


Abstract
Because modulation of P-glycoprotein (Pgp) through inhibition or induction can lead to drug-drug interactions by altering intestinal, central nervous system, renal, or biliary efflux, it is anticipated that information regarding the potential interaction of drug candidates with Pgp will be a future regulatory expectation. Therefore, to be able to utilize in vitro Pgp inhibition findings to guide clinical drug interaction studies, the utility of five probe substrates (calcein-AM, colchicine, digoxin, prazosin, and vinblastine) was evaluated by inhibiting their Pgp-mediated transport across multidrug resistance-1-transfected Madin-Darby canine kidney cell type II monolayers with 20 diverse drugs having various degrees of Pgp interaction (e.g., efflux ratio, ATPase, and calcein-AM inhibition). Overall, the rank order of inhibition was generally similar with IC50 values typically within 3- to 5-fold of each other. However, several notable differences in the IC50 values were observed. Digoxin and prazosin were the most sensitive probes (e.g., lowest IC50 values), followed by colchicine, vinblastine, and calcein-AM. Inclusion of other considerations such as a large dynamic range, commercially available radiolabel, and a clinically meaningful probe makes digoxin an attractive probe substrate. Therefore, it is recommended that digoxin be considered as the standard in vitro probe to investigate the inhibition profiles of new drug candidates. Furthermore, this study shows that it may not be necessary to generate IC50 values with multiple probe substrates for Pgp as is currently done for cytochrome P450 3A4. Finally, a strategy integrating results from in vitro assays (efflux, inhibition, and ATPase) is provided to further guide clinical interaction studies.
P-glycoprotein (Pgp) is a member of the ATP-binding cassette superfamily of transport proteins and is expressed in numerous tissues such as the luminal membrane of the small intestine and blood-brain barrier, and the apical membranes of excretory organs such as liver and kidney (Ayrton and Morgan, 2001). Pgp has broad substrate recognition, which can affect the pharmacokinetics, efficacy, safety, and target organ specificity of drugs. As a result, drug-drug interactions resulting from inhibition or induction of Pgp are a recognized clinical concern (Englund et al., 2004; Balayssac et al., 2005) recently highlighted in the Food and Drug Administration (FDA) concept paper “Drug Interaction Studies—Study Design, Data Analysis, and Implications for Dosing and Labeling” (FDA, 2004).
Despite many years of investigation, considerable uncertainty remains about the number of drug binding sites within Pgp and their mutual relationships. It is postulated that the transmembrane regions of Pgp form a large binding pocket (Sharom et al., 1998; Lugo and Sharom, 2005) composed of amino acid residues from multiple transmembrane segments (Loo and Clarke, 2001, 2002). Recent experiments investigating drug binding (Martin et al., 2000), fluorescent dye uptake (Shapiro and Ling, 1997; Lugo and Sharom, 2005), ATPase activity (Pascaud et al., 1998; Wang et al., 2000), and transport inhibition (Ayesh et al., 1996; Tang et al., 2004) are consistent with multiple (up to four have been speculated) drug binding/transport sites within the Pgp binding pocket. Thus, the Pgp macromolecule is very complex with respect to drug binding and transport.
The recent FDA concept paper on drug interactions recommends that new drug candidates be evaluated as substrates, inhibitors, and inducers of Pgp to assess the potential for clinical drug-drug interactions. The existence of multiple drug binding/transport sites within Pgp raises the question whether multiple probe substrates will be needed to relate in vitro Pgp inhibition results to clinical drug interaction findings, as has been done for cytochrome P450 3A4 (Wandel et al., 1999; Yasuda et al., 2002). Therefore, the objective of this study was to determine whether multiple probe substrates are needed to assess in vitro Pgp inhibition potential by characterizing five potential probe substrates (calcein-AM, colchicine, digoxin, prazosin, and vinblastine) that bind to different sites within the human Pgp protein (Shapiro and Ling, 1997; Martin et al., 2000). To meet this objective, IC50 values for 20 drugs having different interactions with Pgp based on efflux, ATPase, and calcein-AM inhibition assay results (Polli et al., 2001) were determined for the five probes using multidrug resistance-1-transfected Madin-Darby canine kidney type II (MDR1-MDCKII) cell monolayers.
Materials and Methods
Materials. GlaxoSmithKline Chemical Registry (Research Triangle Park, NC) supplied all the test compounds, GF120918 (Elacridar), and [3H]amprenavir (21 Ci/mmol). [G-3H]digoxin (5 Ci/mmol), [7-methoxy-3H]prazosin (70 Ci/mmol), and [G-3H]vinblastine sulfate were purchased from Amersham Biosciences, Inc. (Piscataway, NJ), and [ring C, methoxy-3H]colchicine (60–87 Ci/mmol) was purchased from PerkinElmer Life Sciences, Inc. (Boston, MA). Cell culture reagents were purchased from Invitrogen (Carlsbad, CA). All the other reagents were purchased from Sigma-Aldrich (St. Louis, MO). Transwells (12-well, 11-mm diameter, 0.4-μm pores) were purchased from Corning Costar (Cambridge, MA). The calcein-AM assay kit (Vybrant Multidrug Resistance Kit) was purchased from Molecular Probes (Eugene, OR).
Monolayer Efflux Studies. MDR1-MDCK type II (MDCKII) cells expressing human Pgp were obtained from The Netherlands Cancer Institute (Amsterdam, The Netherlands). Cell culture and transport studies were completed with slight modifications as previously described (Polli et al., 2001; Mahar Doan et al., 2002; Keogh and Kunta, 2006). Briefly, cells were split weekly at a ratio of 1:50 and grown in the absence of antibiotics or selection agent. For transport studies, cells were seeded onto polycarbonate Transwell filter membranes at a density of 300,000 cells/cm2; media were changed the following day; and transport assays were completed 3 days later. Compounds were dissolved at 10 mM in 100% dimethyl sulfoxide (DMSO) and diluted for studies in transport medium [Dulbecco's modified Eagle's medium supplemented with l-glutamine, 25 mM HEPES, pyridoxine hydrochloride, 1% DMSO (v/v) but without sodium pyruvate, and phenol red]. As part of the initial characterization, each probe substrate was tested at a number of concentrations (vinblastine, colchicine, and prazosin 1–10 μM; digoxin 0.043–5 μM; n = 4 test concentrations) and in both the apical-to-basolateral (A→ B) and basolateral-to-apical (B→ A) directions. Each probe substrate had linear flux across a concentration range bracketing the final test concentration used in the inhibition studies. The direction that provided the largest dynamic range was selected as the direction used in the inhibition studies. Based on these initial experiments, the probe substrates were tested at 1 μM (prazosin, vinblastine, and colchicine) or 0.043 μM (digoxin) in A→ B (prazosin) or B→ A (digoxin, vinblastine, and colchicine) directions.
Inhibitors were tested in triplicate at a minimum of eight concentrations generally spanning 0.3 to 100 μM. Monolayer studies were conducted at 37°C in a humidified incubator with shaking (90 rpm) for either 90 min (prazosin, digoxin, and vinblastine) or 240 min (colchicine). Markers for Pgp efflux ([3H]amprenavir, separate set of Transwells) and monolayer integrity [Lucifer yellow (LY) every Transwell along with probe substrate] were included in each experiment. Radiolabeled probes were measured by liquid scintillation counting with Ultima Gold (PerkinElmer) scintillation mixture using a TriCarb T2900 counter (PerkinElmer). The efflux ratio for [3H]amprenavir (test concentration of 3 μM) passed the assay criterion (≥12) and collapsed to unity in the presence of Pgp inhibitor GF120918 showing the functional expression of human Pgp in the monolayers. Amprenavir is a substrate of Pgp but not breast cancer resistance protein or multidrug resistance-associated proteins (Olson et al., 2002; Gupta et al., 2004). LY concentration in the receiver compartments was measured using a SpectraMax Gemini cytofluorimeter (Molecular Devices, Sunnyvale, CA) set to an excitation wavelength of 430 nm and an emission wavelength of 538 nm. Values ≤20 nm/s for LY permeability were considered acceptable for the assay.
Calcein Inhibition Assay. The calcein-AM assay was optimized and performed using the Vybrant Multidrug Resistance Kit (Molecular Probes) and MDR1-MDCKII cells as described (Polli et al., 2001; Mahar Doan et al., 2002). Cells were seeded at 70,000 cells/well (200 μl of culture medium) in 96-well black plates with clear bottoms (Packard Instrument Co., Meridian, CT). The medium was changed 24 h after seeding, and the assay was performed 48 h later. On the day of the study, the medium was aspirated, and monolayers were washed three times with transport buffer. Test drugs were added to monolayers in 50 μl of transport buffer containing 1% DMSO. Test concentrations of each drug (final concentrations of 0.1–100 μM, n = 7, except for GF120918, which was 0.001–10 μM) were selected based on previous work with this assay (Polli et al., 2001). DMSO concentration (1%) was constant in test and control wells (each n = 2). Plates were preincubated at 37°C for 10 min. Calcein-AM was added, and the plates were immediately placed in a SpectraMax Gemini cytofluorimeter (Molecular Devices) for 60 min and read at 15-min intervals at excitation and emission wavelengths of 485 and 530 nm, respectively. Pgp inhibition was quantified using the following equation:  where RFUcomp is fluorescence in the presence of test compound (comp), RFUGF120918 is fluorescence in the presence of 2 μM GF120918 (maximum inhibition), and RFUbackground is fluorescence in absence of the drug (typically 45–65 RFU).
where RFUcomp is fluorescence in the presence of test compound (comp), RFUGF120918 is fluorescence in the presence of 2 μM GF120918 (maximum inhibition), and RFUbackground is fluorescence in absence of the drug (typically 45–65 RFU).
Calculations. The transport rate of each probe was calculated using the following equation:  where J is the transport rate (nmol/cm2/h), V is the receptor volume (ml), C is the receiver drug concentration (nmol/ml), t is time in hours, and A is the membrane surface area (cm2). The permeability coefficient at pH 7.4 (P7.4) for passive membrane transport of LY and probe substrates in the presence of GF120918 was determined using the following equation as previously described (Tran et al., 2004):
where J is the transport rate (nmol/cm2/h), V is the receptor volume (ml), C is the receiver drug concentration (nmol/ml), t is time in hours, and A is the membrane surface area (cm2). The permeability coefficient at pH 7.4 (P7.4) for passive membrane transport of LY and probe substrates in the presence of GF120918 was determined using the following equation as previously described (Tran et al., 2004):  where VD and VR are donor and receiver well volumes, respectively (ml), A is the membrane surface area (cm2), t is the incubation time (s), CR(t) is the measured concentration in the receiver well at time t (nmol/ml), and CD(t) is the measured concentration in the donor well at time t (nmol/ml).
where VD and VR are donor and receiver well volumes, respectively (ml), A is the membrane surface area (cm2), t is the incubation time (s), CR(t) is the measured concentration in the receiver well at time t (nmol/ml), and CD(t) is the measured concentration in the donor well at time t (nmol/ml).
The IC50 values, the concentration of inhibitor required for 50% inhibition of the B→ A transport rates, were calculated with GraFit (version 5.06, Erithacus Software Limited, London, UK) using:  where y is the rate of transport of an appropriate probe (expressed as a percentage of the uninhibited control), Range is the rate of probe in the absence of test compound minus the background transport rate, s is the slope factor, x is the inhibitor concentration (μM), and background is the uninhibited rate of probe transport (expressed as a percentage of the total rate).
where y is the rate of transport of an appropriate probe (expressed as a percentage of the uninhibited control), Range is the rate of probe in the absence of test compound minus the background transport rate, s is the slope factor, x is the inhibitor concentration (μM), and background is the uninhibited rate of probe transport (expressed as a percentage of the total rate).
The K values, the concentration of inhibitor required for a 50% increase in the prazosin A→ B rate, were calculated with GraFit (version 5.06, Erithacus Software Limited) using the Hill equation:  where v is the rate of transport in nmol/cm2/min, Vmax is the maximum rate of A→ B transport of prazosin in nmol/cm2/min, S is the concentration of inhibitor, n is the Hill coefficient, and K is the inhibitor concentration that produces 50% of the rate for A→ B transport of prazosin.
where v is the rate of transport in nmol/cm2/min, Vmax is the maximum rate of A→ B transport of prazosin in nmol/cm2/min, S is the concentration of inhibitor, n is the Hill coefficient, and K is the inhibitor concentration that produces 50% of the rate for A→ B transport of prazosin.
Mass balance was the percentage of original drug mass accounted for at the end of the experiment [sum of the amount in the apical (A) and basolateral (B) chambers]. Mass balance was calculated with the following equation:  where CAt and CBt are the drug concentrations in the A and B chambers at time (t), C0 is the concentration of the donor at time 0, VA and VB are the volumes of the A and B chambers, and VD is the volume of the donor solution added to the appropriate chamber.
where CAt and CBt are the drug concentrations in the A and B chambers at time (t), C0 is the concentration of the donor at time 0, VA and VB are the volumes of the A and B chambers, and VD is the volume of the donor solution added to the appropriate chamber.
Results
Selection of Compounds. The probe substrates calcein-AM, colchicine, digoxin, prazosin, and vinblastine were selected based on literature data suggesting possible differential binding to Pgp, availability of radiolabel material (colchicine, digoxin, prazosin, and vinblastine) or fluorescence assays (calcein-AM), and efflux characteristics in the in vitro assay (Table 1). The 20 diverse drugs tested as inhibitors were selected to explore different interactions with Pgp (e.g., monolayer efflux, ATPase, and calcein-AM assays) and grouped according to the results reported by Polli et al. (2001) (Table 2). Of the selected 20 drugs, 8 belong to category I, in which the drugs exhibited agreement across the three assays; these drugs are inhibitors, unambiguous substrates, or unambiguous nonsubstrates of Pgp. The remaining 12 drugs were selected from category II, which revealed differences among the assays related to membrane permeability and interaction with Pgp. Category II is further subdivided based on the absence (groups IIA, n = 3, nontransported substrates) or presence (group IIB, n = 8, transported substrates) of monolayer efflux. Four of the five probe substrates (colchicine, digoxin, prazosin, and vinblastine) were also included as test inhibitors and belong to either category I (prazosin and vinblastine) or IIB (digoxin and colchicine).
Properties of Pgp probe substrates and inhibitor
Inhibition of Pgp-mediated transport of probe substrates across MDR1-MDCKII cells
The inhibition of probe substrates by test compounds was determined as described under Materials and Methods. A minimum of eight concentrations (n = 3) per test compound were used to determine the IC50 value (calcein-AM, digoxin, vinblastine, and colchicine) using the full four-parameter equation or K value (prazosin) using the Hill equation.
Monolayer Efflux Studies of Probe Substrates. MDR1-MDCKII cells have been shown to be a good in vitro model for determining whether compounds are Pgp substrates (Polli et al., 2001; Tang et al., 2002; Taub et al., 2005). For each probe selected for use in the efflux assay (colchicine, digoxin, prazosin, and vinblastine), a bidirectional concentration-dependent experiment was performed across MDR1-MDCKII cell monolayers (Table 3). The B→ A/A→ B ratios for each probe were greater than 1.0 at each of the four test concentrations between either 1 and 10 μM (colchicine, prazosin, and vinblastine) or 0.043 to 5 μM (digoxin), indicating Pgp-mediated efflux of each probe (Table 3 and data not shown). Because of the low efflux ratio of colchicine under the standard assay conditions, the effect of incubation time on the efflux ratio was further evaluated. An increase in the incubation time from 90 to 240 min increased the B→ A/A→ B ratio from 1.5 to 3.5 (data not shown). Because the efflux ratio was larger at 240 min, this time was used in all the subsequent inhibition studies with colchicine. The efflux ratios for digoxin and vinblastine were greater than 50, whereas those for prazosin and colchicine were less than 4. Addition of 5 μM GF120918, a potent Pgp inhibitor, reduced the B→ A/A→ B ratios of colchicine, prazosin, and digoxin to unity. In contrast, the efflux ratio of vinblastine was reduced to 2.3 in the presence of GF120918, suggesting that a second efflux transporter is present in the MDR1-MDCKII cells. The probe substrates had a range of permeability values (Papp B→ A) with digoxin having the lowest permeability (15.9 nm/s), followed by colchicine (53.4 nm/s), vinblastine (97.4 nm/s), and prazosin (283 nm/s). Finally, mass balance for all the probes was acceptable (>80%), suggesting that there was minimal loss of substrate to plastic surfaces and cells under the experimental conditions.
Transport and permeability of probe substrates across the MDR1-MDCKII cells
Monolayer Pgp-Mediated Inhibition by GF120918. The concentration-dependent inhibitory effect of GF120918 on transport of each probe across MDR1-MDCKII monolayers was tested (Fig. 1). Inhibition studies for digoxin, vinblastine, and colchicine were completed in the B→ A direction because this direction had a large dynamic range as a result of the contribution of Pgp efflux (Table 3). The addition of GF120918 decreased the rate of digoxin, vinblastine, and colchicine B→ A transport (test concentration 1 μM for vinblastine and colchicine and 0.043 μM for digoxin) by 86%, 75%, and 85%, respectively. The GF120918 IC50 values were similar across the three probe substrates ranging from 0.027 to 0.055 μM (Table 2; Fig. 1).
The Pgp inhibition assay of prazosin was completed in both the A→ B and B→ A directions because of the compound's high permeability and modest efflux ratio. Addition of GF120918 increased the A→ B rate of prazosin (test concentration 1 μM) by 1.7-fold (Table 3) and resulted in the IC50 value of 0.05 μM for GF120918 (Table 2), an IC50 value of GF120918 similar to that for the other three probe substrates. In the B→ A direction, GF120918 only inhibited the B→ A rate of prazosin by 51%, yielding an estimated IC50 value of 0.025 μM, similar to that for the A→ B direction and the other probes (Tables 2 and 4). This shows that the IC50 values for prazosin are not influenced by the direction of the assay. Of the four probe substrates tested in the monolayer efflux assay, prazosin had the smallest dynamic range (∼3-fold), which is related to its high intrinsic membrane permeability. However, because this compound has been used extensively to characterize Pgp drug binding/transport pocket (Wang and Georges, 1997; Isenberg et al., 2001), it was examined further as a potential probe substrate. Finally, the IC50 value for GF120918 in calcein-AM was 0.10 μM, a value similar to that observed for the other probe substrates.
Rank order comparison of inhibitors on the Pgp-mediated transport of selected probe substrates across MDR1-MDCKII cells
Inhibition of Pgp-Mediated Efflux of Probe Substrates by a Panel of Inhibitors. The inhibition of Pgp-mediated efflux of the five probe substrates by 20 diverse drugs was measured by determining either the B→ A (colchicine, digoxin, and vinblastine) transport across MDR1-MDCKII monolayers, the A→ B (prazosin) transport across MDR1-MDCKII monolayers, or the increase in calcein fluorescence (calcein-AM) in MDR1-MDCKII cells (Table 2). Furthermore, the inhibition of B→ A transport of prazosin was determined for three inhibitors to confirm consistency of the IC50 values between the directions of the assay. Overall, the rank order of the probe substrates was generally similar across the 20 drugs (Tables 2 and 4), with probe substrates having a notable inhibition by 6 to 9 of the drugs. As expected, GF120918 (category I inhibitor) was the most potent inhibitor (IC50 values below 0.10 μM), followed by cyclosporin A (category IIB3), ketoconazole (category IIA), and verapamil (category IIA); all these drugs are well-established Pgp inhibitors. For GF120918, cyclosporin A, and ketoconazole, there was little difference (within 3- to 5-fold of each) in the IC50 values across the probe substrates (Table 3). In contrast, there were notable differences in the IC50 values of verapamil across the probe substrates. The verapamil IC50 value for digoxin and prazosin was ≤11 μM, between 17 and 34 μM for colchicine and vinblastine, and 60.9 μM for calcein-AM. It is of interest that diphenhydramine, the other category IIA drug, did not show any significant inhibition of the five probe substrates (Table 2), although calcein-AM fluorescence increased 15% at 100 μM diphenhydramine.
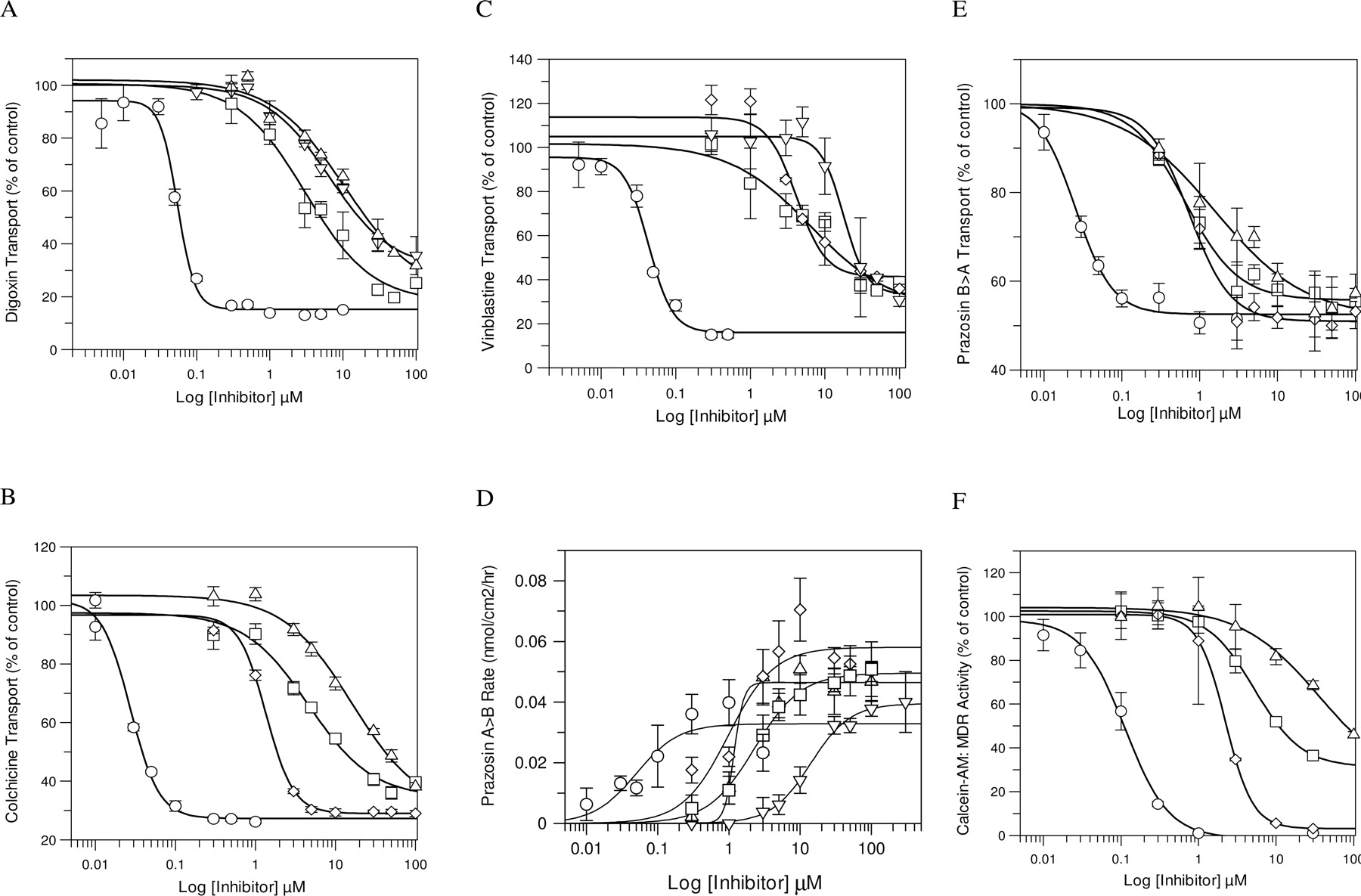
Inhibition of Pgp-mediated transport of probe substrates digoxin (A), colchicine (B), vinblastine (C), prazosin (D, E), and calcein-AM (F). Inhibition was measured by determining the B→ A (digoxin, colchicine, vinblastine, and prazosin) transport across MDR1-MDCKII monolayers, A→ B (prazosin) transport across MDR1-MDCKII monolayers, or efflux of calcein-AM from MDR1-MDCKII cells. Symbols representing the inhibitors are ○ for GF120918, ▿ for quinidine, ▵ for verapamil, ⋄ for cyclosporin A, and □ for ketoconazole. The values represent mean ± S.D. of at least three independent determinations.
Further evaluation revealed that category I nonsubstrates and IIB1/IIB2-transported substrates had little inhibitory activity toward any of the five probe substrates (Table 2). In contrast, there were some notable differences in the inhibition of the five probe substrates by category I substrate drugs. Amprenavir and prazosin had weak inhibition (IC50 values >70 μM) against all the probe substrates. In contrast, the IC50 values for quinidine and vinblastine ranged from 14 to >100 μM across the five substrates and fell into two groupings. For quinidine, the IC50 values were 14 to 23 μM for digoxin, prazosin, and vinblastine and ∼50 μM for calcein-AM and colchicine. The vinblastine IC50 values for colchicine, digoxin, and prazosin were 17 to 30 μM, and those of calcein-AM and vinblastine were >89 μM. It is of note that of the four probe substrates used as inhibitors, only vinblastine was able to show inhibition of its own transport (IC50 value = 89.7 μM), albeit rather weakly compared with the other positive inhibitors.
Discussion
The increased interest in Pgp and potential drug interactions is driving the need to validate the predictability of in vitro Pgp studies against in vivo data. Such in vitro assays will require the identification of specific probe substrates and inhibitors for Pgp (Ayrton and Morgan, 2001). Because a large number of substrates and modulators interact with Pgp, it has been speculated that Pgp has multiple drug binding/transport sites (Shapiro et al., 1999; Martin et al., 2000; Wang et al., 2001; Tang et al., 2004), which makes selection of probe substrates a key consideration when establishing an in vitro inhibition assay. In the present study, the substrates calcein-AM, colchicine, digoxin, prazosin, and vinblastine were selected based on different binding/transport by human Pgp (Shapiro and Ling, 1997; Shapiro et al., 1999; Martin et al., 2000), efflux characteristics, and availability of fluorescent/radiolabel drug. The 20 drugs used as inhibitors were selected based on behavior in the monolayer efflux, ATPase, and calcein-AM assays (see Selection of Compounds under Results). Overall, the rank order profiles of the five probe substrates were generally similar (Table 4), with probe substrates having interaction with six to nine of the drugs; however, there were several notable differences in the IC50 values.
Detailed examination of the relationship between drug category and IC50 values provides further insight into the selection of probe substrates and when a compound may be a potent inhibitor of Pgp. Category I nonsubstrate and category IIB1/2 substrate drugs had little to no inhibition of the probe substrates, consistent with previous calcein-AM and ATPase results showing little to no interaction with Pgp (Polli et al., 2001). This highlights the observation that “competitive” inhibition is not always seen for good Pgp substrates (Lugo and Sharom, 2005; Taub et al., 2005). This is further illustrated through the poor inhibition of the probe substrates on themselves in this study (e.g., colchicine, digoxin, prazosin, and vinblastine). In contrast, category IIA and IIIB3 drugs were good Pgp inhibitors (IC50 values <15 μM) with the rank order of inhibition being GF120918 (I) > cyclosporin A (IIIB3) ≅ ketoconazole (IIA) ≥ verapamil (IIA). There were notable differences in the verapamil IC50 values across the five probes. The IC50 values for digoxin and prazosin were ≤11 μM, and those for calcein-AM, colchicines, and vinblastine were ≥17 μM, which may suggest differential binding of verapamil to Pgp relative to the other inhibitors in this group. This may be explained by GF120918, cyclosporin A, and ketoconazole binding to a central modulatory site (M site, Table 1), thus inhibiting all the substrates (Martin et al., 2000). In contrast, verapamil may exert its inhibitory effect at multiple drug transport/binding sites (R and/or P sites) (Martin et al., 2000), which is consistent with the “two-affinity” model (biphasic binding) proposed for [3H]verapamil binding to Caco-2 membranes overexpressing Pgp (Doppenschmitt et al., 1999).
Examination of category I substrate drugs revealed differences in probe substrate inhibition profiles. Amprenavir and prazosin (as an inhibitor) were weak Pgp inhibitors across the probes; only IC50 values for colchicine were determined. On the other hand, quinidine markedly inhibited digoxin, prazosin, and vinblastine efflux (IC50 values of 11–23 μM) but was up to 3-fold less potent toward colchicine and calcein-AM (IC50 values near 50 μM). Vinblastine (as an inhibitor) inhibited colchicine, digoxin, and prazosin transport with similar IC50 values (17–31 μM); however, inhibition of itself and calcein-AM was up to 5-fold weaker. These data suggest that category I substrate drugs may have different interactions with the Pgp drug binding/transport sites. This is not surprising because previous work has shown differential interactions between Pgp substrates (rhodamine, Hoechst 33342, vinblastine, and prazosin) such that different combinations of substrates can yield stimulation or inhibition of efflux (Dey et al., 1997; Shapiro and Ling, 1997; Martin et al., 2000; Wang et al., 2001; Lugo and Sharom, 2005; Taub et al., 2005). Like [3H]verapamil binding, [3H]vinblastine binding to Caco-2 membranes also fits a two-affinity model, which may explain the differences in IC50 values for the probe substrates.
Considering the importance of Pgp in drug disposition, it is evident that Pgp-mediated drug transport can play a central role in drug-drug interactions (Ayrton and Morgan, 2001; Balayssac et al., 2005). Data derived from the present study and reported by Keogh and Kunta (2006) can serve as a guide to design clinical drug interaction strategies for Pgp. Digoxin proved to be a sensitive probe and is our recommendation as the probe substrate for in vitro Pgp inhibition assays. Its advantages are a large in vitro efflux ratio (>50), limited passive membrane permeability, good mass balance, collapse to unity in the presence of GF120918, and availability of radiolabel drug commercially; these characteristics give digoxin a sufficient dynamic range over which to measure inhibition of Pgp-mediated transport in vitro. Furthermore, IC50 values reported here for GF120918, cyclosporin A, vinblastine, quinidine, and verapamil are in agreement with Ki values reported in Caco-2 or MDCK-MDR1 cells using digoxin as a probe substrate (Tang et al., 2002), highlighting the consistency of digoxin as a probe substrate. Finally, digoxin offers the advantage of being a useful clinical probe substrate, which is mainly eliminated in humans as unchanged drug in the urine, therefore minimizing any confounding metabolism issues.
Vinblastine and colchicine are recommended as alternate in vitro probes. These probes have inhibition profile similar to, but less sensitive, than that of digoxin. One advantage of vinblastine is that it has a larger efflux range than colchicine. Even though prazosin is as sensitive as digoxin, prazosin is not recommended as a probe substrate because of its high membrane permeability and limited dynamic range of the assay (3-fold efflux). Surprisingly, calcein-AM proved to be the least sensitive probe (Table 4). In particular, calcein-AM transport was insensitive to verapamil and quinidine inhibition, relative to the other probe substrates tested. No clear pattern or explanation is available for this behavior. There are conflicting reports in the literature on the IC50 values of these drugs on calcein-AM transport, and our data are consistent with a number of these studies (Tiberghien and Loor, 1996; Wang et al., 2001; Schwab et al., 2003). Finally, translation of in vitro data for these alternative probe substrates to clinical drug interaction studies may be more difficult compared with digoxin because of the limited experience with these agents as clinical Pgp probe substrates and the associated complication of metabolism. However, results reported here can serve as a bridge between the use of these alternate in vitro probes and that of digoxin as a clinical probe substrate.
A comparison of the digoxin in vitro IC50 data from this study with clinically relevant drug-drug interactions (Lanoxin tablets product information) (Englund et al., 2004) revealed a close relationship between the clinical and in vitro findings. For example, quinidine (Angelin et al., 1987; Mordel et al., 1993; Fromm et al., 1999), verapamil (Pedersen et al., 1983; Verschraagen et al., 1999), and cyclosporin A (Okamura et al., 1993) have well-documented effects on digoxin pharmacokinetics in vivo and have established Pgp inhibitory effects in vitro. These Pgp inhibitors belong to categories I (unambiguous substrate), IIA (nontransported substrates), and IIB3 (transported substrates) and have IC50 values between 1 and 15 μM for digoxin. In contrast, none of the category I unambiguous nonsubstrates or category IIB1/2 substrates had interaction with digoxin in vitro, consistent with the in vivo literature. Therefore, as a guide, potential Pgp-mediated drug-drug interactions should be taken into consideration with compounds that belong to categories I (unambiguous substrates only), IIA (nontransported substrates), and IIB3 (transported substrates) that have in vitro IC50 values less than 15 μM (Table 5). Furthermore, the free fraction of drug in plasma and dose (in particular if >100 mg) are other important parameters to consider when evaluating the need for a clinical drug interaction study. As discussed in the FDA Guidance and in the review by Sahi (2005), drug-drug interaction potential can be estimated using [I]/Ki, where I is inhibitor concentration and Ki is the inhibition constant (note that use of total concentration is recommended in the FDA Guidance because this provides the most conservative estimate). If the [I]/Ki ratio is <0.02, the chance of an interaction is remote. In contrast, as the [I]/Ki ratio increases, the chance of an interaction increases. More detailed investigations are required to address the utility of this approach to drug transporters.
Categorizing Pgp inhibition potential by ability to inhibit digoxin B→A transport across MDR1-MDCKII cells
The current interest of the FDA and European regulatory agencies in Pgp drug transport is likely to increase expectations that new drug candidates be evaluated for inhibition of Pgp-mediated transport, in particular for specific therapies such as cardiovascular, oncology, and neurology. Therefore, based on the result of the present study, we recommend the use of digoxin as a standard probe substrate for in vitro Pgp inhibition studies to determine the inhibition potential of drug candidates.
Footnotes
-
This work was supported in part by the Academy of Finland (Grants 200582 and 205139), the Finnish Cultural Foundation, and the University of Kuopio (J.R.).
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.008615.
-
ABBREVIATIONS: Pgp, P-glycoprotein; FDA, Food and Drug Administration; GF120918, N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide; MDR1-MDCK, multidrug resistance-1-transfected Madin-Darby canine kidney; DMSO, dimethyl sulfoxide; A→B, apical to basolateral; B→A, basolateral to apical; LY, Lucifer yellow; K, the concentration of inhibitor required for 50% increase in the prazosin A→B rate; B→A/A→B ratio, Papp B→A/Papp A→B; Papp, apparent permeability.
-
↵1 Current affiliation: Department of Pharmaceutical Chemistry, University of Kuopio, Kuopio, Finland.
-
↵2 Curren affiliation: Pharmaceutical Research and Development, Merck & Co., Inc., West Point, Pennsylvania.
- Received November 27, 2005.
- Accepted January 31, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}