


Abstract
Many drug interactions with drugs used for the therapy of human immunodeficiency virus (HIV) occur at the level of different cytochrome P450 isozymes. Increasing evidence suggests that antiretrovirals may also modify activity and expression of active drug transport systems. Such interactions may alter drug absorption, elimination, and also drug distribution and reach clinical importance if thereby access to the target site is affected. Beyond P-glycoprotein, the family of multidrug resistance-related proteins (MRP/ABCC) substantially contributes to the elimination of numerous drugs and their metabolites. Because the interaction of MRPs with non–HIV protease inhibitor antiretrovirals has not been studied thoroughly, we investigated whether important non-nucleoside reverse transcriptase inhibitors (NNRTI) (delavirdine, efavirenz, and nevirapine), nucleoside reverse transcriptase inhibitors (NRTI) (abacavir, emtricitabine, and lamivudine), and tenofovir as a nonnucleotide reverse transcriptase inhibitor can interact with MRP1, MRP2, and MRP3 in vitro. Inhibition of these ABC transporters was quantified by confocal laser-scanning microscopy using the 5-chloromethylfluorescein diacetate assay. With the exception of abacavir, which had no effect on MRP3, all the test compounds increased intracellular 5-chloromethylfluorescein fluorescence in a concentration-dependent manner, and this effect was observed in all the overexpressing cell lines but not in the parental cell line, indicating inhibition of MRP1, MRP2, and MRP3. In conclusion, the present study provides the first evidence for a significant and concentration-dependent inhibition of MRPs by NNRTI, NRTI, and tenofovir, which was most pronounced for delavirdine, efavirenz, and emtricitabine, suggesting that this might contribute to some of the known drug interactions impairing HIV therapy and also to the superior effectiveness of combination pharmacotherapy.
Infections with the human immunodeficiency virus (HIV) are typically treated with drug combinations consisting of at least three different antiretroviral drugs. Essential components of this highly active antiretroviral therapy (HAART) are the HIV protease inhibitors (HPI), the non-nucleoside and nucleoside reverse transcriptase inhibitors (NNRTI and NRTI), and the nucleotide reverse transcriptase inhibitor tenofovir. Whereas this combination therapy substantially improves the clinical prognosis for patients infected with HIV, it concurrently increases the risk for drug-drug interactions (Piscitelli and Gallicano, 2001; de Maat et al., 2003).
Many drug interactions with antiretrovirals, but by far not all and particularly not those with NRTI, occur at the level of different cytochrome P450 isozymes (Dasgupta and Okhuysen, 2001; Piscitelli and Gallicano, 2001; de Maat et al., 2003). Indeed, increasing evidence suggests that antiretrovirals may also modify activity and expression of active drug transport systems. Such interactions may determine drug absorption, elimination, and also drug distribution and reach clinical importance if thereby access to the target site is affected. For HPI it is already well documented both in vitro and in vivo that they have multiple sites of interaction and inhibit cytochromes P450 (Eagling et al., 1997; Kumar et al., 1999; Malaty and Kuper, 1999; Granfors et al., 2006; Mikus et al., 2006) and ABC transporters like P-glycoprotein (P-gp; MDR1/ABCB1) (Gutmann et al., 1999; Profit et al., 1999; Ding et al., 2004; Sankatsing et al., 2004; Bachmeier et al., 2005; Perloff et al., 2005). Other important ABC transporters belong to the family of multidrug resistance-related proteins (MRP/ABCC) that substantially contribute to the elimination of numerous drugs and their metabolites (Borst et al., 2000). Because the interaction of MRP with non-HPI antiretrovirals has not been studied thoroughly, we investigated whether important NNRTI [delavirdine (N-[2-[4-[3-(1-methylethylamino)pyridin-2-yl]piperazin-1-yl]carbonyl-1H-indol-5-yl] methanesulfonamide), efavirenz (8-chloro-5-(2-cyclopropylethynyl)-5-(trifluoromethyl)-4-oxa-2-azabicyclo[4.4.0]deca-7,9,11-trien-3-one), and nevirapine (1-cyclopropyl-5,11-dihydro-4-methyl-6H-dipyrido [3,2-b:2′,3′-e][1,4] diazepin-6-one)], NRTI [abacavir ([4-(2-amino-6-cyclopropylamino-9H-purin-9-yl)-1-cyclopent-2-enyl]methanol), emtricitabine (4-amino-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]-pyrimidin-2-one), and lamivudine (4-amino-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]-1H-pyrimidin-2-one)], and tenofovir (1-(6-aminopurin-9-yl)propan-2-yloxymethylphosphonic acid) as a nucleotide reverse transcriptase inhibitor can interact with MRP1, MRP2, and MRP3. Inhibition of these ABC transporters was quantified by confocal laser-scanning microscopy using the 5-chloromethylfluorescein diacetate (CMFDA) assay (Bogman et al., 2003).
Materials and Methods
Compounds. The anti-HIV drugs and LY335979 (zosuquidar) were provided by the corresponding manufacturers. Stock solutions of test compounds were dissolved in dimethyl sulfoxide; only tenofovir was dissolved in double distilled water. The dimethyl sulfoxide concentration in the assays never exceeded 1% (v/v), a concentration that was found not to influence the results of the assay.
Cell Lines. As an in vitro model for human MRP1, MRP2, and MRP3, we used Madin-Darby canine kidney II (MDCKII)/MRP1, MDCKII/MRP2, and MDCKII/MRP3 cells. All the cell lines were generated by stable transfection of the corresponding cDNA into MDCKII cells (Evers et al., 1998a,b; Kool et al., 1999) and were provided by Dr. Piet Borst (The Netherlands Cancer Institute, Amsterdam, The Netherlands). MDCKII/Par cells served as a control. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% heat inactivated fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate (Invitrogen, Karlsruhe, Germany).
Confocal Laser-Scanning Microscopy: CMFDA Accumulation Assay. The method for measuring the functional activity of MRP was adopted from the CMFDA assay for MRP2 (Bogman et al., 2003; Lindenmaier et al., 2005) and extended to MRP1 and MRP3. The nonfluorescent lipophilic CMFDA (MobiTec, Göttingen, Germany) passively penetrates the plasma membrane. Inside the cells, cytosolic esterases cleave off its acetate residues, thereby releasing the fluorescent and membrane-impermeable product 5-chloromethylfluorescein (CMF), which can react (e.g., with glutathione) to form fluorescent conjugates. This methylfluorescein-glutathione complex (MF-SG) is then actively secreted by MRP2 (Bogman et al., 2003) and also by MRP1 and MRP3 if present.
Intracellular accumulation of the MF-SG in cells was analyzed with a DM IRE 2 TCS SP II confocal laser-scanning microscope (cLSM) from Leica (Bensheim, Germany) as published previously (Lindenmaier et al., 2005) with minor alterations. In brief, living cells (6 × 105) were seeded on coverslips in a closed miniperfusion chamber (H. Saur, Reutlingen, Germany) directly before the experiment and preincubated for 30 min with or without the test compound in darkness at 37°C in 1 ml of transport buffer consisting of Hank's balanced salt solution and 1 mM pyruvate (Invitrogen) for energy supply. Pilot kinetic experiments had shown that maximum effects are reached within 30 min (data not shown). CMFDA in a final concentration of 50 nM was subsequently added and incubated for 10 min. Data were analyzed according to Lindenmaier et al. (2005). The experiments were performed at least in triplicate on different days. All the compounds were tested in the highest soluble concentration or up to maximum concentration not provoking cytotoxic effects. The selective MRP inhibitor MK571 (BIOMOL Research Laboratories, Plymouth Meeting, PA) served as positive control at a concentration of 20 μM as previously established (Bogman et al., 2003).
Quenching Test. Potential errors in the quantification of CMF caused by the quenching effects of test compounds were excluded in a quenching assay by adding increasing concentrations of individual compounds to aliquots of the cell lysate after incubation with 50 nM CMFDA. Comparison of the fluorescence with control cell lysates without the respective compounds confirmed that none of them showed any quenching effect on the fluorescence of CMF.
Cytotoxicity Assay. Apart from efavirenz, none of the compounds tested exerted cytotoxic effects as evaluated with the Cytotoxicity Detection Kit (Roche Applied Science, Mannheim, Germany). Because of pronounced cytotoxic effects, efavirenz was tested in the cLSM assay only up to 10 μM.
Glutathione Assay. CMF requires the conjugation with glutathione to generate the fluorescent MRP substrate MF-SG. To exclude that the compounds alter intracellular glutathione levels, glutathione concentrations were measured in all the cell lines before and after incubation with the corresponding compounds using the QuantiChrom Glutathione Assay Kit (BioAssay Systems, Hayward, CA). The assay was conducted according to the manufacturer's instruction with 5 × 104 cells/100 μl. None of the compounds altered the intracellular glutathione level in the cell lines used.
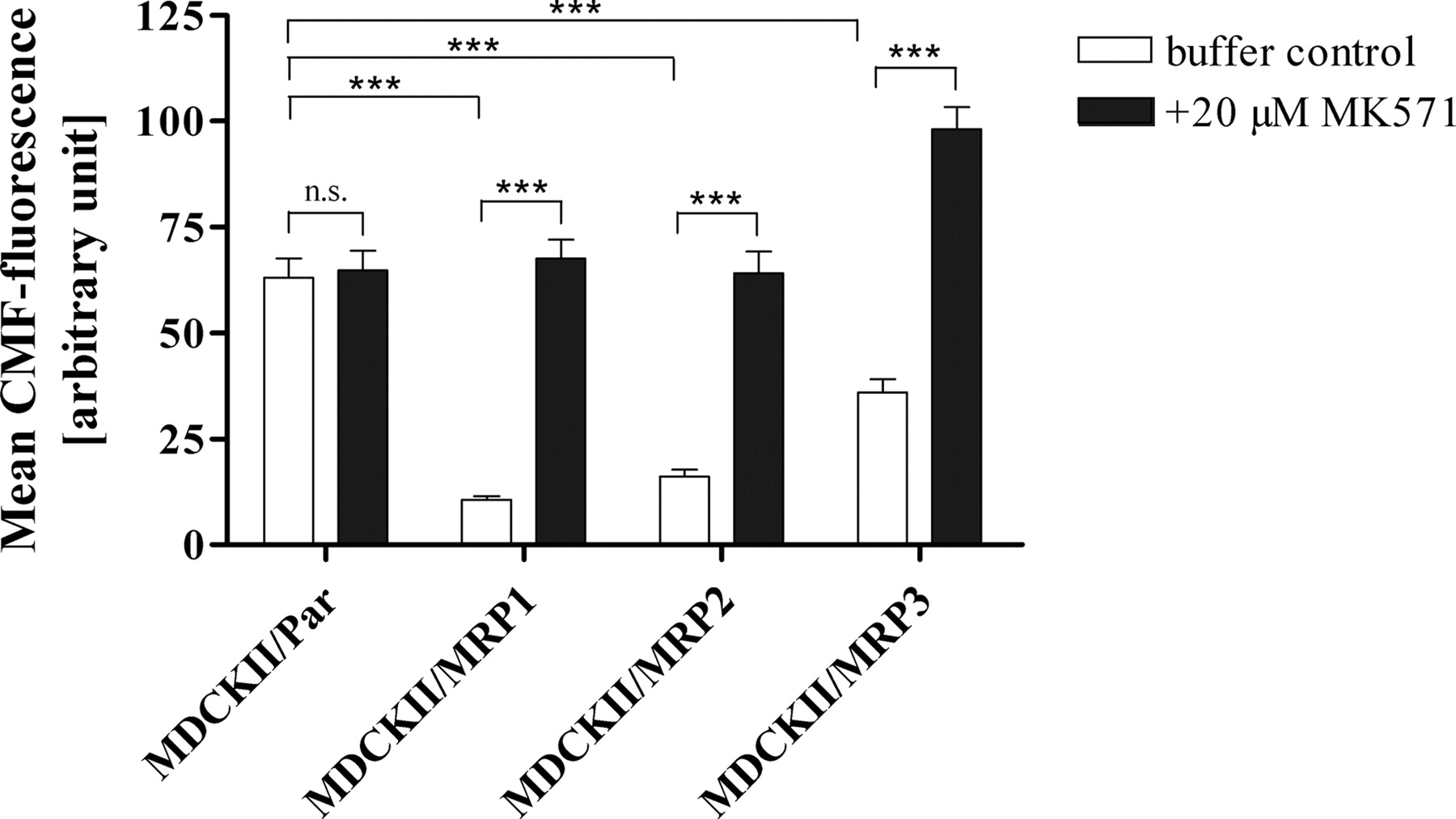
Validation of the cLSM assay evaluating MRP1, MRP2, and MRP3 inhibition. Comparison of the effect of the MRP inhibitor MK571 on intracellular CMF fluorescence in the parental and three overexpressing cell lines. Data are expressed as mean ± S.E.M. (n = 23–27 experiments). The p values were determined by unpaired two-tailed t test, with ***, p < 0.0001 and N.S., p > 0.05.
Statistical Analysis. The p values were calculated by unpaired two-tailed t test or by analysis of variance (ANOVA) with Dunnett's multiple comparison test for post hoc pairwise comparison of the results with the corresponding control (without inhibitor). All the statistical analyses were performed with GraphPad InStat, version 3.05 (GraphPad Software, San Diego, CA). A p value of ≤0.05 was considered significant.
Results
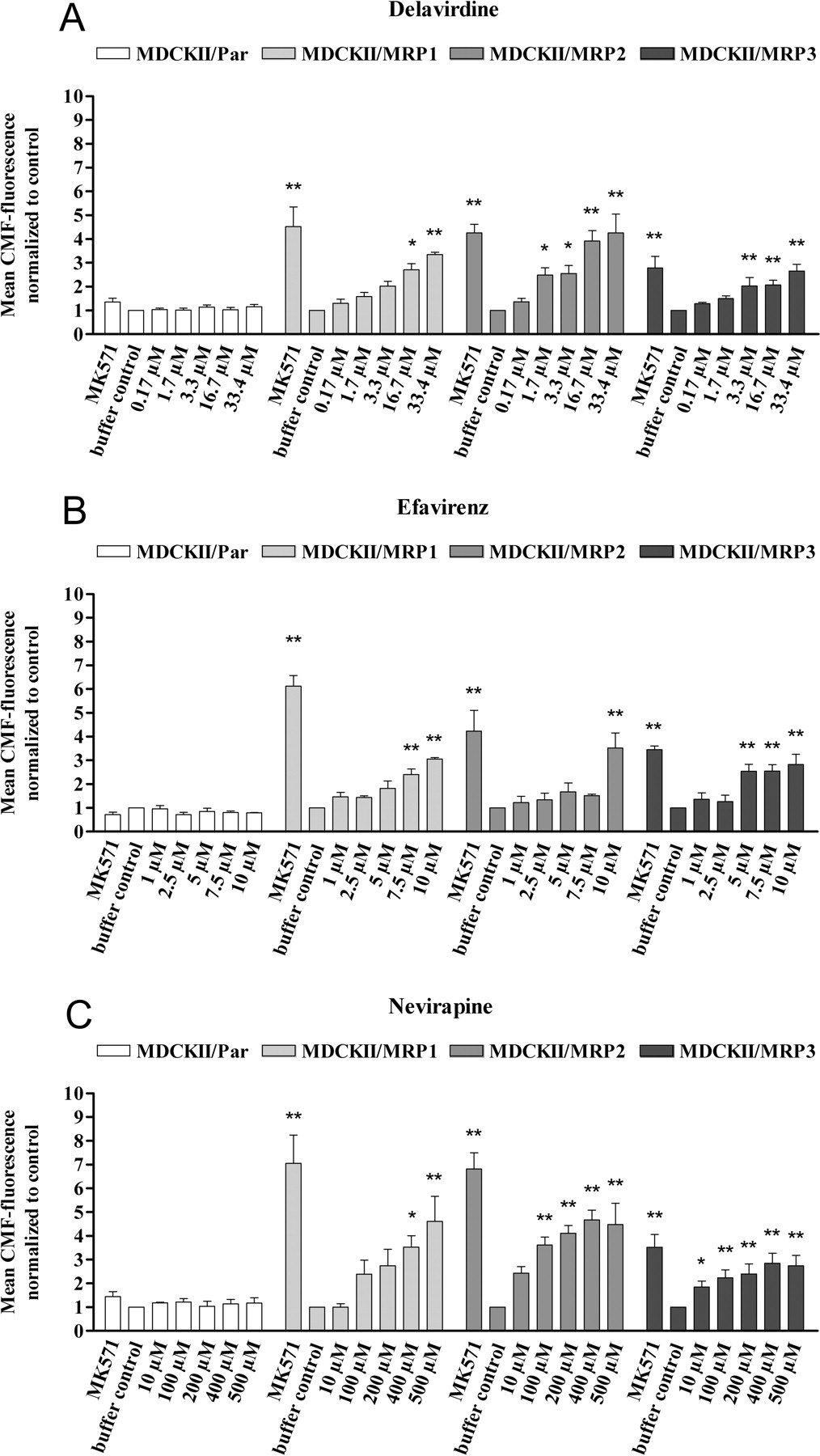
Comparison of the intracellular CMF fluorescence in the four cell lines revealed significantly higher intracellular CMF fluorescence in the parental cell line MDCKII/Par compared with the overexpressing cell lines (p < 0.0001), which did not further increase in the presence of the selective MRP inhibitor MK571 (Fig. 1). In contrast, in all the overexpressing cell lines, the addition of MK571 led to a significant increase in intracellular fluorescence (p < 0.0001), showing the suitability of this cell system for evaluating inhibition of MRP1, MRP2, and MRP3. The increase in fluorescence provoked by MK571 was generally highest in MDCKII/MRP1 and lowest in MDCKII/MRP3 cells, indicating different expression levels of the MRP or different affinities of substrate (MF-SG) or inhibitor (MK571) to the respective MRP. With the exception of abacavir, which had no effect on MRP3, all the test compounds increased intracellular CMF fluorescence in a concentration-dependent manner, and this effect was observed in all the overexpressing cell lines but not in the parental cell line, indicating inhibition of MRP1, MRP2, and MRP3 (Figs. 2 and 3).
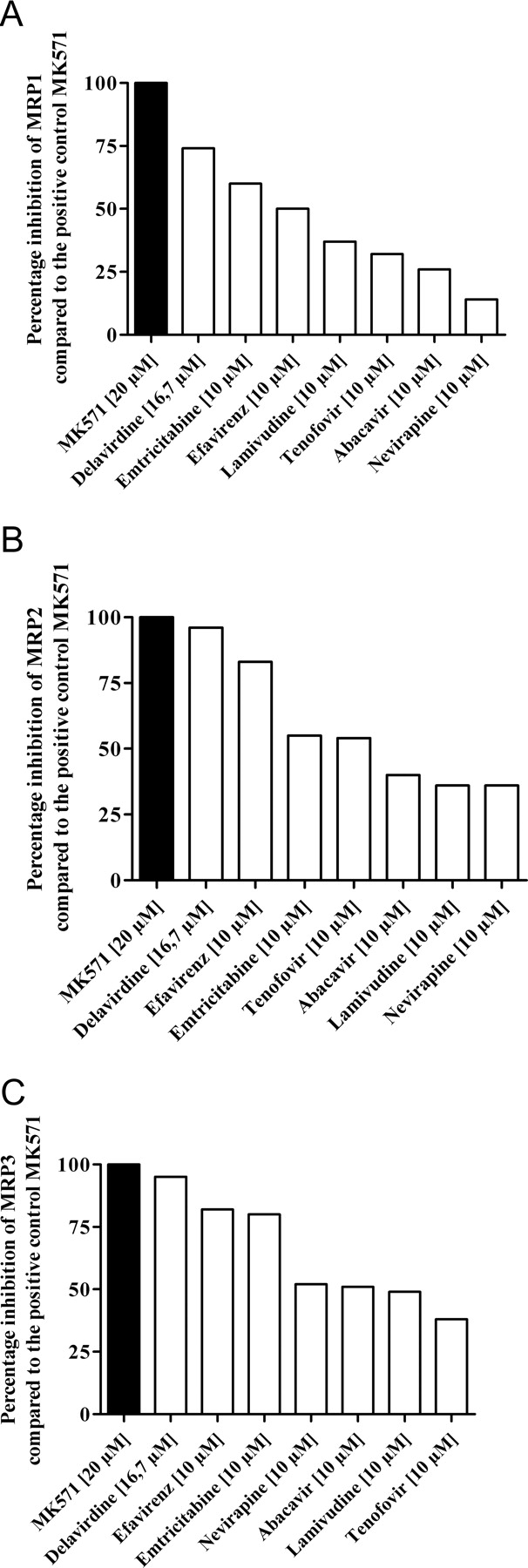
A comparison of the inhibitory potencies is difficult because differences in solubility precluded testing of all the compounds in the same concentration range. However, comparison of the effects observed at 10 μM (16.7 μM for delavirdine) (Fig. 4) revealed that delavirdine, emtricitabine, and efavirenz were the most potent inhibitors in all three cell lines. The P-gp-selective inhibitor LY335979 had no statistically significant influence on the CMF fluorescence in any of the cell lines tested, indicating that P-gp did not influence the assay.
Discussion
Increasing evidence suggests that the challenging drug interactions in patients with HAART are often caused by more than one mechanism (de Maat et al., 2003), stressing the importance of knowing all the potential targets involved and considering their complex interplay for dose individualization. Therefore, we aimed to systematically quantify the modulatory effect of frequently used non-HPI antiretroviral drugs on the important efflux transporters MRP1, MRP2, and MRP3 in vitro. The present study provides the first evidence for a significant and concentration-dependent inhibition of MRP by NNRTI, NRTI, and tenofovir.

cLSM assay assessing the concentration-dependent increase in intracellular CMF fluorescence in MCDKII/Par (control), MDCKII/MRP1, MDCKII/MRP2, and MDCKII/MRP3 cells by NNRTI. Positive control, 20 μM MK571; negative control, buffer without inhibitor. Data are expressed as mean ± S.E.M. (n = 3–5 experiments). The p values (*, p < 0.05; **, p < 0.01) were determined by ANOVA with Dunnett's multiple comparison test for post hoc comparison of the results with the buffer control.
Thus far, only very few clinical studies have addressed this issue. MRP2 inhibition by tenofovir may contribute to the known interaction between tenofovir and didanosine. Coadministration of these two antiretroviral drugs leads to an increase of the area under the didanosine concentration-time curve (AUC) by 44 to 60% (Kearney et al., 2005). This may occur through tenofovir-induced inhibition of the active uptake of didanosine into the proximal tubule cells by the human organic anion transporter 1 (Kearney et al., 2004; Zimmermann et al., 2006) or by inhibition of purine nucleoside phosphorylase, an enzyme involved in the degradation of didanosine (Ray et al., 2004). However, assuming that the MRP2 inhibitor didanosine is also an MRP2 substrate, the increase in didanosine AUC could also be achieved by inhibition of MRP2-mediated efflux in the tubular brush-border membrane or in other tissues. Inhibition of several MRP could also have contributed to the life-threatening toxicity of the MRP substrate vinblastine in a patient with HIV-associated multicentric Castleman's disease who was maintained on lamivudine, abacavir, and nevirapine (Kotb et al., 2006).

cLSM assay assessing the concentration-dependent increase in intracellular CMF fluorescence in MCDKII/Par (control), MDCKII/MRP1, MDCKII/MRP2, and MDCKII/MRP3 cells by NRTI and tenofovir. Positive control, 20 μM MK571; negative control, buffer without inhibitor. Data are expressed as mean ± S.E.M. (n = 3–5 experiments). The p values (*, p < 0.05; **, p < 0.01) were determined by ANOVA with Dunnett's multiple comparison test for post hoc comparison of the results with the buffer control.
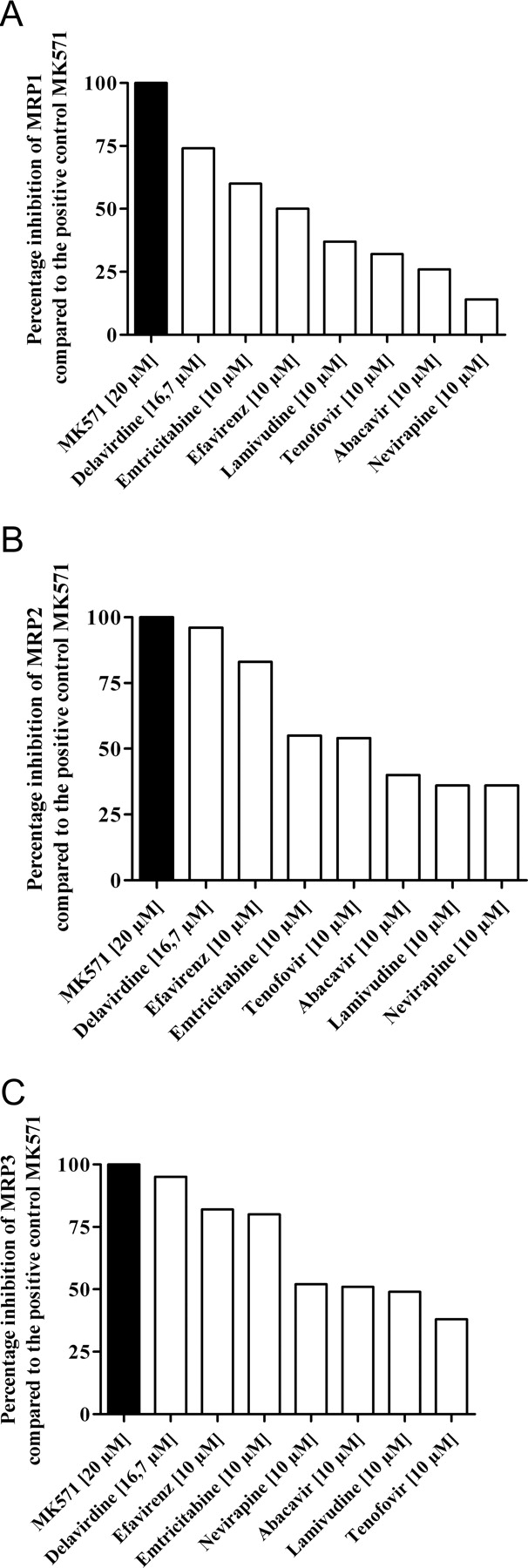
Percentage inhibition of MRP1 (A), MRP2 (B), and MRP3 (C) by anti-HIV drugs at a concentration of 10 μM (16.7 μM for delavirdine) compared with the positive control MK571.
Beyond effects in tissues responsible for drug absorption, metabolism, or elimination, MRP inhibition by NNRTI, NRTI, and tenofovir in leukocytes may also lead to increased intracellular concentrations of other antiretroviral drugs being transported by MRP. For some HPI, it has been shown that they are transported by MRP1 and MRP2 and that their intracellular concentrations depend on the activity of MRP in the leukocytes (Jones et al., 2001; Huisman et al., 2002; Janneh et al., 2005). Therefore, inhibition of MRP-mediated efflux from the target cells could contribute to the superior effectiveness of combination therapy compared with monotherapy.
Limitations include the following. First, because of their tissue distribution and localization in polarized endothelia/epithelia, the three MRP investigated do not play identical roles in drug absorption, distribution, and excretion. MRP2 is localized in apical membranes and may thus lower the bioavailability in the gut and increase the excretion of its substrates into bile and urine. In contrast, MRP1 and MRP3 are expressed basolaterally and therefore exhibit opposite effects to MRP2 on their substrates (Schinkel and Jonker, 2003). Because of the differences in transport direction and localization of the transporters, inhibition of MPR2 in vivo is expected to have other effects than inhibition of MRP1 or MRP3, and it cannot be clarified in an in vitro setting which effect will prevail. Second, the concentrations effective in vitro are partly higher than therapeutic plasma concentrations. Moreover, efavirenz and delavirdine are highly plasma protein-bound, and extrapolation to in vivo situations should therefore consider free and not total plasma concentrations. However, compounds were tested in overexpressing cell lines in which much higher concentrations are needed for inhibition than in cells or tissues with normal MRP activity because many inhibitors are also substrates of the respective transporter. Moreover, after p.o. administration, concentrations achieved in the intestine are much higher than plasma concentrations, possibly enabling MRP inhibition in the gut. Third, for all these reasons, the ultimate proof of clinical relevance can only be obtained in a clinical study. In vitro data may, however, be helpful to discover an interaction, to understand a mechanism, to detect the need for such a trial, and to plan it appropriately by selecting the most relevant compounds. In many cases, this is not an easy task because in vitro evidence was obtained in different studies using cell systems and markers that are hardly comparable. Therefore, we believe that the merit of this study is to provide comprehensive information on the interaction of the commonly used antiretrovirals with a family of important drug transporters and that this information was gathered under identical assay conditions. This allows defining ranking orders of inhibition that are more meaningful and reproducible than absolute concentrations (e.g., IC50 values), which strongly depend on assay conditions (Weiss and Haefeli, 2006). Therefore, it can only be addressed in appropriate clinical studies whether the inhibition of MRP1, MRP2, or MRP3 is clinically relevant.
In conclusion, it has become increasingly evident that the mutual interaction between antiretroviral agents is often a complex interplay of modified activities of several targets. The results of this in vitro study clearly show MRP1, MRP2, and MRP3 inhibition, particularly by delavirdine, efavirenz, and emtricitabine, suggesting that this may contribute to some of the known drug interactions impairing HIV therapy and also to the superior effectiveness of HAART.
Acknowledgments
We thank Dr. P. Borst for providing the cell lines MDCKII/MRP1, MDCKII/MRP2, and MDCKII/MRP3. We also thank GlaxoSmithKline (Brentford, UK) for abacavir and lamivudine; Gilead Sciences (Foster City, CA) for tenofovir and emtricitabine; Boehringer Ingelheim Pharmaceuticals (Ingelheim, Germany) for nevirapine; Pharmacia (Kalamazoo, MI) for delavirdine; Bristol-Myers Squibb (München, Germany) for efavirenz; and Eli Lilly and Company (Bad Homburg, Germany) for LY335979.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.012765.
-
ABBREVIATIONS: HIV, human immunodeficiency virus; HAART, highly active antiretroviral therapy; HPI, HIV protease inhibitor; NNRTI, nonnucleoside reverse transcriptase inhibitor(s); NRTI, nucleoside reverse transcriptase inhibitor(s); P-gp, P-glycoprotein; MRP, human multidrug resistance-associated protein; CMFDA, 5-chloromethylfluorescein diacetate; LY335979, zosuquidar; MDCKII, Madin-Darby canine kidney II; CMF, 5-chloromethylfluorescein; MF-SG, methylfluorescein-sulfoglutathione; cLSM, confocal laser-scanning microscope/microscopy; MK571, 3-[[[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]-[[3-dimethylamino)-3-oxopropyl]thio]methyl]thio]-propanoic acid; ANOVA, analysis of variance; AUC, area under the curve.
- Received September 21, 2006.
- Accepted December 12, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}