


Abstract
The amino acid sequences of the human UDP-glucuronosyltransferases (UGTs) 1A9 and 1A10 are 93% identical, yet there are large differences in their activity and substrate selectivity. For example, the regioselectivity in propranolol glucuronidation, the regioselectivity in dobutamine glucuronidation, and the glucuronidation rate of α- and β-estradiol differ greatly between UGT1A9 and UGT1A10. To identify the residue responsible for the activity differences, we divided the N-terminal half of the two UGTs into five comparable segments by inserting four unique restriction sites at identical positions in both genes and constructing chimeras in which segments of UGT1A9 were individually replaced by the corresponding segments from UGT1A10. Activity analyses of the resulting mutants, 910A [1A10(1–83)/1A9(84–285)], 910B [1A9(1–83)/1A10(84–147)/1A9(148–285)], 910C [1A9(1–147)/1A10(148–181)/1A9(182–285)], 910D [1A9(1–181)/1A10(182–235)/1A9(236–285)], and 910E [1A9(1–235)/1A10(236–285)] indicated that more than one residue is responsible for the differences between UGT1A9 and UGT1A10. We next prepared four double chimeras, in which two of the above UGT1A9 segments were replaced simultaneously by the corresponding UGT1A10 segments. However, none of the double chimeras glucuronidated either estradiol, propranolol, or dobutamine at rates that resembled those of UGT1A10. On the other hand, studying the kinetics of 1-naphthol glucuronidation yielded more focused results, indicating that residues within segment B (84–147) contribute directly to the Km value for this substrate. Further mutagenesis and activity assays suggested that Phe117 of UGT1A9 participates in 1-naphthol binding. In addition, it appears that residues within segment C of the N-terminal domain, mainly at positions 152 and 169, contribute to the higher glucuronidation rates of UGT1A10.
UDP-glucuronosyltransferases (UGTs) (EC 2.4.1.17) catalyze the glucuronidation of many endogenous and exogenous compounds. The human UGTs are approximately 530 amino acid long membrane proteins, of which the first 25 residues, the signal sequence, are removed after the transfer of the newly synthesized polypeptides to the endoplasmic reticulum. A single transmembrane helix is predicted close to the C terminus of the proteins, and their membrane topology is such that most of the mass of the proteins is on the luminal side of the endoplasmic reticulum membrane (Radominska-Pandya et al., 1999; Ouzzine et al., 2003). There are 19 functional human UGTs that are divided into three subfamilies, 1A, 2A, and 2B, based on the sequence homology and gene structure (for UGTs of other subfamilies, see Mackenzie et al., 2005, 2009). The aglycone substrate selectivity of the UGTs is very complex, and most of them can bind and glucuronidate compounds that differ significantly in their chemical structure (Tukey and Strassburg, 2000; Ouzzine et al., 2003). There is often partial overlap in the substrate selectivity of individual UGTs, but there are also unique or specific substrates that are glucuronidated by only a single UGT (Court, 2005).
The UGTs of subfamily 1A share exons 2 to 5 and, as a consequence, their amino acid sequences from Glu286 (UGT1A9 numbering, Fig. 1) to the end of the protein are identical. The N-terminal halves of the UGT1As are encoded by individual exon 1s and are variable, but highly homologous, particularly among UGTs 1A7 to 1A10. Only 16 residues of UGT1A10 are unique to this enzyme and are not identical to amino acids in one or more of the corresponding residues in either UGTs 1A7, 1A8, or 1A9 (Fig. 1). Mature UGT1A9 and UGT1A10 (after the cleavage of the signal sequence) differ by 33 amino acids out of 502 and, as may be expected, there are overlaps in their substrate selectivity (Luukkanen et al., 2005). Nevertheless, we have previously found interesting differences in their activity with different substrates, including dobutamine (Alonen et al., 2005), propranolol (Sten et al., 2006), estradiol (Itäaho et al., 2008), and dopamine (Itäaho et al., 2009). With dobutamine and dopamine, the regioselectivity of UGT1A10 deviates from that of UGT1A7, 1A8, and 1A9; UGT1A10 favors the hydroxyl at the para position, whereas the other isoforms favor the hydroxyl at the meta position (Alonen et al., 2005; Itäaho et al., 2009). A similar situation is seen with propranolol and estradiol; UGT1A10 favors R-propranolol and β-estradiol, whereas UGT1A9 favors S-propranolol and epiestradiol (Sten et al., 2006; Itäaho et al., 2008). There are also differences in enzyme kinetics between UGT1A9 and UGT1A10 with many common substrates such as 1-naphthol, entacapone, p-nitrophenol, and 4-methylumbelliferone (Uchaipichat et al., 2004; Luukkanen et al., 2005; Xiong et al., 2006).
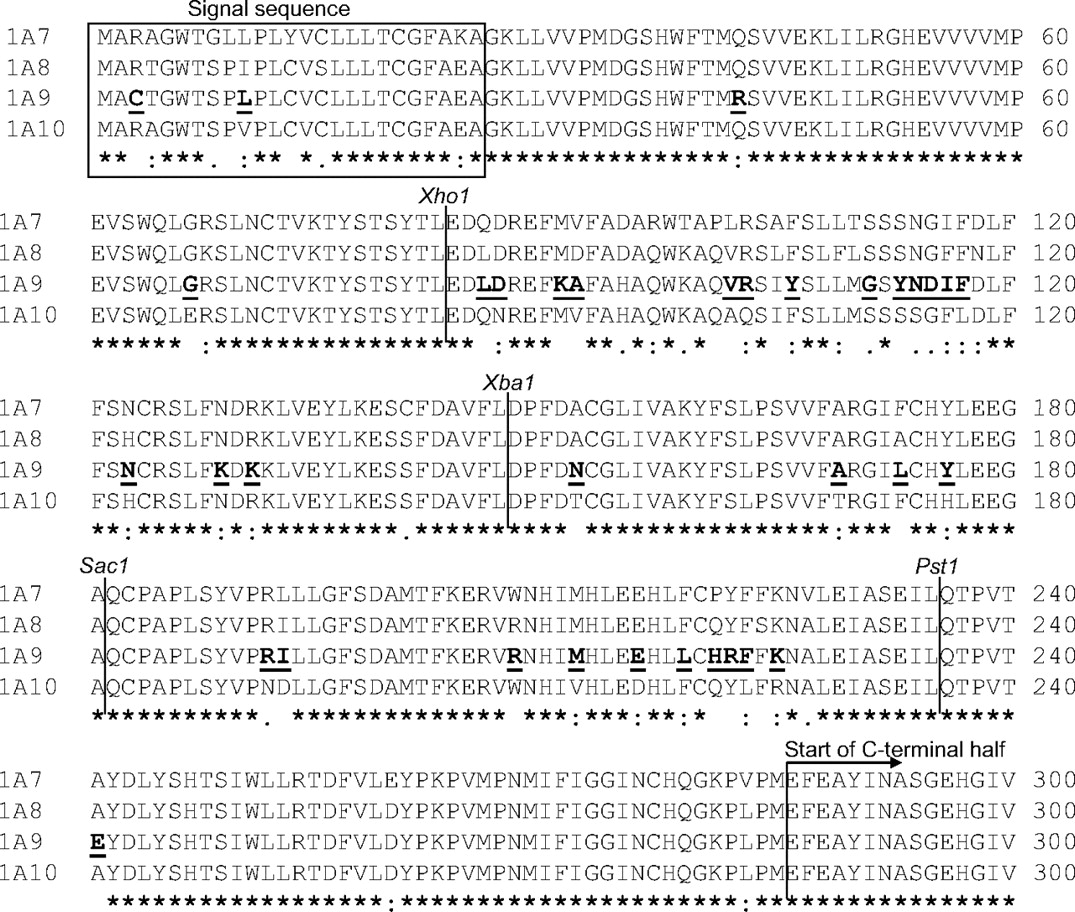
Sequence alignment of the first exons of UGT 1A7, 1A8, 1A9, and 1A10. The locations of the four new restriction sites that were inserted into UGT1A9 and UGT1A10 are indicated as well as the residues in UGT1A9 that differ from the corresponding amino acids in UGT1A10.
Several mutagenesis studies were conducted previously to identify amino acids that confer variable (even if partly overlapping) substrate specificity on individual UGT isoforms. In an elegant work on the differences between UGTs 1A3 and 1A4, Kubota et al. (2007) found that the residues at positions 36 and 40 contribute to the substrate selectivity of these enzymes. The corresponding residues in UGT1A9 and UGT1A10, Met33 and His37, are the same in both UGT1A9 and UGT1A10 and, therefore, cannot explain the activity differences between them. Lewis et al. (2007) constructed UGT2B7-UGT2B15 chimeras to locate the substrate binding-domains of these enzymes. They found that amino acids within the 61 to 194 region were important for the substrate selectivity of UGT2B15 and may be involved in substrate binding. Nishiyama et al. (2008) obtained comparable results with UGT1A8-UGT1A9 chimeras and concluded that amino acids within the 69 to 132 segment are important for the substrate specificity of UGT1A9. More specifically, Fujiwara et al. (2009) observed that amino acids 42 and 152 are important in determining the differences in substrate specificity of UGT1A9 and UGT1A8.
The aim of this study was to find out which and how many amino acids play major roles in the activity differences, including regio- and stereoselectivity as well as kinetics, between UGT1A9 and UGT1A10. To this end, we constructed different mutants, mainly aimed to confer the higher activity of UGT1A10 onto UGT1A9 (gain of function), to increase the credibility and significance of the findings. Although we failed to turn UGT1A9 into a UGT1A10-like enzyme, the results of this study yield valuable information on substrate selectivity of the human UGTs.
Materials and Methods
Chemicals and Reagents.
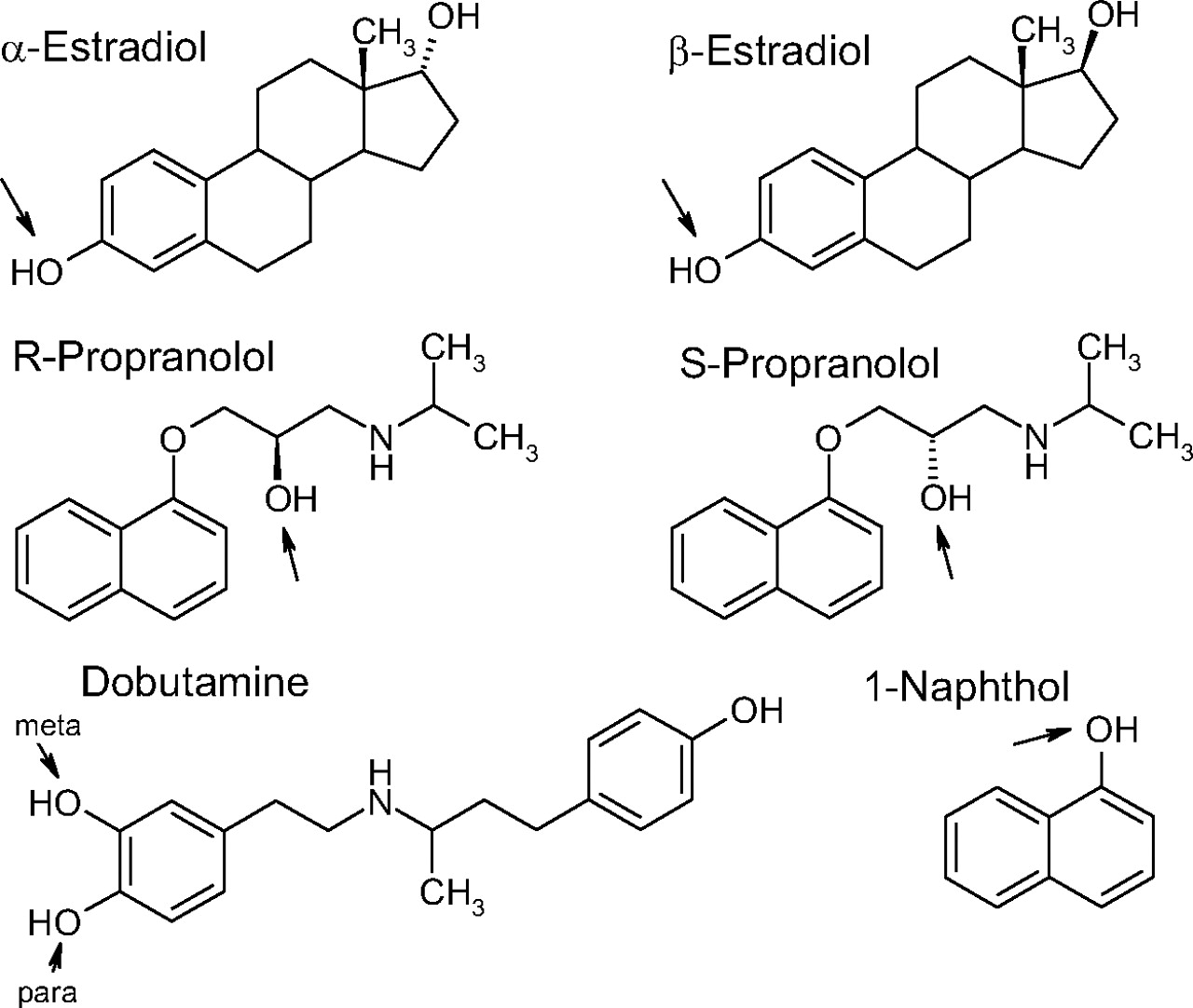
Dobutamine hydrochloride was purchased from Eli Lilly & Co. (Indianapolis, IN) and UDP-glucuronic acid (UDPGA) (trisodium salt and triammonium salt), S-propranolol, R-propranolol, 1-naphthol, 17β-estradiol, and 17α-estradiol (epiestradiol) were obtained from Sigma-Aldrich (St. Louis, MO and Steinheim, Germany). The structures of all of the aglycone substrates are presented in Fig. 2. Sodium dihydrogen phosphate dihydrate and disodium hydrogen phosphate were from Fluka (Buchs, Switzerland and Steinheim, Germany), ammonium acetate was from Riedel-de Haën (Seelze, Germany), acetic acid, and HPLC-grade methanol were from Mallinckrodt Baker (Deventer, Holland), and acetonitrile was from Rathburn Chemicals (Walkerburn, UK).

Chemical structures of the substrates used in this study. The glucuronidation sites that were examined here are marked with arrows.
Recombinant UDP-Glucuronosyltransferases and Mutagenesis.
Recombinant UGTs 1A9 and 1A10 were produced in our laboratory as His-tagged proteins in baculovirus-infected insect cells (Kurkela et al., 2003; Kuuranne et al., 2003). Mutagenesis was done by polymerase chain reaction, and the correctness of the resulting mutants was confirmed by DNA sequencing of the entire fragment that was amplified by polymerase chain reaction and subcloned into the previously sequenced vectors. The constructed chimeric and point mutants were expressed in baculovirus-infected insect cells as described previously (Kurkela et al., 2003; Kuuranne et al., 2003). Protein concentrations were determined by the BCA method (Pierce Biotechnology Inc., Rockford, IL). The relative expression level of the recombinant UGTs was determined using a monoclonal antibody, tetra-His (QIAGEN, Hilden, Germany), directed to the C-terminal His-tag that they carry, as detailed earlier (Kurkela et al., 2007). The relative expression level values are listed in Table 1. All of the activity rates that are presented in this study are normalized values with respect to the expression level of UGT1A10.
Mutations made to UGT1A9 and the relative expression levels of the mutant and control enzymes
Propranolol Glucuronidation Assays.
The samples were incubated in duplicate at 37°C for 60 min in the presence of 0.5 to 4 mg/ml protein, 2 mM UDPGA, 500 μM S-propranolol or R-propranolol, and 2% dimethyl sulfoxide (DMSO) in a total volume of 100 μl. The reactions were terminated by adding 10 μl of chilled 4 M perchloric acid and cooling the tubes in a cold block followed by centrifugation for 6 min at 16,100g to remove the precipitated proteins. Twenty microliters of the supernatant was injected to the HPLC system. Agilent 1100 series HPLC equipment (Agilent Technologies, Waldbronn, Germany) was used with a Zorbax Eclipse Plus C18 (150 × 4.6 mm; 5 μm) column (Agilent) and fluorescence detector (excitation at 230 nm and emission at 342 nm). A mixture of 50 mM phosphate buffer (pH 3.0) and methanol was used as the mobile phase. For S-propranolol, 30% methanol was used. The retention time of the glucuronide was 31.2 min, and the total run time was 40 min. For R-propranolol 45% methanol was used. The retention time of the glucuronide was 5.5 min and the total run time was 10 min. For both isomers the flow rate was 1 ml/min, and the column temperature was 40°C.
Estradiol Glucuronidation Assays.
The samples were incubated in triplicate at 37°C for 60 min in the presence of 0.5 to 4 mg/ml protein, 2 mM UDPGA, 100 μM epiestradiol or β-estradiol, and 5% DMSO in a total volume of 100 μl. The reactions were terminated by adding 10 μl of chilled 4 M perchloric acid. After centrifugation, 80 μl of the supernatant was injected into the high-performance liquid chromatograph equipped with a fluorescence detector (excitation at 216 nm and emission at 316 nm). A Chromolith SpeedRod RP18e column (50 × 4.6 mm; Merck, Darmstadt, Germany) was used, and the mobile phase consisted of 60% 25 mM phosphate buffer (pH 3.0) and 40% methanol. The flow rate was 1 ml/min for 0 to 9.5 min and 2 ml/min for 10 to 19.5 min, and the column temperature was 30°C. The retention times were 4.8 and 5.7 min for β-estradiol-3-glucuronide and epiestradiol-3-glucuronide, respectively, and the total run time was 20 min.
Dobutamine Glucuronidation Assays.
The samples were incubated in duplicate at 37°C for 60 min in the presence of 0.5 to 4 mg/ml protein, 2 mM UDPGA, 1 mM dobutamine, and 2% DMSO in a total volume of 100 μl. The reactions were terminated by adding 10 μl of chilled 4 M perchloric acid. After centrifugation, 40 μl of the supernatant was injected into the high-performance liquid chromatograph equipped with a fluorescence detector (excitation at 285 nm and emission at 313 nm). The reaction products were analyzed by HPLC with a Hypersil BDS-C18 column (250 × 4.0 mm, 5 μm) and 20 mM ammonium acetate (pH 4.5) and methanol as the mobile phase. The separation of glucuronides was achieved with isocratic flow of 10% methanol for 5 min followed by a linear gradient of 10 to 35% methanol for 5 to 25 min. The column was washed with 35% methanol for 5 min and with 35 to 10% methanol for 1 min. The column was equilibrated with 10% methanol for 4 min, and the total run time was 35 min. Retention times were 11.9 min for phenolic dobutamine glucuronide, 14.9 min for dobutamine-para-glucuronide, and 17.9 min for dobutamine-meta-glucuronide. The flow rate was 1 ml/min, and the column temperature was 40°C.
The estradiol screening assays were done in triplicate, as were many of the assays with propranolol and dobutamine. However, because of the very large number of samples some of the screening assays with propranolol and dobutamine were done in duplicate. The results of the duplicated samples were mostly close to each other with 1 to 15% difference, except for the low activity sample 910A, for which they were 30%.
1-Naphthol Glucuronidation Assays.
The samples were prepared in triplicate, and they were incubated at 37°C in the presence of 2 mM UDPGA and 2% DMSO in a total volume of 100 μl. The protein concentration and the reaction time varied, within the linear range, from 0.05 to 0.5 mg/ml protein and from 10 to 30 min, respectively. Eight substrate concentrations were selected for each enzyme after preliminary studies, and the ranges used for each enzyme preparation are presented in Table 2. The reactions were terminated by adding 10 μl of chilled 4 M perchloric acid. After centrifugation, 40 μl of supernatant was injected to the high-performance liquid chromatograph equipped with a fluorescence detector (excitation at 285 nm and emission at 335 nm). A Hypersil BDS-C18 column (150 × 4.6 mm, 5 μm) was used for the separation, and the mobile phase consisted of 58% 50 mM phosphate buffer (pH 3) and 42% methanol. The column temperature was 40°C, the flow rate was 1 ml/min, and the total run time was 15 min. The retention time of 1-naphthylglucuronide was 4.2 min.
Enzyme kinetic parameters for the glucuronidation of 1-naphthol
Vmax values were normalized with respect to UGT1A10 (see Table 1 for relative expression levels). All the values are expressed as best fit values ± S.E. of the estimate.
Enzyme Kinetic Analyses.
The normalized initial velocity data from the enzyme kinetic studies were fitted to the Michaelis-Menten equation [v = Vmax × S/(Km+ S), where v is the initial velocity of the reaction, S is the substrate concentration, Vmax is the maximal velocity, and Km is the substrate concentration at 0.5 Vmax], to the substrate inhibition equation [v = Vmax × S/(Ks + S + S2/Ki), where Ki is the constant describing the substrate inhibition interaction], or to the biphasic equation [v = Vmax1 × S/(Km1+ S) + Vmax2 × S/(Km2 + S)] by nonlinear regression using GraphPad Prism (version 5 for Windows; GraphPad Software, Inc., San Diego, CA).
Modeling of the Active Site.
A homology model for human UGT1A9 was constructed using Modeler 9v6 with a standard modeling scheme (Sali and Blundell, 1993). For the C-terminal domain, the human UGT2B7 (Protein Data Bank code 2o6l) was used as a template and for the N-terminal domain, UGT72B1 (Protein Data Bank code 2vce) from Arabidopsis thaliana, GtfB (Protein Data Bank code 1iir) from Mycolatopsis orientalis, and macrolide glycosyltransferase (Protein Data Bank code 2iya) from Streptomyces antibioticus were used. The modeling of the segments of poor homology was aided with secondary structure predictions by PredictProtein (Rost et al., 2004). Full details of the model construction are given elsewhere (L. Laakkonen and M. Finel, submitted for publication). Eventually, 1-naphthol was docked manually to this resultant model of UGT1A9.
Results
Segmentation of the N-Terminal Halves and Construction of Chimeras.
At the outset of this study, we selected an assumption-free approach to finding the location of the residue or residues that play a central role in determining the activity differences between UGT1A9 and UGT1A10. In line with this approach, we divided the N-terminal half of each of the two enzymes into five exchangeable segments of comparable length by inserting four unique restriction sites at identical positions along the exon 1-encoded part of the enzymes. The restriction sites were generated by silent mutations so that no amino acid was changed at this stage. The inserted restriction sites were Xho1 (in the codons for Leu83-Glu84), Xba1 (codons for Phe146-Leu147-Asp148; this mutagenesis also eliminated the internal BamH1 site of UGT1A10), Sac1 (codons for Gly180-Ala181-Gln182), and Pst1 (codons for Gln235 and Thr236) (Fig. 1).
The four new unique restriction sites were used to generate chimeras in which each one of the five N-terminal segments of UGT1A9 was separately replaced by the corresponding segment from UGT1A10 (Fig. 3). The chimeric mutants generated in this way were named 910 (combination of UGT1A9 and UGT1A10) and a letter (A–E), denoting the segment originating from UGT1A10, namely 910A [1A10(1–83)/1A9(84–285)], 910B [1A9(1–83)/1A10(84–147)/1A9(148–285)], 910C [1A9(1–147)/1A10(148–181)/1A9(182–285)], 910D [1A9(1–181)/1A10(182–235)/1A9(236–285)], and 910E [1A9(1–235)/1A10(236–285)] (Fig. 3). A list of all the mutations that were involved in each of these chimeras is given in Table 1.

A schematic diagram showing the different single and double chimeras that were constructed for this study.
Regio- and Stereoselectivity of the UGT1A9-UGT1A10 Chimeras.
The activity of the different 910 chimeras, alongside the parent enzymes, was examined using compounds whose glucuronidation varied considerably between UGT1A9 and UGT1A10. We selected R-and S-propranolol, 17-β and 17-α estradiol (the latter is also called epiestradiol), and dobutamine (Fig. 2) for this large screen because earlier studies showed large differences in either stereoselectivity (Sten et al., 2006; Itäaho et al., 2008) or regioselectivity (Alonen et al., 2005).
The propranolol glucuronidation activities of the different chimeras are shown in Fig. 4A (the figure also contains results with point mutants that are described below). None of the single-segment chimeras displayed a UGT1A10-like high rate of R-propranolol glucuronidation, but the activity of 910B and 910C differed most from that of the parent UGT1A9 (Fig. 4A), nor did any of the single-segment chimeras glucuronidate estradiols such as UGT1A10, particularly considering the very high turnover rate of UGT1A10 toward the 3-hydroxy of β-estradiol (Fig. 4B). Mutants 910B and 910C revealed a qualitative change in the stereoselectivity pattern toward UGT1A10, but because the rates were only a fraction of that exhibited by UGT1A10, the observation is inconclusive. Once again, 910D and 910E resemble closely UGT1A9 in their estradiol glucuronidation activity, whereas the activity of 910A was too low to consider.

Regio- and stereoselectivity of UGT1A9, UGT1A10, and the different mutants in the glucuronidation of propranolol (A), estradiol (B), and dobutamine (C). The samples were incubated in duplicate or triplicate for 60 min in the presence of 0.5 to 4 mg/ml protein, 2 mM UDPGA, and either 500 μM propranolol, 100 μM estradiol, or 1 mM dobutamine. The error bars show the S.E.M. in triplicate incubations. See Materials and Methods for further details.
The fact that none of the single-segment chimeras resembled UGT1A10 was perhaps most clearly demonstrated by the dobutamine glucuronidation results (Fig. 4C). In this case, even 910C and 910D exhibited UGT1A9-like regioselectivity. 910B deviated most from the parent enzyme, even if it did not exhibit as clear a preference for the para position as UGT1A10 (Fig. 4C). The dobutamine glucuronidation rate of 910C was significantly higher than that of UGT1A9 (normalized rates) toward both catecholic hydroxyls of this substrate but with practically a UGT1A9-like ratio between them. The dobutamine glucuronidation activities of 910A and 910E were similar to that of UGT1A9, whereas 910D and 910B exhibited very low activity toward this substrate (Fig. 4C). It may be noted here that the dobutamine assays, like the results with other substrates, demonstrated that the single amino acid difference between UGT1A9 and UGT1A10 in segment E, Glu241 versus Ala241 (Fig. 1), has no significant effect on the enzymatic activity. Accordingly, segment E was not included in the double-segment chimeras.
Double-Segment Chimeras.
Our original plan was to follow the primary chimera examinations with “zooming in,” namely further mutagenesis within the segment from UGT1A10 of the single chimera UGT1A9 mutant that exhibits a UGT1A10-like activity. Because no such chimera was identified, we concluded that residues within at least two different segments are responsible for the activity differences between UGT1A9 and UGT1A10 (Fig. 3). Accordingly, we constructed four double-segment chimeras, namely 910AB [1A10(1–147)/1A9(148–285)], 910BC [1A9(1–83)/1A10(84–181)/1A9(182–285)], 910BD [1A9(1–83)/1A10(84–147)/1A9(148–181)/1A10(182–235)/1A9(236–285)], and 910CD [1A9(1–147)/1A10(148–235)/1A9(236–285)] (see Table 1 for the resulting mutations) and analyzed their activity.
All four double-segment chimeras exhibited low propranolol glucuronidation activity. Nevertheless, it is worth noting that two double chimeras with segment B, 910BC and 910BD (but not 910AB), resembled UGT1A10 in their preference for R-propranolol and the ratio of glucuronides for the two propranolol enantiomers (Fig. 4A). The estradiol glucuronidation activities of 910BC and 910BD were also more similar to the corresponding activities in UGT1A10 than these activities in any other single or double chimeras (Fig. 4B; Table 1). In the case of dobutamine glucuronidation, none of the double chimeras exhibited high activity, but the regioselectivity of 910BC and perhaps also of 910BD was interesting. It seemed that the combination of segments C and B from UGT1A10 (and to a lesser extent of D and B) stimulates UGT1A9 activity toward the para-hydroxyl of dobutamine, while lowering activity toward the meta-hydroxyl, yielding regioselectivity that is much more similar to that of UGT1A10 than to that of UGT1A9 (Fig. 4C).
Enzyme Kinetics of the UGT1A9-UGT1A10 Chimeras.
The activity screenings described above (Fig. 4, A–C) were performed in the presence of a single concentration for each substrate. To learn more about the substrate affinity and maximal turnover rate of each enzyme we turned to kinetic analyses. To keep the study within reasonable size, we selected one substrate, 1-naphthol, for these analyses. It is a substrate for both UGT1A9 and UGT1A10, but the kinetics of its glucuronidation by the two enzymes vary considerably. There are both significant differences between their respective Km and Vmax values and different types of enzyme kinetics. The glucuronidation of 1-naphthol by UGT1A9 follows a biphasic equation, whereas in the case of UGT1A10 it follows a Michaelis-Menten equation.
The 1-naphthol glucuronidation kinetic analyses of the single-segment chimeras revealed that the activity (low) and affinity (high) of 910A and 910E were rather similar to that of the parent UGT1A9. On the other hand, 910D and 910CD exhibited much higher Km values than UGT1A9, or even UGT1A10, whereas 910C had intermediary Km and Vmax values (note that the Vmax values are normalized with respect to the relative expression level of the enzyme in each sample) (Figs. 5 and 6; Table 2). The highest 1-naphthol glucuronidation rate of the chimeras was observed with 910CD and the lowest with 910B. The Km most similar to that of UGT1A10 was observed with 910B, even if the Vmax in this case was very low (Table 2). In addition, two double-segment chimeras that contained segment B, 910BC and 910BD, exhibited Km values close to that of UGT1A10 and their activity rates were higher than that of UGT1A9 (Figs. 5 and 6; Table 2). The kinetic parameters for 910AB were not determined because of its very low activity toward 1-naphthol.

Kinetics of 1-naphthol glucuronidation by UGT1A9, UGT1A10, and the five single chimeras. Initial rates were fitted to either Michaelis-Menten or biphasic kinetics equations by nonlinear regression. The assays were done in triplicate, and the error bars show the S.E.M. A magnification of the lower rates part of the curves is shown in the inset. See Table 2 for kinetic constants.

Kinetics of 1-naphthol glucuronidation by UGT1A9, UGT1A10, and three double chimeras. Initial rates were fitted to either Michaelis-Menten or biphasic kinetics equations by nonlinear regression. The assays were done in triplicate, and the error bars show the S.E.M. A magnification of the lower rates part of the curves is shown in the inset. See Table 2 for kinetic constants.
Mutations within Segment B of UGT1A9 and Their Effect on Regio- and Stereoselectivity.
Although none of the chimeras exhibited activities as high as those of UGT1A10, the results suggest that residues within segment B may contribute to the activity differences between UGT1A9 and UGT1A10 more than amino acids in other segments. This conclusion prompted us to prepare five point mutants within segment B of UGT1A9, namely D87N, V102A, R103Q, N114S, and F117L. In each of these mutants, the indicated amino acid of UGT1A9 was replaced by the corresponding residue from UGT1A10. The selected positions are those of segment B in which UGT1A10 has an amino acid that is not found in any of its three closest homologs, UGT1A7 to UGT1A9 (Fig. 1). UGTs 1A7 and 1A8 were taken into account when we selected the amino acids to be mutated because they are very close homologs of both UGT1A9 and UGT1A10 (Fig. 1), whereas their activities with some of the test substrates resemble that of UGT1A9 more closely than that of UGT1A10 (see Discussion).
Four of the five point mutants, 9D87N, 9V102A, 9R103Q, and 9N114S, had very little effect on either the activity rate or the regio- and stereoselectivity of UGT1A9 (Fig. 4, A–C). The fifth mutant, 9F117L, like the parent UGT1A9, was almost inactive in estradiol glucuronidation (Fig. 4B). With dobutamine, the 9F117L mutation had a clear effect on the regioselectivity, but because of the poor glucuronidation rate it was difficult to determine whether it was a change toward the formation of more dobutamine-para-glucuronide or an inhibition of the formation of dobutamine-meta-glucuronide (Fig. 4C). In the case of propranolol glucuronidation, the 9F117L mutation exerted strong inhibition of the UGT1A9-preferred activity, S-propranolol glucuronidation, but without stimulation of R-propranolol glucuronidation, the UGT1A10-like activity (Fig. 4A).
While doing the point mutagenesis we also constructed a triple mutant, 910B(115–117), in which the F117L mutation was accompanied with the corresponding UGT1A10-based mutations in the two positions that precede it, namely 9D115G-I116F-F117L. Mutant 910B(115–117) was more active than 9F117L with all substrates studied, and it exhibited an exceptionally high rate of S-propranolol glucuronidation, but without the stereoselectivity of UGT1A10 (Fig. 4A). However, in contrast to S-propranolol, the effect of 910B(115–117) on the glucuronidation rate of estradiols was very mild (Fig. 4B). For dobutamine glucuronidation, 910B(115–117) exhibited rates that were much higher than those of UGT1A9, whereas the regioselectivity was an intermediate between UGT1A9 and UGT1A10 (Fig. 4C).
Enzyme Kinetics of the Segment B Mutants.
The kinetic analyses of 1-naphthol glucuronidation by the five point mutants in segment B revealed an interesting difference between F117L and the other ones, particularly with respect to the Km values (Table 2; Fig. 7). Mutations D87N, V102A, R104Q, and N114S, did not affect the kinetic model or the affinity of UGT1A9 for 1-naphthol much (Table 2; Fig. 7). However, in 9F117L, the kinetic model was changed from biphasic to (mild) substrate inhibition, and the Km value highly resembled that for UGT1A10 (Table 2). In addition, in this mutant the Vmax rose to approximately 10 times higher than the corresponding value in UGT1A9 (Table 2). The triple mutant, 910B(115–117), exhibited a similar type of kinetics and Km value similar to that of 9F117L, whereas its Vmax value was four times higher (Table 2).

Kinetics of 1-naphthol glucuronidation by UGT1A9, UGT1A10, the point mutants, and the triple mutant in segment B. Initial rates were fitted to either substrate inhibition, Michaelis-Menten, or biphasic kinetics equations by nonlinear regression. The assays were done in triplicate, and the error bars show the S.E.M. See Table 2 for kinetic constants.
Combinations of Single Chimeras and Point Mutations.
Next, six new mutants were constructed to examine the interactions between segment B and C. Four of these mutants contained the entire segment B from UGT1A10 in combination with a single-point mutation in one of the four residues of segment C, in which UGT1A9 differs from UGT1A10 (Fig. 1; Table 1). The goal was to identify which of these four amino acids is responsible for the relatively high and UGT1A10-like activity that was exhibited by 910BC (Figs. 4, A–C, and 6). In parallel, we prepared two mutants that contained F117L together with either the entire segment C or the entire segment D from UGT1A10. The regioselectivity and stereoselectivity of 910B-L173F and 910B-Y176H with propranolol, dobutamine, and estradiol were similar to those of 910B, meaning mainly very low glucuronidation rates (Figs. 4, A–C). Mutant 910D-F117L was similar to 9F117L, whereas mutants 910B-N152T, 910B-A169T, and 910C-F117L exhibited regio- and stereoselectivity that resembled those of UGT1A10. Among the latter three mutants, 910B-N152T had the highest activity with all the three substrates (Fig. 4).
Kinetic analyses of 1-naphthol glucuronidation by the six “chimera plus a point mutation” mutants revealed that all of them had much higher Km values than UGT1A9 (Table 2). Mutant 910B-N152T was the most active among all of the mutants that contained segment B from UGT1A10, and its Km for 1-naphthol was similar to those of 910B and UGT1A10 (Table 2). Nonetheless, mutant 910C-F117L was the most similar to UGT1A10 when both 1-naphthol affinity and glucuronidation rates were considered (Fig. 8; Table 2).

Kinetics of 1-naphthol glucuronidation by UGT1A9, UGT1A10, the chimeras having an additional point mutation in another segment, and the double mutant 9F117L-N152T. Initial rates were fitted to either substrate inhibition, Michaelis-Menten, or biphasic kinetics equations by nonlinear regression. The assays were done in triplicate, and the error bars show the S.E.M. A magnification of the lower rates part of the curves is shown in the inset. See Table 2 for kinetic constants.
Double Mutant 9F117L-N152T.
Finally, based on the results from mutants 910B-N152T and 910C-F117L (see above), we prepared the double mutant, 9F117L-N152T, hoping that this combination will largely confer UGT1A10-like activity on UGT1A9. However, the results were disappointing. The stereoselectivity of this mutant with estradiol was qualitatively similar to that of UGT1A10, but its activity rate was very low (Fig. 4B). It also exhibited low activity in the glucuronidation of propranolol and dobutamine and with the latter substrates its regio- and stereoselectivity resembled that of UGT1A9 more than that of UGT1A10 (Fig. 4, A and C). Regarding 1-naphthol glucuronidation, the Vmax value of 9F117L-N152T was high, nearly as high as that of UGT1A10, but its Km value was also very high (Table 2; Fig. 8). Hence, the affinity of 9F117L-N152T for 1-naphthol seems to be much lower than that of UGT1A10, even if not as low as that of 910D-F117L (Fig. 8; Table 2).
Modeling.
The one available crystal structure from a UGT enzyme, residues 285 to 446 of the human UGT2B7, harbors the UDPGA binding site within the C-terminal domain (Miley et al., 2007). There is currently no structural information on the N-terminal domain of these enzymes, as would be needed for deeper understanding of substrate binding and glucuronidation. During the course of the experimental work described above, we constructed a homology model for selected human UGTs (L. Laakkonen and M. Finel, submitted for publication). Using this model, we docked 1-naphthol to the aglycone binding site of UGT1A9 (Fig. 9) to analyze the activity data, as well as the model itself. The core of the modeled N-terminal domain comprises a seven-stranded β sheet that is surrounded by four helices. In addition to these conserved structural elements of the N-terminal domain, there are two long and highly variable interstrand loops, which are predicted to fold into several short helices. These variable loops correspond closely to segments B and D, as defined in this study (Fig. 3), and segment B is highlighted in the overall image of the structural model (Fig. 9, left side). In accordance with the experimental results, the model shows that segments A and E are the farthest away from the active site, without any direct contacts with the bound substrate.

A model of UGT1A9 (left, overview) and a close look at the active site (right). In both parts of the figure, the N-terminal domain is drawn as a cartoon and is on the top of the figure. Segment B is colored black in the overview (left). The C-terminal domain is depicted as a trace at the bottom of the image. The active site histidine (His37 in UGT1A9), bound UDPGA, and Phe117 are shown as ball-and-stick molecules (left). Docked 1-naphthol is shown in the close-up view at the active site in this model (right).
The “catalytic His,” His37, and the sugar donor, UDPGA, are highlighted, along with the docked 1-naphthol, in the close-up view at the substrate binding site in the model (Fig. 9, right side). Previous studies of related plant and bacterial protein structures showed that the binding site of UDP-sugar and the position of the catalytic histidine are essentially invariable in the related glycosyltransferase structures (L. Laakkonen and M. Finel, submitted for publication). Accordingly, 1-naphthol was docked between the side chain of His37 and the anomeric carbon of the glucuronic acid moiety of UDPGA. Because of the rigidity of the 1-naphthol structure and the presence of a single reactive group within this substrate, it may be assumed that it only possesses rotational freedom at the active site. Depending on the rotational orientation of the bound aglycone (Supplemental Fig. S1), segments B, C, and D come to close proximity with it. The two most extreme orientations for 1-naphthol point to either segments C and D (169–173 and 188–192) or to segment B (residues 116–119). In the latter position, the substrate comes in contact with Phe117 (Fig. 9), in full agreement with the activity data.
Discussion
In an attempt to identify and locate the molecular basis of the differences in substrate selectivity between human UGTs 1A9 and 1A10, we constructed different mutations aimed to confer UGT1A10-like activity on UGT1A9. We focused on residues that differ between the two human UGTs and at the later stage further “homed in” on residues in which UGT1A10 also differs from UGT1A7 and UGT1A8 (Fig. 1). We started by making the minimalist assumption that a single amino acid difference, one of the 33 different residues between the mature UGT1A9 and UGT1A10, could lead to the activity differences between them. The chimera approach was adopted to eliminate most of these 33 different residues in the first round of mutagenesis and then to focus on the central residue. Test substrates, including two pairs of stereoisomers (Fig. 2), were selected on the basis of our previous observations and to allow broad examination of activity differences, including regio- and stereoselectivity.
The activity results for the first five chimeras revealed that none of them highly resemble UGT1A10 (Fig. 4; Table 2), strongly suggesting that the functional differences between UGT1A9 and UGT1A10 could not be explained by a single amino acid difference. Nevertheless, these results implied that segment A (residues 1–83 of which residues 26–83 are present in the mature UGTs) with its two variable residues, Arg42 and Gly67, and segment E, which contains a single variable residue, Glu241 do not contribute to the activity differences between UGT1A9 and UGT1A10. The results also suggested that even segment D (182–235) with 10 variable residues (Fig. 1) does not contribute significantly to the substrate selectivity differences between the two UGTs.
Segment A contains the catalytic His residue, His37, which is assumed to activate the aglycone substrates before their nucleophilic attack on UDP-glucuronic acid (Brazier-Hicks et al., 2007; Miley et al., 2007; Patana et al., 2008). Hence, His37 of either UGT1A9 or UGT1A10 is expected to face the substrate binding site of the enzyme (Fig. 9) and be crucial for activity but not to the selectivity differences between these enzymes. Segment A also contains the residue at position 42 that was recently reported to be involved in a substrate selectivity difference between UGT1A9 and UGT1A8 (Fujiwara et al., 2009). Our results with chimera 910A do not lead to the same conclusion. Nonetheless, the other residue these authors depicted, the one at position 152 (Fujiwara et al., 2009), also affected activity in some of our assays in the current study, when combined with segment B (Figs. 4, A–C, and 8).
Construction of several double-segment chimeras was undertaken because it was reasoned that if the minimal number of “crucial residues” for the differences between UGT1A9 and UGT1A10 is two and if they are located within different segments, then a double chimera may contain the correct combination and will yield a UGT1A10-like activity. In this round of mutagenesis we concentrated mainly on segments B, C, and D, and the results with two such double-segment chimeras, 910BC and 910BD, were quite promising (Fig. 4, A–C).
We then turned to identifying the most important residues in segment B (84–147) because this segment, when present in combination with either segment C or segment D, changed the activity of UGT1A9 toward that of UGT1A10 (Figs. 4, A–C, and 6; Table 2). In addition, segment B includes the residues and part of the regions that were previously reported to play a major role in substrate binding (Dubois et al., 1999; Lewis et al., 2007; Nishiyama et al., 2008). Within segment B there are 16 residues that differ between UGT1A9 to UGT1A10, but only 5 of them are unique to UGT1A10 compared with UGT1A7, 1A8, and 1A9. Because the regioselectivity of UGT1A7 and UGT1A8 in dobutamine glucuronidation resembled that of UGT1A9 rather than that of UGT1A10 (Alonen et al., 2005) and to a large extent their estradiol glucuronidation also, at least with UGT1A7 (Itäaho et al., 2008), we concentrated on these 5 unique residues when making the point mutations. Of the resulting five point mutants, only 9F117L exhibited some UGT1A10-like properties, particularly its affinity for 1-naphthol (Table 2; Fig. 7). According to our sequence alignment, Phe117 of UGT1A9 corresponds to Ser121 of UGT2B17 and Tyr121 of UGT2B15, the two residues that were previously suggested to determine the regioselectivity of these UGTs in the glucuronidation of steroids (Dubois et al., 1999). Still, the sequence alignment between UGTs 1A7–1A0 and 2B15/2B17 in this region is ambiguous, because it requires the insertion of three gaps closely upstream of this position in UGTs 1A7 to 1A10 (data not shown).
The possibility that Phe117 is responsible for the high affinity of UGT1A9 for 1-naphthol and replacing it with Ala, as found in UGT1A10, changes the interactions of the enzyme with the aglycone substrate, is further supported by the results from mutant 910C-F117L (Fig. 8). Based on these observations and on our recent homology model for UGT1A1 (L. Laakkonen and M. Finel, submitted for publication), we built a model of the active site of UGT1A9 with a bound 1-naphthol molecule. In this model Phe117 is found close to the catalytic His, His37, and it may stack with the aromatic ring of 1-naphthol (Fig. 9).
Although Phe117 of UGT1A9 seems to face the substrate binding site, it is not the only residue that affects substrate binding and glucuronidation rate. The triple mutant 9D115G-I116F-F117L increased the turnover rate significantly compared with 9F117L but did not change the regio- and stereoselectivity of UGT1A9 (Fig. 4, A–C). The effect of this triple mutant on the Km for 1-naphthol (Fig. 7; Table 2) is probably due to the F117L mutation alone, suggesting that Asp115 and Ile116 of UGT1A9 do not directly interact with the substrate. In the model, this segment is predicted to be helical, which precludes three consecutive residues from pointing to the same direction.
Mutant 910B-N152T also seemed to increase the UGT1A10-like activity, and it may thus be suggested that the residue at position 152 faces the active site. Such a suggestion is in agreement with the finding of Fujiwara et al. (2009). Assuming that both Phe117 and Asn152 of UGT1A9 affect substrate specificity and interact with the bound substrate, we prepared the double mutant 9F117L-N152T and assayed its activity. The results were somewhat disappointing because the affinity of the substrates for this mutant seemed to be particularly low (Fig. 8; Table 2). Very low substrate affinity for this double mutant, as seen with 1-naphthol, may also explain the very low activity of this double mutant in the glucuronidation of propranolol, estradiol, and dobutamine. Hence, although these results do not mean that Asn152 of UGT1A9 does not face the substrate binding pocket, they strongly suggest that one or two other residues are needed there to support the binding of the test substrates in the same way they bind to UGT1A10. Several other amino acids were previously reported to affect the substrate selectivity of UGT1A10, including Phe90 and Phe93 (Xiong et al., 2006; Starlard-Davenport et al., 2007), Glu139 (Dellinger et al., 2006), and Ile211 (Martineau et al., 2004). However, the latter residues cannot be responsible for the activity differences between UGT1A9 and UGT1A10 because they are identical in the two UGTs (Fig. 1).
In summary, residues between Leu86 and Tyr176 of UGT1A9 appear to determine the substrate selectivity differences between UGT1A9 and UGT1A10. Within this region, residues at positions 115, 116, 117, 152, and 169 exerted a significant impact on the catalytic properties that were studied here. The results indicate that interactions between several amino acids, rather than the presence of a single residue in a “strategic” location within the active site, govern the complex substrate selectivity of the UGTs.
Acknowledgments.
We are grateful to Sanna Sistonen and Johanna Mosorin for skillful technical assistance and to Mika Kurkela, Anna Alonen, Taina Sten, and Nina Sneitz for conducting different preliminary assays.
Footnotes
This study was supported by funding from the Sigrid Juselius Foundation (to M.F.); and the Magnus Ehrnrooth Foundation (to L.L.).
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.109.031229.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
- UGT
- UDP-glucuronosyltransferase
- UDPGA
- UDP-glucuronic acid
- HPLC
- high-performance liquid chromatography
- DMSO
- dimethyl sulfoxide.
- Received November 17, 2009.
- Accepted January 20, 2010.

- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}