


Abstract
Studies were performed to determine the human enzymes responsible for the biotransformation of atomoxetine to its major metabolite, 4-hydroxyatomoxetine, and to a minor metabolite,N-desmethylatomoxetine. Utilizing human liver microsomes containing a full complement of cytochrome P450 (P450) enzymes, averageKm and CLint values of 2.3 μM and 103 μl/min/mg, respectively, were obtained for 4-hydroxyatomoxetine formation. Microsomal samples deficient in CYP2D6 exhibited average apparent Km and CLint values of 149 μM and 0.2 μl/min/mg, respectively. In a human liver bank characterized for P450 content, formation of 4-hydroxyatomoxetine correlated only to CYP2D6 activity. Of nine expressed P450s examined, 4-hydroxyatomoxetine was formed at a rate 475-fold greater by CYP2D6 compared with the other P450s. These results demonstrate that CYP2D6 is the enzyme primarily responsible for the formation of 4-hydroxyatomoxetine. Multiple P450s were found to be capable of forming 4-hydroxyatomoxetine when CYP2D6 was not expressed. However, the efficiency at which these enzymes perform this biotransformation is reduced compared with CYP2D6. The formation of the minor metabolite N-desmethylatomoxetine exhibited average Km and CLint values of 83 μM and 0.8 μl/min/mg, respectively. Utilizing studies similar to those outlined above, CYP2C19 was identified as the primary enzyme responsible for the biotransformation of atomoxetine toN-desmethylatomoxetine. In summary, CYP2D6 was found to be the primary P450 responsible for the formation of the major oxidative metabolite of atomoxetine, 4-hydroxyatomoxetine. Furthermore, these studies indicate that in patients with compromised CYP2D6 activity, multiple low-affinity enzymes will participate in the formation of 4-hydroxyatomoxetine. Therefore, coadministration of P450 inhibitors to poor metabolizers of CYP2D6 substrates would not be predicted to decrease the clearance of atomoxetine in these individuals.
Atomoxetine (Fig. 1) (formally known as tomoxetine; LY139603) is under development as a therapeutic agent for the treatment of attention deficit hyperactivity disorder in children and adults. Atomoxetine enhances norepinephrine function through a highly selective blockade of the presynaptic norepinephrine transporter and has low affinities for other neuronal transporters or neurotransmitter receptor sites (Wong et al., 1982; Gehlert et al., 1993). This is an interesting and potentially important new drug since it is likely to be the first approved treatment for attention deficit hyperactivity disorder that is not a psychostimulant. Studies with this compound in healthy human volunteers (Farid et al., 1985) showed that the clearance of atomoxetine exhibited a bimodal distribution, suggesting that an enzyme that exhibits a genetic polymorphism was involved in the metabolism of atomoxetine. The study further reported that in both extensive and poor metabolizers of atomoxetine,para-hydroxyatomoxetine (later definitively identified as 4-hydroxyatomoxetine) was the major oxidative metabolite; however, the formation of this metabolite was decreased in poor metabolizers. A minor metabolite to the overall metabolism of atomoxetine,N-desmethylatomoxetine, was also detected (Farid et al., 1985). Identification of the cytochromes P450 (P450s1) responsible for the metabolism of atomoxetine and knowing the interindividual differences in the expression and catalytic activities of those P450s may help explain the population variability observed in the metabolic clearance of atomoxetine. Therefore, the studies described herein utilize in vitro techniques to identify the enzyme(s) responsible for the formation of both 4-hydroxyatomoxetine andN-desmethylatomoxetine.

Structure of atomoxetine, 4-hydroxyatomoxetine, and N-desmethylatomoxetine.
Materials and Methods
Atomoxetine, 4-hydroxyatomoxetine (Fig. 1),N-desmethylatomoxetine (Fig. 1), LY110086 (internal standard for the N-desmethylatomoxetine assay), and LY127809 (internal standard for the 4-hydroxyatomoxetine assay) were synthesized by Eli Lilly & Co. (Indianapolis, IN). Quinidine, NADPH, sulfaphenazole, and coumarin were purchased from Sigma-Aldrich (St. Louis, MO). Furafylline and S-mephenytoin were obtained from Ultrafine Chemicals (Manchester, UK). Ketoconazole was obtained from Sigma-RBI (Natick, MA). Diethyldithiocarbamate (DDC) and coumarin was obtained from Aldrich Chemical Co. (Milwaukee, WI). Monoclonal antibodies to CYP2B6, CYP2D6, CYP2C, and CYP3A4/5 in ascites fluid were obtained from PanVera Corp. (Madison, WI), and control ascites fluid obtained from ICN Pharmaceuticals Biochemical Division (Aurora, OH).
Human liver samples designated HLA through HLT were obtained from the Medical College of Wisconsin (Milwaukee, WI), Medical College of Virginia (Richmond, VA), or Indiana University School of Medicine (Indianapolis, IN) under protocols approved by the appropriate committee for the conduct of human research. Hepatic microsomes were prepared by differential centrifugation (van der Hoeven and Coon, 1974) and characterized for their relative levels of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 via immunoquantification or through the use of form-selective catalytic activities (Ring et al., 2001). Microsomes prepared from human β-lymphoblastoid cells engineered to express CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 were obtained from GENTEST (Woburn, MA).
For kinetic analyses, the conversions of atomoxetine toN-desmethylatomoxetine or 4-hydroxyatomoxetine by human liver microsomes or microsomes containing expressed P450s were accomplished under initial rate conditions. Incubation mixtures contained substrate, microsomes, and NADPH (1 mM) in 100 mM sodium phosphate buffer, pH 7.4. The reactions were stopped with an equal volume of either acetonitrile (N-desmethylatomoxetine) or methanol (4-hydroxyatomoxetine). The denatured protein was removed by centrifugation, and the supernatant was analyzed for metabolite formation. Enzyme kinetic parameters were determined following fit of the data to the Michaelis-Menten model of enzyme kinetics (Segel, 1975) using nonlinear regression analysis (WinNonlin version 1.5; Statistical Consultants, Cary, NC) (Ring et al., 2001).
Correlation analyses were performed (JMP; SAS Institute, Cary, NC) between the rates of metabolite formation and the enzymatic activities or immunoquantified levels of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A in a human liver microsomal bank of up to 20 samples (Ring et al., 2001). The rates of formation of 4-hydroxyatomoxetine by this human liver microsomal bank were determined following incubations with 1 μM atomoxetine or 100 μM atomoxetine plus the CYP2D6 inhibitor quinidine (10 μM) (Newton et al., 1995). The rates of formation ofN-desmethylatomoxetine were determined following incubations with 10 μM atomoxetine for the correlation studies.
Expressed P450s were examined for their ability to form either 4-hydroxyatomoxetine or N-desmethylatomoxetine following incubation with either 1 μM atomoxetine to examine 4-hydroxyatomoxetine formation or 10 μM atomoxetine to examineN-desmethylatomoxetine formation. The incubation times varied from 1 to 60 min at 37°C to assure detectable metabolite formation.
Inhibitors or substrates utilized as competitive inhibitors of P450-mediated reactions were examined for their impact on 4-hydroxyatomoxetine formation by two human liver microsomal samples deficient in CYP2D6 (HLK and HLN). These compounds and the P450 primarily inhibited included coumarin (500 μM) for CYP2A6, sulfaphenazole (10 μM) for CYP2C9, S-mephenytoin (250 μM) for CYP2C19, quinidine (10 μM) for CYP2D6, or ketoconazole (2 μM) for CYP3A. Two mechanism-based inhibitors were also examined for their ability to inhibit this reaction: furafylline (10 μM) for CYP1A2 or DDC (300 μM) for CYP2E1 (Pearce et al., 1992; Wrighton et al., 1993; Newton et al., 1995; Gorski et al., 1997). These compounds, except for coumarin, were also examined for their effect onN-desmethylatomoxetine formation. In addition, inhibitory monoclonal antibodies to the CYP2C, CYP2D6, CYP3A4/5, or CYP2B6 were used with human liver microsomal samples HLC, HLJ, or HLM to assess their effect on N-desmethylatomoxetine formation (Ring et al., 2001).
Analyses of 4-hydroxyatomoxetine levels were determined by liquid chromatography tandem mass spectrometry. A Prodigy ODS (3) column (50 × 2 mm, 5 μl) (Phenomenex, Torrance, CA) was utilized with a gradient mobile phase containing 100 mM formic acid in water and 100 mM formic acid in isopropanol, water, and methanol (10:10:80) to separate 4-hydroxyatomoxetine from the internal standard. Positive ion electrospray with selected reaction monitoring was used for analysis. The transition of m/z 272.1 →m/z 44 at 0.1 s was monitored for 4-hydroxyatomoxetine.
Analyses of N-desmethylatomoxetine levels were determined by gas chromatography/mass spectrometry utilizing selected ion monitoring. Samples containing N-desmethylatomoxetine and internal standard were extracted utilizing Isolute HCX solid phase extraction cartridges (130 mg and 3 ml) (Jones Chromatography, Inc., Lakewood, CO) conditioned with 2 ml of methanol followed by 2 ml of 0.1 M phosphate buffer. Samples were applied to solid phase extraction cartridges and washed with 2 ml of acetonitrile followed by 2 ml of hexane/ethyl acetate (50:50). The analyte and internal standard were eluted with 2 ml of 2% ammonium hydroxide in methylene chloride:methanol (80:20). Samples were dried at 50°C under nitrogen and derivatized with 2% heptafluorobutryic acid anhydride in hexane. Derivatized samples were dried, solubilized in hexane, and analyzed forN-desmethylatomoxetine formation by gas chromatography/mass spectral analysis. A Restek Rtx-5 mass spectrometry column (15 m, 0.25 mm i.d., and 0.25 μm df) (Restek Corp., Bellefonte, PA) was used to separate N-desmethylatomoxetine from the internal standard. Negative chemical ionization with methane as the reagent gas was used for detection in selective ion monitoring mode.
Results
4-Hydroxyatomoxetine Studies.
The apparent kinetic parameters for 4-hydroxyatomoxetine formation were examined utilizing four different preparations of human liver microsomes, HLC, HLM, HLK, and HLN (Ring et al., 2001). Using microsomal samples containing either the full complement of P450s (HLC and HLM) or samples that were CYP2D6-deficient (HLK and HLN), Eadie-Hofstee transformations of the data examining the formation of 4-hydroxyatomoxetine were monophasic in nature (data not shown). Kinetic analyses yielded apparent Kmvalues of 2.3 and 2.2 μM for HLC and HLM, respectively, and apparentKm values of 153 and 144 μM for CYP2D6-deficient HLK and HLN, respectively (Table1). The calculated CLint(Vmax/Km) for the microsomal samples containing the full complement of enzymes were at least 160-fold greater than that calculated for the CYP2D6-deficient microsomal samples (Table 1).
Enzyme kinetic analyses of the formation of 4-hydroxyatomoxetine by human liver microsomes or expressed P450s
The rates of formation of 4-hydroxyatomoxetine were determined in a P450-characterized bank of 20 human liver microsomal samples (Ring et al., 2001) at a substrate concentration of 1 μM (Table2). The formation rates of this metabolite by these microsomal samples were then correlated to previously determined form-selective activities or immunoquantified levels for nine P450s. The only significant (p< 0.05) correlation observed was between the formation of 4-hydroxyatomoxetine and the catalytic activity for CYP2D6, the 1′-hydroxylation of bufuralol (r = 0.73). The formation rates of 4-hydroxyatomoxetine by this microsomal bank were also examined in the presence of 10 μM quinidine, a potent CYP2D6 inhibitor, at an atomoxetine concentration of 100 μM, which reflects the Km value for this biotransformation observed in CYP2D6-deficient microsomes. The only activity found to be a significant regressor was that for CYP2D6.
4-Hydroxyatomoxetine or N-desmethylatomoxetine formation rates (picomoles per minute per milligram) by a bank of human liver microsomal samples2-a
The abilities of nine cDNA-expressed P450s to form 4-hydroxyatomoxetine following incubations with 1 μM atomoxetine were determined. Expressed CYP1A2, CYP2B6, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 were able to form 4-hydroxyatomoxetine at detectable levels. However, the rate of formation of this metabolite by CYP2D6 (928 pmol/min/mg of protein) was 475-fold greater than that observed for the other enzymes.
To further identify enzymes other than CYP2D6 that may form 4-hydroxyatomoxetine, chemical inhibitors of the P450s were examined for their ability to inhibit the formation of 4-hydroxyatomoxetine utilizing microsomes from CYP2D6-deficient human livers HLK and HLN (Fig. 2). Both quinidine (CYP2D6) and sulfaphenazole (CYP2C9) had little effect on the formation rates of 4-hydroxyatomoxetine compared with control. S-Mephenytoin (CYP2C19) and ketoconazole (CYP3A) exhibited moderate inhibition (20–30%) of the formation of 4-hydroxyatomoxetine. The inhibitors of CYP1A2 (furafylline), CYP2A6 (coumarin), and CYP2E1 (DDC) had the greatest effect on formation of 4-hydroxyatomoxetine, inhibiting the formation of this metabolite by CYP2D6-deficient microsomes from 34 to 67%.

Inhibition of 4-hydroxytomoxetine formation by human liver microsomes deficient in CYP2D6, HLK, and HLN by form-selective inhibitors or substrates of CYP2D6 (quinidine), CYP2C9 (sulfaphenazole), CYP2A6 (coumarin), CYP2C19 (S-mephenytoin), CYP3A (ketoconazole), CYP1A2 (furafylline), and CYP2E1 (DDC).
Finally, the apparent kinetic parameters for 4-hydroxyatomoxetine formation were determined with cDNA-expressed CYP1A2, CYP2B6, CYP2C19, and CYP2E1 (Table 1). The apparent Kmvalues obtained were 399 μM for CYP1A2, 29 μM for CYP2B6, 96 μM for CYP2C19, and 5.0 μM for CYP2E1.
N-Desmethylatomoxetine Studies.
The apparent kinetic parameters for the enzyme(s) responsible forN-desmethylatomoxetine were examined utilizing human liver microsomes HLC, HLM, and HLK. Using microsomal samples containing either the full complement of P450s (HLC and HLM) or a sample that was CYP2D6-deficient (HLK), Eadie-Hofstee transformations of the data examining the formation of N-desmethylatomoxetine were monophasic in nature (data not shown). Therefore, the kinetic parameters for the formation of N-desmethylatomoxetine by HLC, HLM, and HLK were estimated utilizing the Michaelis-Menten equation, resulting in apparent Kmvalues of 91, 45, and 113 μM, respectively (Table3). The calculated CLint for all three microsomal samples was similar, ranging from 0.75 to 0.86 μl/min/mg. (Table 3).
Enzyme kinetic analyses of the formation of N-desmethylatomoxetine by human liver microsomes
The rates of formation of N-desmethylatomoxetine were determined by the bank of 20 characterized human liver microsomal samples following incubations with 10 μM atomoxetine (Table 2) for use in correlation analyses. Significant correlations were obtained with CYP2B6, CYP2C19, and CYP2D6 and N-desmethylatomoxetine formation (multivariate r = 0.96).
The expressed cDNA enzymes that formedN-desmethylatomoxetine following incubations with 10 μM atomoxetine were CYP2C19, CYP2B6, CYP1A2, CYP3A4, and CYP2C9 (Fig.3). The expressed P450s able to formN-desmethylatomoxetine were then examined as potential coregressors in multivariate correlation analyses. Significant correlations with the formation of this metabolite were observed with both CYP2B6 and CYP2C19 form-selective catalytic activities.
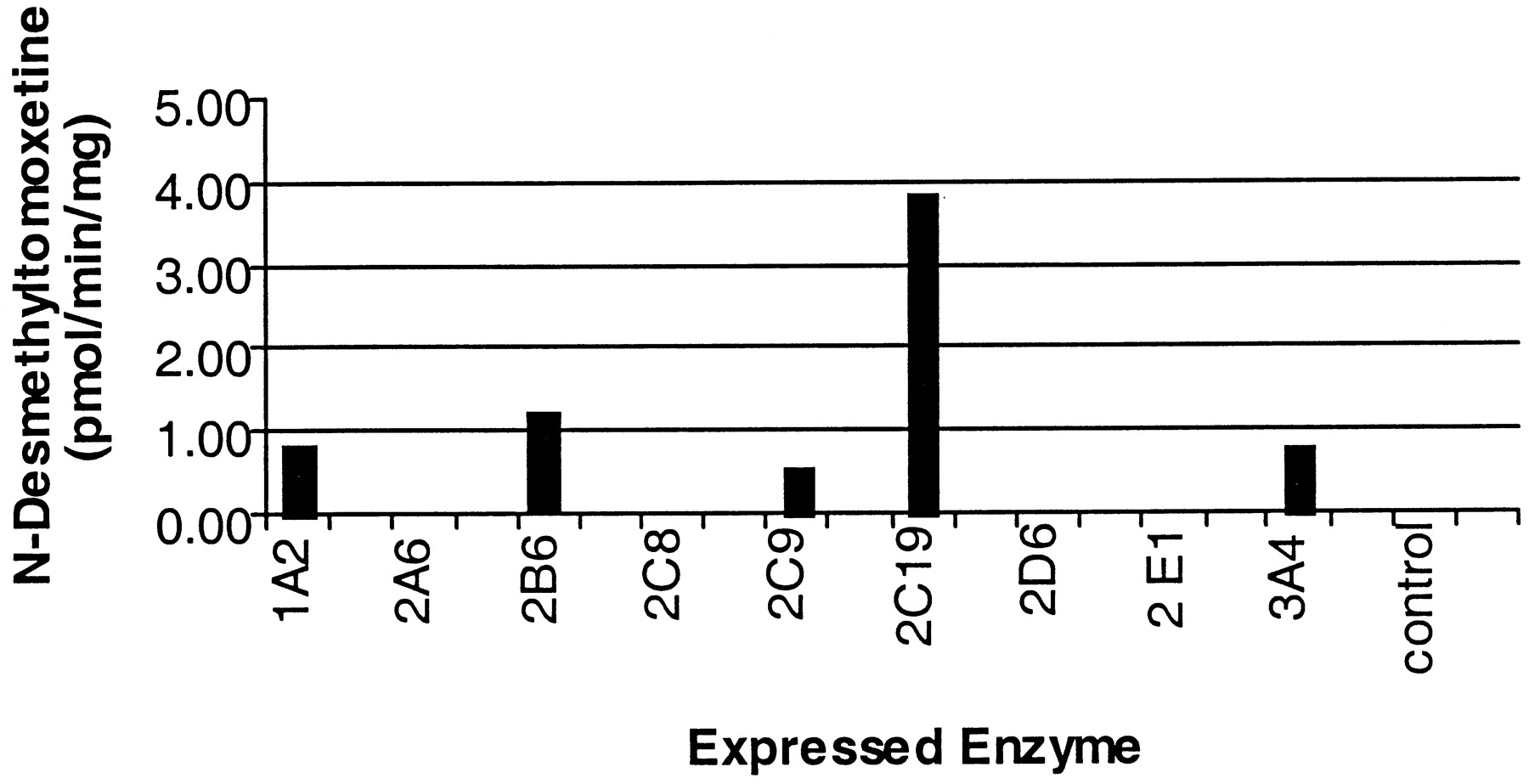
Formation of N-desmethylatomoxetine by expressed P450s following incubation with 10 μM atomoxetine.
Monoclonal antibodies recognizing the CYP2C subfamily of enzymes, CYP2D6, CYP3A4/5, or CYP2B6, were examined for their ability to inhibitN-desmethylatomoxetine formation by HLC, HLJ, and HLM. The antibody to the CYP2C subfamily inhibitedN-desmethylatomoxetine formation by HLC, HLJ, and HLM by ≥ 34, 38, and 34%, respectively. Antibodies to CYP2D6, CYP2B6, or CYP3A4/5 did not significantly inhibit formation of this metabolite in any of the livers examined.
Due to the ability of several expressed P450s to formN-desmethylatomoxetine, chemical inhibitors of the P450s were also examined for their ability to inhibit the formation of this metabolite by microsomes from human livers HLC, HLJ, and HLM at an atomoxetine concentration of 10 μM. The inhibitors examined included sulfaphenazole (CYP2C9), S-mephenytoin (CYP2C19), quinidine (CYP2D6), ketoconazole (CYP3A), DDC (CYP2E1), and furafylline (CYP1A2). The only inhibitors found to decrease the formation ofN-desmethylatomoxetine were S-mephenytoin and furafylline. Furafylline exhibited inhibition by ≥22%, 46%, and ≥18% in HLC, HLJ, and HLM, respectively. Based on limits of detection, it was determined that inhibition byS-mephenytoin was ≥13% in HLC and 46% in HLJ. Inhibition by S-mephenytoin of this metabolite formed by HLM was not determined.
Discussion
The major oxidative metabolite of atomoxetine in vivo is known to be 4-hydroxyatomoxetine (Farid et al., 1985). In addition, a bimodal distribution in atomoxetine clearance was observed upon administration of atomoxetine to normal volunteers. Thus, the first studies reported here focused on the identification of the enzymes responsible for the formation of 4-hydroxyatomoxetine. Utilizing microsomal samples containing a complete complement of P450 enzymes, the apparent meanKm value for the formation of 4-hydroxyatomoxetine was 2.3 μM. The large CLint value obtained in these kinetic studies confirmed the in vivo studies (Farid et al., 1985), which reported that the formation of 4-hydroxyatomoxetine was the primary route of metabolism of atomoxetine. Correlating the rates of formation of 4-hydroxyatomoxetine utilizing sub-Kmconcentrations of atomoxetine with the form-selective catalytic activities/immunoquantified levels in a characterized human liver microsomal bank indicated that CYP2D6 was the primary enzyme responsible for the metabolism of atomoxetine to 4-hydroxyatomoxetine. These results were verified when it was observed that expressed CYP2D6 formed this metabolite at a rate 475-fold greater than that observed for the other eight P450s examined.
The conclusion that CYP2D6 is responsible for the formation of 4-hydroxyatomoxetine is important due to the incidence of genetic polymorphism of this enzyme in the population. The poor metabolizer (PM) phenotype is found in about 7% of the Caucasian population and <1% of the Asian population (Guengerich, 1995). In those patients with the extensive metabolizer phenotype for CYP2D6, coadministration of drugs that inhibit CYP2D6 activity may result in alterations in atomoxetine pharmacokinetic parameters. Maximum inhibition of CYP2D6-mediated atomoxetine metabolism by coadministration with drugs known to significantly inhibit CYP2D6, such as paroxetine, would essentially change the extensive metabolizer phenotype to that of a poor metabolizer phenotype, and clearance of atomoxetine, upon maximal inhibition, should reflect that of a CYP2D6-deficient patient. This underscores the importance of understanding the enzymes responsible for routes of atomoxetine metabolism in patients deficient in CYP2D6 or in whom CYP2D6 is inhibited. To examine this question, studies identifying other enzymes that form 4-hydroxyatomoxetine and those that form the minor metabolite, N-desmethylatomoxetine, were performed.
It is known that the formation of 4-hydroxyatomoxetine remains the major route of atomoxetine metabolism in CYP2D6 poor metabolizers, albeit at a reduced rate from that observed in extensive metabolizers of CYP2D6 substrates (Farid et al., 1985). Kinetic experiments were performed in vitro examining the formation of 4-hydroxyatomoxetine in two human liver microsomal samples known to be deficient in CYP2D6. The average apparent Km and CLint values for the formation of this metabolite were 149 μM and 0.2 μl/min/mg, respectively. This CLint value is at least 160-fold lower than the CLint values calculated for those samples containing a full complement of drug-metabolizing enzymes (Table 1). These results suggest that although enzymes other than CYP2D6 can form 4-hydroxyatomoxetine, the efficiency at which 4-hydroxyatomoxetine formation occurs by these additional enzymes is reduced, and the amount of metabolite formed will be less than that mediated by CYP2D6.
The formation of 4-hydroxyatomoxetine following incubations with microsomes utilizing concentrations of atomoxetine at 100 μM and including a CYP2D6 inhibitor quinidine with P450 activities in a microsomal bank unexpectedly correlated to CYP2D6 activity. Quinidine, reported to inhibit CYP2D6-mediated reactions in vitro by 80 to 90% (Newton et al., 1995), was added to the incubations in an attempt to eliminate CYP2D6 in the formation of 4-hydroxyatomoxetine. However, it was apparent that sufficient CYP2D6 activity remained to form the 4-hydroxyatomoxetine, which was significant enough to mask the contribution of other enzymes in the formation of this metabolite when examined at a very high concentration of atomoxetine.
In addition to a high rate of 4-hydroxyatomoxetine formation by expressed CYP2D6, expressed CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2E1, and CYP3A4 were able to form 4-hydroxyatomoxetine albeit with a lower rate of turnover. Therefore, inhibitors of various P450s were examined for their ability to effect the formation of this metabolite in microsomes deficient in CYP2D6. Quinidine (a CYP2D6 inhibitor) and sulfaphenazole (CYP2C9 inhibitor) did not significantly effect the metabolite formation. Inhibition from 20 to 67% of 4-hydroxyatomoxetine formation was observed following incubations with inhibitors of CYP2C19, CYP3A, CYP1A2, CYP2A6, and CYP2E1. This moderate inhibition by several different P450 inhibitors of 4-hydroxyatomoxetine formation by microsomes deficient in CYP2D6 suggests that multiple P450s contribute to the formation of 4-hydroxyatomoxetine when CYP2D6 is absent. Kinetic analyses of 4-hydroxyatomoxetine formation by expressed CYP1A2, CYP2B6, CYP2C19, and CYP2E1 resulted inKm values of <100 μM for all but CYP1A2 (Table 2). These results confirm that multiple P450s participate in the formation of 4-hydroxyatomoxetine when CYP2D6 is absent or inhibited.
In both CYP2D6 extensive metabolizers and PMs, theN-desmethyl route of atomoxetine metabolism is minor, accounting for <10% of the total dose of atomoxetine in these patients (data not shown). Formation ofN-desmethylatomoxetine exhibited an averageKm value of 83 μM in incubations with three human liver microsomal preparations. It should be noted that one of these three microsomal samples was deficient in CYP2D6 yet exhibited a Km value similar to that observed by the other two, indirectly suggesting that CYP2D6 is not involved in the formation of N-desmethylatomoxetine. Correlation studies and cDNA-expressed enzymes identified CYP2C19, CYP2B6, and others as playing a role inN-desmethylatomoxetine formation. However, only inhibitory monoclonal antibodies to CYP2C but not CYP2B6 were able to inhibit the formation of this metabolite. These results suggest a primary role for CYP2C19 in this biotransformation. Further confirmation was seen with the ability of chemical inhibitors of CYP2C19-mediated metabolism to inhibit N-desmethylatomoxetine formation by human liver microsomes. In summary, these results indicate that CYP2C19 is the primary enzyme involved in N-desmethylatomoxetine formation at pharmacological concentrations of atomoxetine.
In total, the results of the studies reported here demonstrate that the bimodal distribution of atomoxetine clearance observed in vivo (Farid et al., 1985) is due to the primary involvement of the polymorphically expressed CYP2D6 in the formation of the major metabolite of atomoxetine, 4-hydroxyatomoxetine. In patients with compromised CYP2D6 catalytic activity, 4-hydroxyatomoxetine remains the major metabolite (Farid et al., 1985), which was determined in these studies to be formed by multiple enzymes with a relatively low affinity for atomoxetine. In addition, in all patients formation ofN-desmethylatomoxetine, a minor route of atomoxetine metabolism (Farid et al., 1985), was found to be mediated primarily by CYP2C19. These results indicate that in CYP2D6 PMs, multiple enzymes would be expected to metabolize atomoxetine albeit at a reduced rate. Therefore, due to the many different enzymes involved in the formation of the major metabolite 4-hydroxyatomoxetine, it is unlikely that coadministration of additional P450 substrates and/or inhibitors to CYP2D6 PMs would markedly affect the clearance of atomoxetine.
Acknowledgments
We thank Dr. William Wheeler for the synthesis of 4-hydroxyatomoxetine.
Footnotes
- Abbreviations used are::
- P450
- cytochrome P450
- PM
- poor metabolizers
- DDC
- diethyldithiocarbamate
- Received October 11, 2001.
- Accepted November 27, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}