potential of selected drug candidates before proceeding to development.
Acknowledgments
We thank Drs. Ronald White, Anthony Lu, and Mitchell Cayen for support and critical review of the manuscript.
Footnotes
-
Send reprint requests to: Amin A. Nomeir, Ph.D., Director, Exploratory Drug Metabolism, Department of Drug Metabolism and Pharmacokinetics, Mail Stop 2880, Schering-Plough Research Institute, 2015 Galloping Hill Rd., Kenilworth, NJ 07033. E-mail:amin.nomeir{at}spcorp.com
- Abbreviations used are::
- HLM
- human liver microsomes
- NCE
- new chemical entity
- NRS
- NADPH-regenerating system
- TAO
- troleandomycin
- HPLC
- high-performance liquid chromatography
- P450
- cytochrome P450
- Received October 3, 2000.
- Accepted February 2, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}