


Abstract
The objective of this study was to investigate whether cyclosporin A (CsA) is a modulator for breast cancer resistance protein (BCRP). The interactions between CsA and BCRP were evaluated by using both membrane- and cell-based assays. CsA inhibited BCRP or BCRP R482T mutant-associated ATPase with an IC50 of 26.1 and 7.3 μM (31,388 and 8779 ng/ml), respectively, indicating that CsA is a modulator for BCRP and its R482T mutant. The apparent permeability (Papp) of CsA was not affected by the BCRP-specific inhibitor Ko143 in both apical-to-basolateral (A-to-B) and basolateral-to-apical (B-to-A) directions in hBCRP- or mBcrp-transfected MDCKII cells, whereas CsA at 50 μM significantly increased the A-to-B transport and decreased B-to-A transport of BCRP substrates, [3H]estrone-3-sulfate ([3H]E3S) and [3H]methotrexate ([3H]MTX), in hBCRP- and mBcrp1-trasfected MDCKII cells. Similar to cellular transport studies, CsA did not exhibit ATP-dependent uptake in BCRP-expressed membrane vesicles but inhibited the ATP-mediated E3S and MTX uptake in the same vesicles. The inhibitory constant (Ki) of CsA toward BCRP was 6.7 μM (8507 ng/ml) and 7.8 μM (9380 ng/ml) when using E3S or MTX, respectively, as a BCRP substrate. The inhibitory potency of CsA on BCRP wild type or its R482T mutant was lower than that on P-glycoprotein. The present studies demonstrate that CsA is an inhibitor but not a substrate for BCRP, and has low potential to cause drug-drug interactions with BCRP substrate drugs due to its weak inhibitory effect on BCRP and BCRP R482T mutant at its normal therapeutic blood concentrations (200–400 ng/ml) (Blood 91:362–363, 1998).
Cyclosporin A (CsA) is a noncytotoxic immunosuppressant that was first discovered in 1970. It is an antibiotic produced by the fungus Tolypocladium inflatum Gams. CsA initially was used for immunosuppression following organ and marrow transplantation. Subsequently, it has been applied in virtually all branches of medicine where autoimmune or inflammatory processes play a role in the pathology (Laupacis et al., 1982).
CsA is a substrate and inhibitor of P-glycoprotein (P-gp) (Coley et al., 1989; Silbermann et al., 1989; Saeki et al., 1993). It has been used as one of the first-generation multidrug resistance (MDR) modulators to reverse MDR and improve chemotherapy (Tan et al., 2000). As a P-gp inhibitor, CsA is involved in drug-drug interactions (Sparreboom and Nooter, 2000). It can increase the plasma concentration and decrease the clearance of P-gp substrates, such as digoxin and etoposide (Carcel-Trullols et al., 2004; Englund et al., 2004; Shibayama et al., 2004), and it can also increase the brain penetration of P-gp substrates such as nimodipine and verapamil (Liu et al., 2003; Sasongko et al., 2005).
Breast cancer resistance protein (BCRP), also known as MXR, ABCP and ABCG2, was initially cloned from highly doxorubicin-resistant MCF7 AdVp human breast cancer cells and mitoxantrone-resistant S1-M1-80 human colon carcinoma cells in 1998 (Doyle et al., 1998; Miyake et al., 1999). Subsequently, it has been demonstrated to confer resistance to quite a few anticancer drugs such as mitoxantrone, doxorubicin, daunorubicin, topotecan, irinotecan, etoposide, flavopiridol, methotrexate (MTX), and imitinib, and other therapeutic agents, such as zidovudine, pantoprazole, cimetidine, sulfasalazine, nitrofuratoin, and several statins (Xia et al., 2005b). BCRP mutations have been found in some cancer cell lines (Honjo et al., 2001). The mutation has either threonine (T) or glycine (G), instead of arginine (R), at the amino acid position 482. The R482 of BCRP locates in the third transmembrane domain and may alter substrate specificity upon mutation. The mutant BCRP confers resistance to daunorubicin, doxorubicin, epirubicin, and idarubicinol (Xia et al., 2005b). Interestingly, these mutations were only found in drug-resistant human tumor cell lines (Honjo et al., 2001), but not in human individuals (Honjo et al., 2002; Zamber et al., 2003). To date, BCRP has become one of the three major ATP-binding cassette (ABC) membrane efflux transporters besides P-gp and multidrug resistance-associated protein (MRP), conferring drug resistance in cancer and inflammation chemotherapies (Silbermann et al., 1989; Litman et al., 2001; Merino et al., 2004; van der Heijden et al., 2004a,b). Besides being present in drug-resistant cancer and T-cells, BCRP is also endogenously expressed at a high level in human placenta and to a lesser extent in liver, small intestine and colon, ovary, veins, capillaries, kidney, adrenal, and lung, with little to no expression in brain, heart, stomach, prostate, spleen, and cervix (Doyle et al., 1998; Litman et al., 2001; Maliepaard et al., 2001; Scheffer and Scheper, 2002). In addition, BCRP is expressed in the human jejunum at levels considerably higher than many other ABC transporters (Taipalensuu et al., 2001). BCRP has been demonstrated to be present on the apical membrane of intestinal epithelium and to limit the oral absorption of topotecan in mouse and human (Jonker et al., 2000; Kruijtzer et al., 2002a). Given the liver and intestinal localization pattern, BCRP, similar to P-gp, may act as a barrier to uptake and limit the oral bioavailability of drugs as well as mediating hepatobiliary excretion of drugs (Jonker et al., 2000; Jorritsma et al., 2002; Kruijtzer et al., 2002b).
Since CsA is a commonly used drug for treating autoimmune or inflammatory diseases and reversing the MDR of anticancer reagents (Laupacis et al., 1982), and BCRP has broad substrate specificity (Xia et al., 2005b), it is essential to clearly understand the interactions between CsA and BCRP to better interpret the enhanced cytotoxicity of anticancer drugs and drug-drug interactions caused by CsA clinically. The effect of CsA on BCRP has been debatable (Wierdl et al., 2003; Xia et al., 2004; Ejendal and Hrycyna, 2005; Qadir et al., 2005). CsA has been demonstrated as a broad-spectrum modulator for multidrug resistance proteins such as P-gp, BCRP, MRP1, and lung cancer resistance protein by cell-based uptake and cytotoxicity studies (Qadir et al., 2005). Gupta et al. (2006) have demonstrated that CsA is not a BCRP substrate, but it inhibits BCRP substrate efflux with an IC50 of 4.3 ± 1.9 μM 5(171 ± 2285 ng/ml) using flow-cytometric efflux assay in BCRP-expressing HEK293 cells (Pawarode et al., 2007). Wierdl et al. (2003) demonstrated that CsA was a weak BCRP inhibitor and its inhibitory potency depended upon the amount of BCRP expression. CsA showed higher IC50 values in high protein-expressing cells than in the low-expressing ones. However, Ejendal and Hrycyna (2005) showed that CsA was not an inhibitor or substrate of BCRP by ATPase assay and cell accumulation studies of [3H]CsA. All the controversial results could be due to the different assay systems and the chosen substrates. In the present studies, we characterized the modulations of BCRP by CsA using both cellular transport studies and membrane-based assays. We demonstrated that CsA was not a substrate of BCRP in BCRP-overexpressed membrane vesicles, or human BCRP (hBCRP)- or murine Bcrp1 (mBcrp1)-transfected MDCKII cells. We also demonstrated that CsA reduced the ATPase activities of both wild-type BCRP and BCRP R482T mutant with an IC50 of 26.1 and 7.3 μM (31,388 and 8779 ng/ml), respectively, and inhibited BCRP-mediated efflux of estrone-3-sulfate (E3S) and MTX with a Ki of 6.7 and 7.8 μM (8507 and 9380 ng/ml), respectively. Ki was measured to avoid the potentially different inhibitory potency values caused by experimental conditions since Ki is an intrinsic constant, which reflects the affinity of the inhibitor for the functional protein (such as a transporter, enzyme, or receptor) and is independent of substrate and incubation conditions (protein concentration or incubation time, etc.). Compared with the effect on daunorubicin-stimulated P-gp ATPase activity, CsA had less potency on the inhibition of daunorubicin-stimulated BCRP R482T mutant ATPase activity. Knowledge of the inhibitory potency of CsA on BCRP is valuable to understand the drug-drug interactions caused by CsA and to assist in setting the clinic dose regimen of CsA as a MDR reversing reagent.
Materials and Methods
Human BCRP or BCPR R482T mutant-expressed cell membranes and human BCRP-expressing and control membrane vesicles were obtained from Solvo Biotechnology, Inc. (Budaörs, Hungary). The control membranes were not used in the assays since the BCRP-expressing membranes have specific BCRP activities (personal communication with Solvo). MDR1 (P-gp)-expressed membrane was obtained from BD Gentest (Woburn, MA). All chemicals were analytical grade and purchased from Sigma-Aldrich (St. Louis, MO). [3H]CsA, [3H]MTX, and [3H]E3S were obtained from American Radiolabeled Chemicals (St. Louis, MO), Moravek Biochemicals (Brea, CA), and PerkinElmer Life and Analytical Sciences (Boston, MA), respectively.
ATPase Activity Assay. BCRP, BCRP R482T mutant, or P-gp-associated ATPase activities were measured according to the method of Sarkadi et al. (1992). The liberation of inorganic phosphate from ATP was quantified using a sensitive colorimetric assay originally described by Chifflet et al. (1988). Cell membrane expressing human BCRP or P-gp (5 μg/μl) was thawed rapidly in a 37°C water bath before diluting to a concentration of 1 μg/μl in ice-cold ATPase assay medium (50 mM KCl, 2 mM dithiothreitol, 50 mM MOPS-Tris, pH 7.0) containing 2 mM EGTA, 2 mM ouabain, and 5 mM sodium azide. The experiment was carried out in a 96-well microtiter plate in triplicate. Cell membrane (20 μl of 1 μg/μl solution), with or without 360 μM sodium orthovanadate, was mixed with 20 μl of test compound serially diluted in the assay medium and preincubated at 37°C for 5 min. The reaction was initiated by adding 20 μl of 15 mM Mg-ATP. The final protein amount in the assay was 0.02 mg and the ATP concentration was 5 mM. The assay plate was placed in a 37°C incubator for 20 min after shaking at room temperature for 2 min. The reaction was terminated by the addition of 30 μl of stopping medium [10% sodium dodecyl sulfate with 1 drop of Antifoam]. The phosphate standards were constructed by mixing 20 μl of KH2PO4 standard (0.0, 0.15, 0.45, 1.5, 3.0, 4.5, 6.0, and 7.5 mM) with 20 μl of assay buffer, after addition of 30 μl of stopping medium and 20 μl of 15 mM Mg-ATP. The released phosphate or phosphate standards were measured by a modified colorimetric reaction assay described previously (Pavek et al., 2005). The sodium dodecyl sulfate-containing samples were supplemented with 160 μl of the detection reagent (5 ml of 35 mM ammonium molybdate in 15 mM zinc acetate, pH 5.0, mixed with 20 ml of 10% ascorbic acid, pH 5.0) for 20 min at 37°C, and the reaction product was measured by absorbance at 800 nm with a SpectraMax Plus384 Spectrophotometer (Molecular Devices, Sunnyvale, CA). The difference in ATPase activity in the presence or absence of sodium vanadate in the assay buffer containing EGTA, ouabain, and sodium azide was the BCRP- or P-gp-mediated ATPase activity (Ozvegy et al., 2001). The IC50 of CsA on BCRP, mutant BCRP or P-gp ATPase activities was calculated by XL-Fit (ID Business Solutions Ltd., Cambridge, MA).
Membrane Vesicle Transport Assay. ATP-dependent transport of [3H]CsA, [3H]E3S, or [3H]MTX into inside-out BCRP membrane vesicles was measured by a modified rapid filtration method that was adapted to a 96-well plate format (List et al., 2001). The assay was conducted at 37°C in a total volume of 50 μl of assay buffer (50 mM KCl, 7 mM MgCl2, and 50 mM MOPS-Tris, pH 7.0) containing 10 μg of membrane vesicle protein, test compounds, and 10 mM Mg-ATP or 10 mM Mg-AMP. BCRP-mediated uptake was measured for 4 min and stopped by 200 μl of ice-cold washing buffer (70 mM KCl and 40 mM MOPS-Tris, pH 7.0). The uptake buffer mixture was then transferred to a PerkinElmer Unifilter GF/B plate and followed by five more 250-μl washes using the cell harvest. Tritium was counted on a PerkinElmer Top Count NXT Microplate Scintillation and Luminescence Counter. ATP-dependent uptake of the test compound was calculated by subtracting the uptake in the presence of Mg-AMP from the uptake measured in the presence of Mg-ATP. The uptake rate was calculated based on the total protein content of the membrane vesicles. Ki values were calculated using a computer-generated curve-fitting program, SigmaPlot 8.0 (SYSTAT Software Inc., Chicago, IL).
Cell Culture. Polarized MDCKII cells stably expressing hBCRP or mBcrp1 (ABCG2) cDNA were kindly provided by Dr. Alfred Schinkel (Netherlands Cancer Institute, Amsterdam, The Netherlands) (Jonker et al., 2000). The MDCKII cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37°C with 5% CO2 under humidifying conditions. Cells were seeded on microporous polycarbonate membrane filters (0.33-cm2 growth area and 0.4-μ pore size Transwell; Costar, Corning, NY) at a density of 1 × 105 cells/well in 0.2 ml of complete medium. MDCKII cells were grown for 3 days with medium replacements every day.
Transport Studies. Bidirectional transport studies were performed at 37°C in air. Before each experiment, the confluent cell monolayers on Transwell inserts were washed and equilibrated for 30 min with transport media [Hanks' balanced salt solution containing 10 mM HEPES and 10 mM glucose, pH 7.4]. The experiment was initiated by adding a solution containing the test compound to either the apical (for A-to-B transport) or basolateral (for B-to-A transport) compartment. When applicable, inhibitors were present in the transport medium of the donor side from the preincubation period throughout the permeability study. At 15, 30, 45, and 60 min, the sample aliquots of receiving solutions were withdrawn from the basolateral side (for A-to-B transport) or from the apical side (for B-to-A transport) and replaced immediately with an equal amount of fresh transport medium except at the 60-min time point (the end of the incubation). The samples were mixed with 5 ml of scintillation cocktail and the radioactivity was determined in a liquid scintillation spectrophotometer (Beckman-Coulter, Fullerton, CA). The cell integrity was monitored by transepithelial electric resistance.
Transport Calculation. The cumulative amount of drug (Q) on the receiver side was plotted as a function of time. The steady-state flux J was then estimated from the slope (dQ/dt). The apparent permeability coefficient (Papp) of unidirectional flux for the test compound was estimated by normalizing the flux J (mol/s), against the nominal surface area A (0.33 cm2) and the initial drug concentration in the donor chamber C0 (mol/ml), or Papp = J/(A · C0). The basolateral/apical ratio equals the Papp value for B-to-A transport (Papp, B-to-A) divided by the Papp value for A-to-B transport (Papp, A-to-B).
All the data are expressed as mean ± S.D. of three individual incubations. Tests of significance of difference between mean values were made using a two-tailed unpaired Student's t test. A probability of less than 0.05 (p < 0.05) was considered to be statistically significant.

Effects of CsA on BCRP (A)- or BCRP R482T mutant (B)-associated ATPase activities. The vanadate-sensitive ATPase activity of BCRP-expressed cell membrane was determined as described under Materials and Methods. The maximum concentration of tested CsA was 50 μM in the assays because of its low solubility. The IC50 of CsA on BCRP- or BCRP mutant-associated ATPase activities was calculated by XL-Fit. Each data point represents the mean of triplicate determinations (±S.D.).

The effect of CsA on the daunorubicin-stimulated P-gp and BCRP R482T mutant ATPase activity. A, P-gp and BCRP R482 mutant ATPase activities in the absence or presence of 200 μM daunorubicin. The basal ATPase activities of P-gp and BCRP mutant were 3.8 and 37.4 nmol/min/mg, respectively. In the presence of 200 μM daunorubicin, the ATPase activities of P-gp and BCRP mutant were increased to 9.8 and 90.4 nmol/min/mg. B, the inhibitory effects of CsA on daunorubicin-stimulated P-gp and BCRP mutant ATPase activities. Daunorubicin was tested at 200 μM and the maximum concentration of tested CsA was 50 μM in the assays because of its low solubility. The IC50 values of CsA on daunorubicin-stimulated P-gp and BCRP mutant ATPase activities were 9.8 and 29.2 μM (11,785 and 35,116 ng/ml), respectively. The IC50 of CsA on P-gp or BCRP mutant-associated ATPase activities was calculated by XL-Fit. Each data point represents the mean of triplicate determinations(±S.D.).
Results
Effects of CsA on BCRP ATPase Activities. CsA reduced vanadate-sensitive ATPase activities of wild-type BCRP and BCRP R482T mutant with the IC50 of 26.1 and 7.3 μM (31,388 and 8779 ng/ml), respectively (Fig. 1). The inhibitory effects of CsA on BCRP and BCRP R482T mutant-associated ATPase activities indicate that CsA is a modulator for both BCRP and BCRP R482T mutant.
Effects of CsA on Daunorubicin-Stimulated P-gp or BCRP R482T Mutant ATPase Activities. To compare the inhibitory potency of CsA on P-gp and mutant BCRP, the effects of CsA on daunorubicin-stimulated P-gp or BCRP R482T mutant ATPase activities were evaluated. The basal ATPase activities of P-gp and BCRP mutant were 3.8 and 37.4 nmol/min/mg. Daunorubicin at a concentration of 200 μM could stimulate P-gp or BCRP R482 mutant ATPase activities by 2.6- and 2.4-fold, respectively (Fig. 2A). CsA inhibited daunorubicin-stimulated P-gp or BCRP mutant ATPase in a concentration-dependent manner. The solubility of CsA allowed using 50 μM as the highest concentration in these assays. The IC50 of CsA on daunorubicin-stimulated BCRP mutant ATPase activity was 29.2 μM (35,116 ng/ml), which is 3.0-fold lower than that on daunorubicin-stimulated P-gp ATPase activity (Fig. 2B), indicating that CsA may have less inhibitory potency on BCRP mutant than on P-gp.

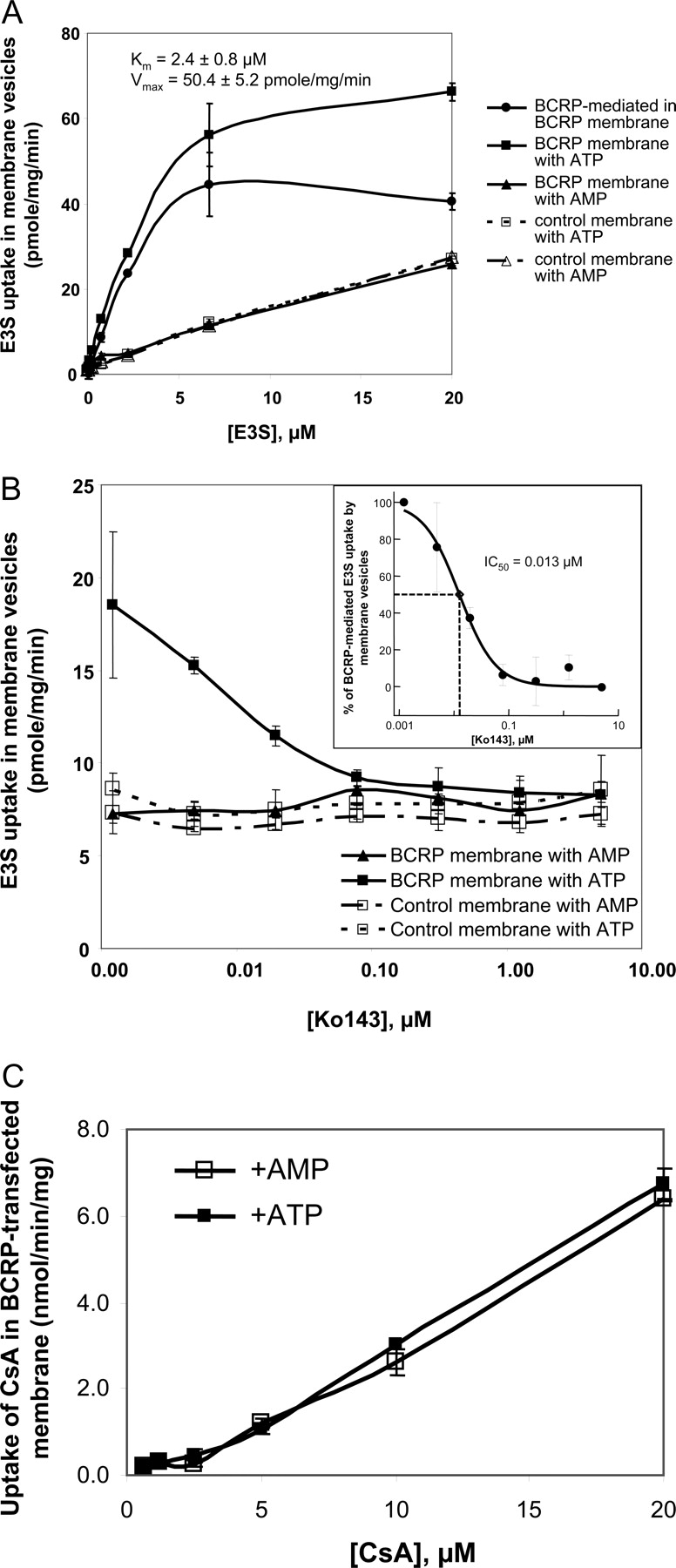
A, E3S uptake in BCRP-expressing or control membrane vesicles. The Km of E3S was approximately 2.0 μM. B, the inhibitory effect of Ko143 on E3S uptake in BCRP-expressing or control membrane vesicles. E3S was tested at 2.0 μM, which was close to Km. The inset is the determination of the IC50 of Ko143 on BCRP-mediated E3S uptake in BCRP-expressing membrane vesicles. The IC50 curve was fitted by XL-Fit. C, CsA uptake in BCRP-expressing membrane vesicles. [3H]CsA at different concentrations was incubated with BCRP membrane vesicles (10 μg) in the assay buffer containing 10 mM ATP or AMP for 4 min. Each data point represents the mean of triplicate determinations (±S.D.).
CsA Is Not a Substrate for BCRP. The known BCRP substrate, E3S (Suzuki et al., 2003), and the BCRP highly selective inhibitor, Ko143 (Allen et al., 2002; van der Heijden et al., 2004a) were used to confirm the activity of BCRP in the transfected membrane vesicles. There were no difference of E3S uptake among the BCRP-expressing membrane vesicles in the presence of AMP and the control membrane vesicles in the presence of AMP or ATP (Fig. 3A, p > 0.05). The uptake of E3S in the BCRP-expressing membrane vesicles was significantly different in the presence of ATP and AMP (Fig. 3A, p < 0.05). The ATP-dependent uptake of E3S was saturable (Fig. 3A, Km = 2.4 μM and Vmax = 50.4 pmol/mg/min). The BCRP selective inhibitor, Ko143, only inhibited E3S uptake in BCRP-expressing membrane vesicles in the presence of ATP but not in the membranes in the presence of AMP or in the control membrane vesicles (Fig. 3B). The IC50 of Ko143 on the ATP-mediated E3S uptake in BCRP-expressing membrane vesicles was approximately 0.013 μM (Fig. 3B, inset). All these results demonstrated that BCRP functions well in the BCRP-expressing membrane vesicles. There was no difference in [3H]CsA uptake in BCRP-overexpressed cell membrane vesicles in the presence or absence of ATP (Fig. 3C), suggesting that CsA is not a substrate of BCRP. To further prove this observation, the transport of CsA in hBCRP-, mBcrp-, and vector control-transfected MDCKII cells was also assessed. Ko143 did not significantly increase A-to-B transport and decrease B-to-A transport of CsA in vector control, hBCRP-, or mBcrp1-transfected MDCKII cells (Fig. 4, p > 0.05), indicating that CsA is not a substrate for BCRP or mBcrp. The lower Papp values of CsA from the A-to-B direction compared with those from the B-to-A direction in all three transfected MDCKII cells (Fig. 4) could be caused by the endogenous P-gp and MRP efflux functions. The different Papp values of CsA from the B-to-A direction among three transfected MDCKII cells (Fig. 4) were probably due to the expression level differences of other transporters, such P-gp, MRP, and OATP (Su et al., 2004).
CsA Is an Inhibitor of BCRP. The Papp value of E3S from the A-to-B direction was 4.5-fold less than that from the B-to-A direction in both hBCRP- and mBcrp-transfected MDCKII cells, whereas they did not show any significant difference in vector control-transfected MDCKII cells (p > 0.05), indicating that hBCRP and mBcrp function well in the transfected cell lines. At a concentration of 50 μM, CsA diminished the directional transport of E3S in both hBCRP- and mBcrp-transfected MDCK cells, suggesting that CsA inhibited BCRP-mediated E3S efflux (Fig. 5A).
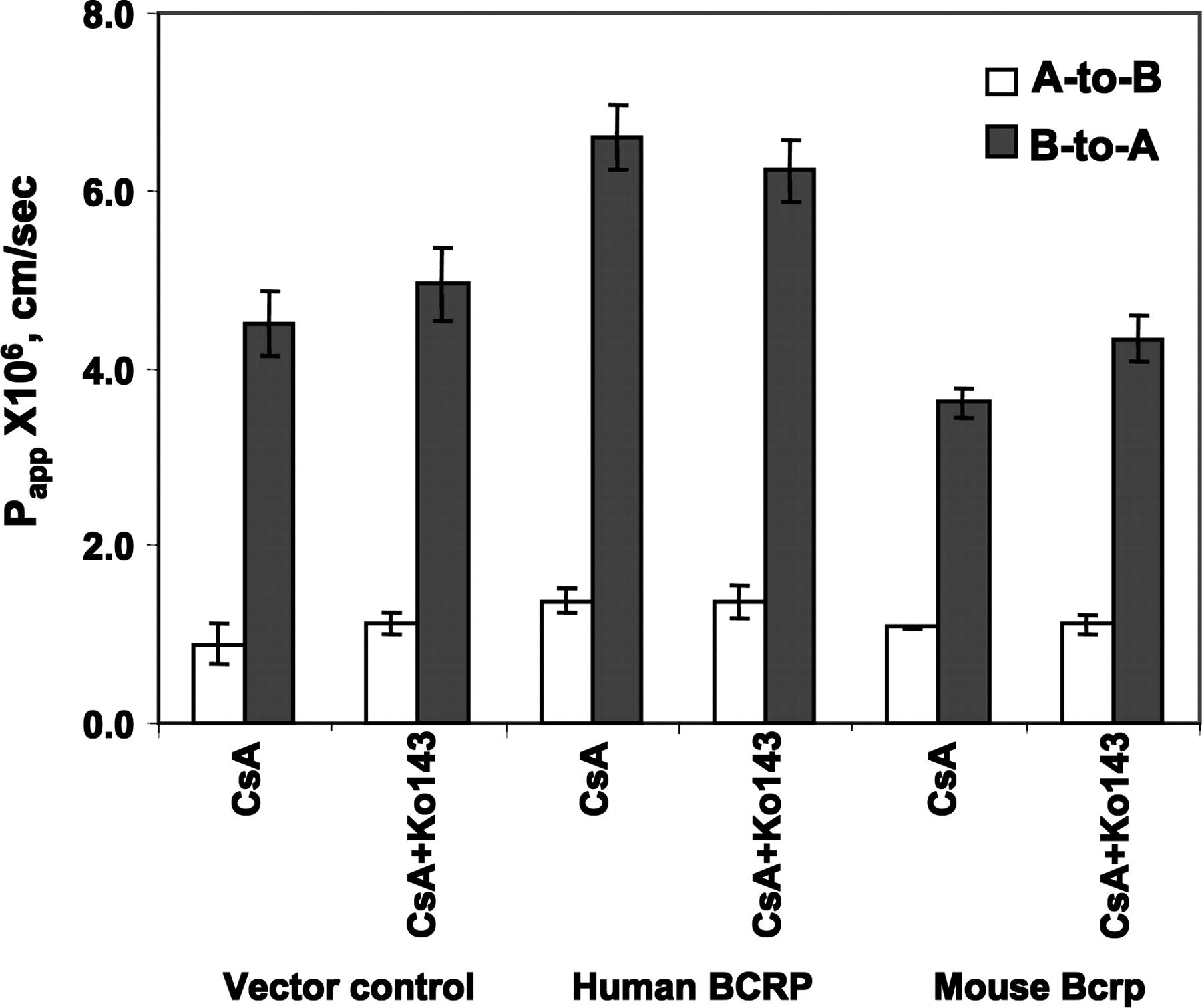
Transport of CsA in hBCRP or mBcrp1-transfected MDCKII cells. CsA and Ko143 were used at a concentration of 0.22 μM and 1 μM, respectively. Each bar represents the mean of triplicate determinations (± STD).
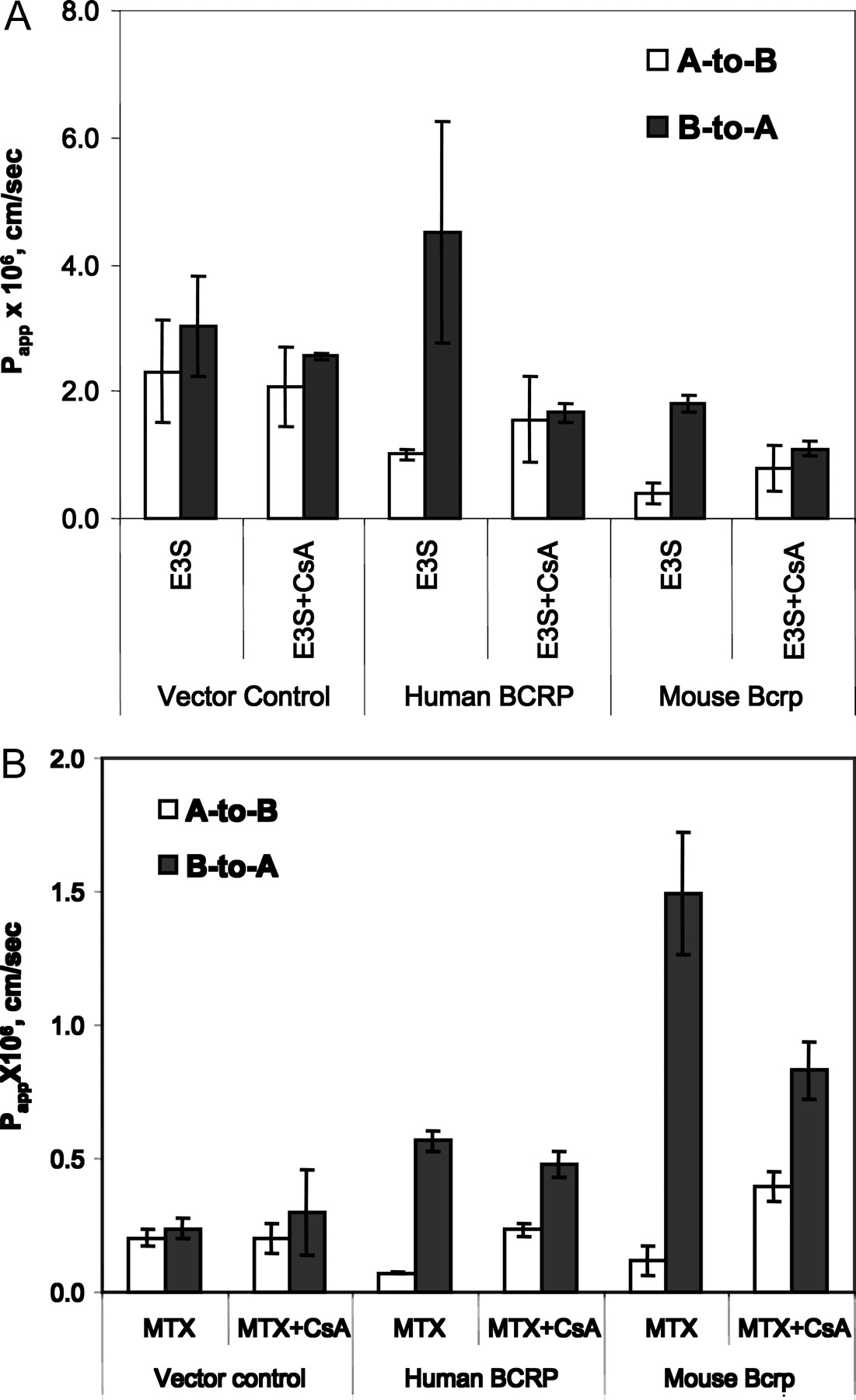
The effect of CsA on the transport of E3S and MTX in vector control-, hBCRP-, or mBcrp1-transfected MDCKII cells. [3H]E3S, [3H]MTX, and CsA were used at a concentration of 35 nM, 60 nM, and 50 μM, respectively, during the transport study. Each bar represents the mean of triplicate determinations (±S.D.).
MTX has been shown as a wild-type BCRP substrate in membrane vesicle assays (Volk and Schneider, 2003), in cell accumulation studies (Volk et al., 2002), and in the present transport studies (Fig. 6). In vector control-transfected MDCKII cells, MTX did not show any significant difference of transports from either A-to-B or B-to-A directions (Fig. 6). However, in hBCRP- and mBcrp1-transfected MDCKII cells, the Papp value of MTX from the A-to-B direction was 7.9-fold and 12.5-fold less, respectively, than that from the B-to-A direction, indicating that BCRP mediates MTX efflux in transfected cells. With 50 μM CsA coincubation in hBCRP- and mBcrp1-transfected MDCKII cells, the A-to-B transport rate of MTX was significantly increased and the B-to-A transport rate was significantly decreased (Fig. 5B, p < 0.05).
To further investigate the inhibitory potency of CsA on BCRP, we exploited the BCRP-expressing membrane vesicular transport assay by using E3S and MTX as BCRP substrates. The Km of E3S and MTX to BCRP was 2.5 μM and 1.9 mM, respectively (Fig. 6, A and B). CsA was an uncompetitive inhibitor of BCRP, and the inhibition constant (Ki) of CsA for BCRP-mediated E3S and MTX efflux was 6.7 μM (8507 ng/ml) and 7.8 μM (9380 ng/ml), respectively, which was calculated by the Eadie-Hofstee plot (Fig. 6, A and B).

The inhibitory effect of cyclosporin A on the BCRP-mediated membrane vesicular uptake of estrone-3-sulfate (A) and methotrexate (B). [3H]Estrone-3-sulfate (1.0, 1.5, 3.0, or 6.0 μM) or [3H]methotrexate (0.375, 0.75, 1.5, or 3.0 mM) was incubated with BCRP membrane vesicles (10 μg) in the assay buffer containing 10 mM ATP or AMP for 4 min at 37°C in the presence or absence of cyclosporin A (0, 5, 10, or 20 μM). ATP-dependent uptake of the test compound was calculated by subtracting the uptake in the presence of Mg-AMP from the uptake measured in the presence of Mg-ATP. Each data point represents the mean of triplicate determinations (±S.D.). The data points were fitted by Eadie-Hofstee plot from SigmaPlot (version 8.0).
Discussion
BCRP, one of the ABC transporter family members, demonstrated a baseline level of ATPase activity in BCRP-overexpressed mammalian or insect cell membranes, which is 2- to 3-fold higher than that of P-gp (Ozvegy et al., 2001, 2002). Similar to the P-gp ATPase activity, both BCRP and BCRP R482T mutant ATPase activities were vanadate-sensitive and could be stimulated or inhibited by its substrates or inhibitors (Ozvegy et al., 2001, 2002; Xia et al., 2004). Although the BCRP R482T mutant was only found in the drug-resistant human tumor cell lines (Honjo et al., 2001) but not in human individuals (Honjo et al., 2002; Zamber et al., 2003), it is still of interest to know whether CsA can be a modulator of this mutant because CsA is a commonly used MDR-reversing reagent in anticancer therapy. CsA reduced vanadate-sensitive ATPase activities of wild-type BCRP and BCRP R482T mutant with an IC50 of 26.1 and 7.3 μM (31,388 and 8779 ng/ml), respectively (Fig. 1). Because the ATPase assay is not a transport functional assay and cannot distinguish substrates from inhibitors, the inhibitory effects of CsA on BCRP ATPase activities (Fig. 1) indicate that CsA is a modulator of BCRP and BCRP R482T mutant activity. The lower IC50 of CsA on the BCRP R482T mutant ATPase activity than that on the wild-type BCRP ATPase activity (Fig. 1) suggests that CsA binding to the BCRP R482T mutant is stronger than that to the wild-type BCRP.
The functional interactions between CsA and BCRP transporter were evaluated using both membrane vesicle- and cellular-based assays. BCRP membrane vesicular transport assay is a high-through-put assay to identify BCRP substrates or inhibitors. Because BCRP-mediated transport needs ATP as an energy source and ATP cannot pass through the lipid membrane because of its hydrophilicity, only inside-out membrane vesicles can bind to ATP and pump a substrate into the vesicles. By rapid filtration, the membrane vesicles can stay in a filter membrane and the substrate trapped inside the vesicles can be measured by sensitive analytical techniques such as LC/MS/MS, fluorescence spectrometry, and scintillation spectrophotometry. The difference in the uptake in the presence or absence of ATP is attributed to BCRP-mediated transport. The membrane vesicular assay is an effective in vitro system to assess kinetic constants such as Km for substrates and Ki for inhibitors (Xia et al., 2004). CsA did not show any ATP-dependent uptake in BCRP-expressed membrane vesicles (Fig. 3C), indicating that CsA is not a substrate of BCRP. Since the membrane vesicular transport assay may give a false-negative result for compounds with high passive permeability and high lipophilicity, hBCRP-MDCKII and mBcrp1-MDCKII cells were chosen to further confirm the findings from membrane vesicular transport studies (Figs. 4). Combined with the BCRP highly selective inhibitor, Ko143 [IC50 0.033 μM for BCRP, >50 μM for P-gp, > 100 μM for MRP2, and 61.3 μM for OATP2 (Xia et al., 2005a)], we have demonstrated that CsA is not a substrate for mBcrp and BCRP in either mBcrp1- or hBCRP-transfected MDCKII cells (Fig. 4) and is consistent with published observations (Saeki et al., 1993; Gupta et al., 2006).
CsA has been used to reverse MDR by abrogating P-gp in cancer treatments (List et al., 2001). CsA also inhibits BCRP- or mBcrp1-mediated E3S and MTX efflux hBCRP-MDCKII and mBcrp1-MDCKII cells (Figs. 5 and 6). As an inhibitor for efflux pumps, CsA can be potentially applied in clinic to alleviate MDR function in cancer cells and improve the drug absorption and disposition of efflux pump substrates. To get clinical benefits from the combination chemotherapy by overcoming MDR with CsA, determination of the inhibitory potency is valuable for the dose regimen. Since the IC50 values of CsA on BCRP inhibition are dependent on BCRP protein expression (Wierdl et al., 2003), we measured the Ki of CsA on BCRP using a low-affinity substrate (MTX) and a high-affinity substrate (E3S) in membrane vesicular transport assays. The Ki of CsA for wild-type BCRP was 6.7 μM (8507 ng/ml) and 7.8 μM (9380 ng/ml), determined by using human BCRP-expressing membrane vesicles and E3S and MTX as BCRP substrates, respectively (Fig. 6). The Southwest Oncology Group reported (List et al., 2001; Doyle and Ross, 2003) that CsA, administered by 72-h continuous intravenous infusion at a dosage of 5 mg/kg per day concurrently with daunorubicin, has no clinical benefit in the daunorubicin chemotherapy for relapsed acute myeloid leukemia, whereas CsA at a dosage of 16 mg/kg significantly improved median survival and overall survival rates in P-gp-positive AML but not in P-gp-negative AML patients. It is suspected that the lack of survival benefits in the P-gp-negative patients may be due to the presence of other efflux transporters, such as BCRP, in AML patients (Doyle and Ross, 2003). A possible reason for this could be that the median whole-blood CsA concentration of 1774 ng/ml, when CsA was i.v. infused at 16 mg/kg per day in AML patients, was higher than the CsA Ki for P-gp of 1.1 μM (1311 ng/ml) (Shiraki et al., 2000), but lower than the Ki of 6.7 μM (8507 ng/ml) or 7.8 μM (9380 ng/ml) for BCRP (Fig. 6). Daunorubicin is known to be a more selective substrate for BCRP R482T mutant (Honjo et al., 2001) than wild-type BCRP. The inhibitory effect of CsA on daunorubicin-simulated BCRP R482T mutant ATPase activity was 3.0-fold less than that on P-gp ATPase activity (Fig. 2B), indicating that CsA has less inhibitory potency on BCRP mutant than on P-gp.
In conclusion, we have used BCRP-expressing membrane and cell lines to investigate the inhibitory mechanism and potency of CsA on BCRP. CsA is not a substrate but an uncompetitive inhibitor of BCRP. CsA can modulate both wild-type BCRP and BCRP R482T mutant activity, with Ki of 6.7 to ∼7.8 μM (8507–9380 ng/ml) for the wild-type BCRP using E3S and MTX as substrates. Therefore, CsA may not cause BCRP-related drug-drug interactions at its normal therapeutic blood concentrations (200–400 ng/ml, [I]/Ki <0.04) (Emilia et al., 1998). To exploit CsA-mediated BCRP inhibition for reversing MDR in cancer chemotherapy, the pharmacological concentration of CsA may need to be higher than 6.7 to 7.8 μM (8507–9380 ng/ml) to obtain therapeutic benefits upon coadministration with BCRP substrate drugs.
Acknowledgments
We thank Dr. Alfred Schinkel from The Netherlands Cancer Institute (Amsterdam, The Netherlands) for his kindness in providing us Ko143 and BCRP-transfected cell lines. We also thank Dr. Suresh K. Balani from Millennium Pharmaceuticals, Inc. for scientific comments on the manuscript.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.011866.
-
ABBREVIATIONS: CsA, cyclosporin A; P-gp, P-glycoprotein; MDR, multidrug resistance; BCRP, breast cancer resistance protein; MTX, methotrexate; ABC, ATP-binding cassette; MRP, multidrug resistance-associated protein; B-to-A, basolateral-to-apical; A-to-B, apical-to-basolateral; h, human; m, mouse; E3S, estrone-3-sulfate; MOPS, 3-(N-morpholino)propanesulfonic acid; Papp, apparent permeability.
- Received July 6, 2006.
- Accepted January 9, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}