


Abstract
Paclitaxel and docetaxel are metabolized by liver microsomal monooxygenases into inactive metabolites further eliminated from the body via the bile route. In spite of their close chemical structure, the two drugs are oxidized by two different enzymes; CYP2C8 catalyzes the 6-hydroxylation on the taxane ring of paclitaxel, whereas CYP3A4 oxidizes docetaxel on the tert-butyl group of the lateral chain in C13. Since paclitaxel and docetaxel differ only by two substitutions, the role of individual modifications was investigated; the regioselectivity of hydroxylation was assessed by high-pressure liquid chromatography/mass spectrometry, and enzymes implicated in individual reactions were identified using human liver microsomes and recombinant P450 expressed in Ad293 cells. The biotransformation of docetaxel, 10-deacetylpaclitaxel, and 10-deacetylbaccatin III was steadily increased (2- to 5-fold) by the addition of an acetyl group in position 10, suggesting that the presence of a hydrophobic group in position 10 stimulated hydroxylation by P450 proteins. The absence of the lateral chain at C13 in baccatin III severely impaired the metabolism supported by CYP3A4. The presence of a tert-butyl group in the lateral chain of docetaxel favored the hydroxylation on the tert-butyl by CYP3A4, whereas the presence of a phenyl group in the lateral chain facilitated the oxidation on the taxane ring by CYP2C8. Collectively, these data strongly suggested that the structure of the lateral chain and the nature of substituent in position 10 play an important role in determining the regioselective oxidation by P450 proteins and modulate the reaction rate by human liver microsomes.
Taxoids are natural compounds isolated from the bark or leaves of yew trees and screened for their antiproliferative properties. Taxol or paclitaxel was initially selected on the basis of its cytotoxic properties upon various cell lines and tumor-bearing mice; its biological action was attributed to the capacity to bind tubulin and to stabilize microtubules, thus modifying the cytoskeleton architecture and blocking the cell cycle in phase G2/M (Wani et al., 1971; Schiff et al., 1979). However, the limited availability of paclitaxel led to the development of analogs, which retained the biological activity on tubulin assembly but were readily synthesized in large quantities. This led to the discovery of docetaxel (Taxotere) a semisynthetic compound derived from 10-deacetylbaccatin III with an affinity for microtubules 2 times higher than paclitaxel (Ringel and Horwitz, 1991). Paclitaxel and docetaxel are widely used with success in patients with various tumors, including ovarian or breast carcinomas resistant to conventional chemotherapy (Rowinski et al., 1992).
The therapeutic efficiency of taxoids is the result of the balance between their biological action on tubulin and rate of degradation. The elimination of paclitaxel and docetaxel from the body occurs mainly through the biliary route. Five metabolites are isolated from the bile of a patient given paclitaxel and three are hydroxylated (Monsarrat et al., 1993). The complete chemical structure of these metabolites has been established by mass spectrometry and NMR spectrometry (Monsarrat et al., 1993, 1998; Harris et al., 1994a; Monegier et al., 1994; Royer et al., 1995); the major metabolite formed is a monohydroxylated derivative at C6 of the taxane ring not present in rat bile (Monsarrat et al., 1990). Hydroxylated product on the phenyl at C3′ is also observed in human bile, as well as the dihydroxylated molecule at C3′ of the lateral chain and C6 of the taxane ring. Two distinct monooxygenases are involved in the hydroxylation of paclitaxel in the human liver; the formation of the C3′-derivative is clearly assigned to CYP3A4, whereas the 6-hydroxylated derivative is the major metabolite produced by a CYP2C protein identified as CYP2C8 (Cresteil et al., 1994; Harris et al., 1994b; Rahman et al., 1994).
In contrast with paclitaxel, the biotransformation of docetaxel is similar in animals and humans (Vuilhorgne et al., 1995). The chemical structure of purified metabolites was elucidated by mass spectrometry and NMR spectrometry and demonstrates that the metabolism of docetaxel involves an initial oxidation on the tert-butyl of the lateral side chain before its cyclization, both reactions catalyzed by CYP3A4 (Monegier et al., 1994; Marre et al., 1996; Royer et al., 1996;Shou et al., 1998). These metabolites were also isolated from feces collected from patients given docetaxel (Sparreboom et al., 1996). No hydroxylation on the taxane ring of docetaxel or on the phenyl group of the lateral chain has been reported in in vitro and in vivo studies. Thus, relatively minor modifications on the taxoid molecule have major consequences on the site of oxidation and the identity of the P4501 isoform implicated in the reaction. Furthermore, the oxidative metabolism of paclitaxel and docetaxel strikingly reduced their biological activity. Thus, the 6-hydroxypaclitaxel is more than 30-fold less potent than paclitaxel in the in vitro inhibition of cell proliferation (Harris et al., 1994a;Kumar et al., 1995; Sparreboom et al., 1995). Similarly, the hydroxylated metabolite of docetaxel shows poor in vitro cytotoxicity in P388 cells and in B16 tumor-bearing mice (Vuilhorgne et al., 1995). Consequently, the biotransformation of taxoids at the hepatic level leads to the inactivation of the active parent molecule and consequently to a reduction of its therapeutic effect.
The purpose of the present study is to investigate the metabolism of paclitaxel and docetaxel analogs to define the structural elements orientating the hydroxylation on the taxoid molecule and determining the kinetic parameters for individual analogs. Since the only structural differences between paclitaxel and docetaxel are the replacement of the phenyl ring of the side chain at C13 by at-butyl-oxy group and the absence of the acetyl in position 10, we choose to explore separately the role of these modifications on the biotransformation of taxoids by P450 (for structure see Fig.1). Incubations were conducted with human liver microsomes to assess the overall biotransformation and with recombinant human P450 to ascribe individual reaction to a single P450 isoform.
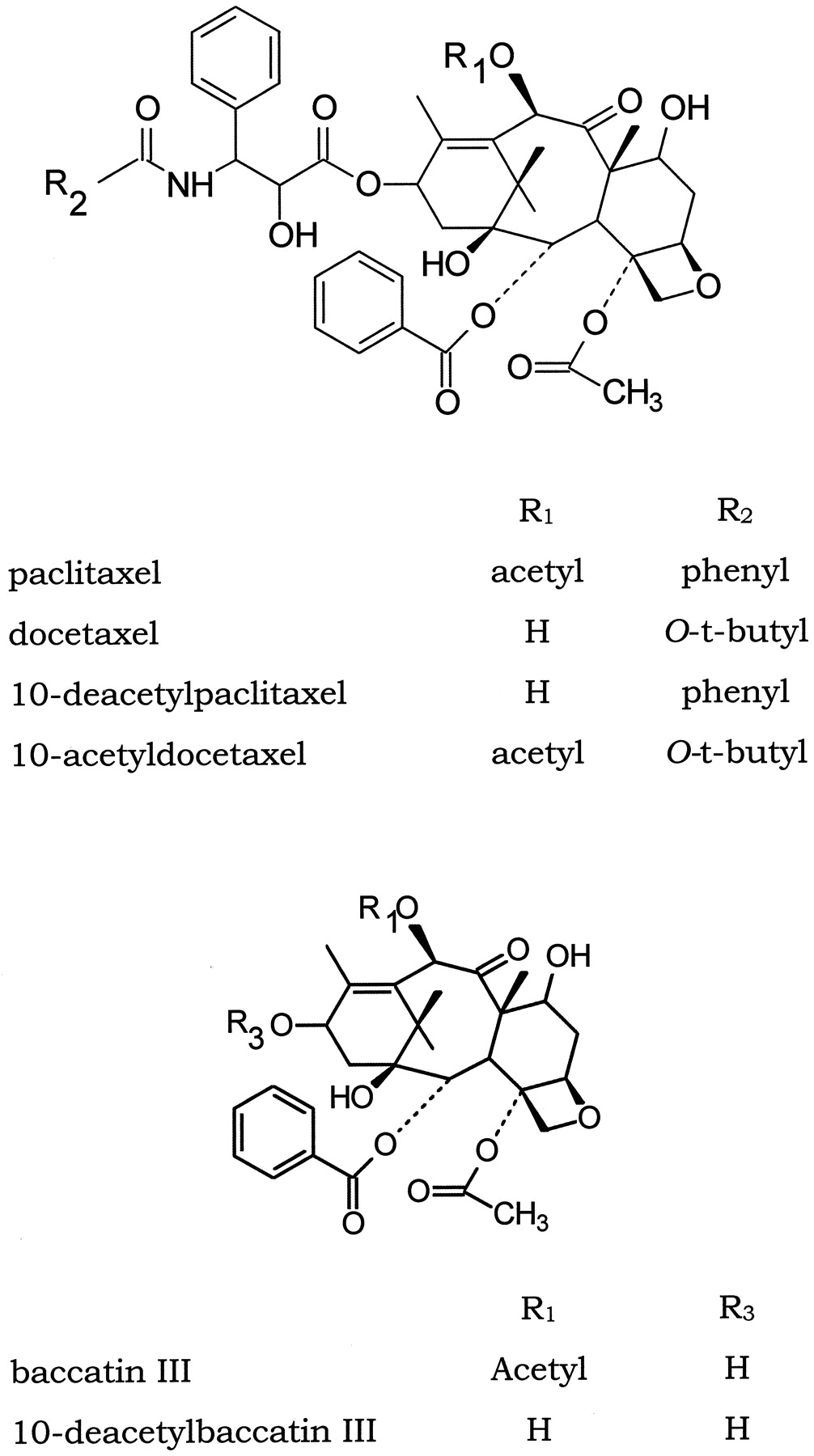
Chemical structure of taxoids.
Experimental Procedures
Materials.
Paclitaxel was kindly provided by Bristol-Myers Squibb Co. (Wallington, CT) and docetaxel by Rhone Poulenc Rorer (Antony, France). 10-Deacetylpaclitaxel, 10-acetyldocetaxel, 10-deacetylbaccatin III, and baccatin III were isolated or synthesized as previously reported (Chauvière et al., 1981; Denis et al., 1988; Mangatal et al., 1989). All chemicals were from the highest commercially available grade.
Microsome Preparation.
Human hepatic tissues were collected and microsomes prepared from frozen samples, as stated previously (Cresteil et al., 1979). The total cytochrome P450 content, the protein content, and the concentration of individual P450 isoforms were estimated as reported elsewhere (Cresteil et al., 1985, 1994).
Expression of Recombinant P450.
COS-1 cells or Ad293 cells were maintained at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 4500 mg/l d-glucose, 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The preparation of stable transfectants expressing CYP1A1, 1A2, 3A4, and 3A7 in Ad293 cells was detailed previously (Lacroix et al., 1997;Sonnier and Cresteil, 1998). A similar procedure was developed to generate stable transfectants of CYP2C8, 2C9, and 3A5. Full-length cDNA encoding CYP2C8, 2C9, and 3A5 were kindly provided by Dr. F. J. Gonzalez (NCI, Bethesda). Briefly, CYP3A5 cDNA was removed from pUC9 and 2C8 and 2C9 cDNAs from pUV-1 as EcoRI fragments and inserted in the correct orientation in the EcoRI site of pMT2 originated from the Genetics Institute (Cambridge, MA). Fifty micrograms of P450-pMT2 plasmid was transfected in COS-1 cells for transient expression by the calcium phosphate procedure, as reported byLacroix et al. (1997). Stable transfectants were obtained by transfection of Ad293 cells with 60 μg of P450-pMT2 and 3 μg of pIBW-3 plasmid containing the neomycin resistance gene. Seventy-two hours after transfection, selection was initiated with geneticin sulfate (1 mg/ml culture medium) for 2 to 3 weeks. Resistant clones derived from single colonies were harvested and grown individually in culture medium containing 50 μg/ml geneticin for 2 to 3 additional weeks. Microsomes were prepared and their content in P450 protein was estimated by Western blot with antibodies purchased from Daiichi Chemicals (Tokyo, Japan) against the rat CYP2C6 and reacting with all human CYP2C proteins, from Daiichi Chemical for CYP3A4/5 or from Affiniti (Exeter, UK) for anti-CYP3A5. Clones selected showed the highest content in P450 protein and activity toward reference compounds; 6β-hydroxylation of testosterone for CYP3A5 and dealkylation of methoxy trifluoromethylcoumarin for CYP2C. When tested with antibodies to CYP3A4/5 reacting with the two proteins, comparable intensities were obtained indicating that Ad293-3A4 and Ad293-3A5 expressed similar amount of CYP3A proteins. Immunochemically determined P450 level and activity expressed per milligram of microsomal protein were comparable in stable transfectants and liver microsomes.
Although Ad293 cells contain an appreciable NADPH-cyt P450 reductase activity, additional copies of the reductase gene could substantially stimulate the monooxygenase activity of CYP3A genes, as stated elsewhere (Ding et al., 1997). The full-length cDNA encoding the human NADPH-cyt P450 reductase was excised from pUC19-HredL, provided by Dr. P. Urban (Gif, France) and inserted into the EcoRI site of pMT2 to give pMT2-Red. Fifty micrograms of pMT2-Red was transfected into stable transfectants expressing CYP3A4, 3A5, or 3A7 together with 3 μg of pTK-Hyg, a selection marker bearing the hygromycin resistance gene (CLONTECH, Palo Alto, CA). After selection with 200 μg/ml of the culture medium hygromycin B (Invitrogen, Carlsbad, CA) for 4 weeks, resistant clones were grown and screened for their reductase activity; clones showing the highest activity were selected for further experiments.
Metabolism of Taxoids.
Microsomal proteins corresponding to 0.3 nmol of P450 were incubated in a final volume of 1 ml of 50 mM sodium phosphate, pH 7.4, 5 mM MgCl2, 10% glycerol with a NADPH-generating system consisting in 0.1 mM NADP, 1 mM glucose 6-phosphate. Taxoids (10 mM dissolved in methanol) were added to a final concentration of 50 μM except when otherwise indicated, and the reaction was initiated by the addition of glucose-6-phosphate dehydrogenase. After 30 min at 37°C, the reaction is stopped by the addition of 2.5 ml of ethyl acetate to extract the parent molecule and its metabolites. Extraction is repeated three times; organic phases were pooled and evaporated to dryness under a nitrogen stream. The residues, dissolved in 200 μl of acetonitrile/water (70:30, v/v), were analyzed by HPLC and HPLC/MS. ForKm determinations, taxoid concentrations ranged from 0 to 250 μM; double reciprocal plots allowed the calculation of apparent Kmand Vm. For structural determinations, incubations were carried out for 1 h and were scaled up by 10 to obtain a sufficient amount of metabolites.
When incubated with stable transfectants, 50 μM taxoids was added to 4 ml of culture medium without fetal calf serum in 75-cm2 tissue culture flasks containing cells at near confluence. After 24 h at 37°C, cells were scraped with a rubber policeman and maintained in the culture medium to minimize loss of material, and 5 ml of ethyl acetate was added for extraction. The extraction step was repeated three times, and organic phases were pooled before evaporation.
Characterization and Quantification of Taxoids Metabolites.
Metabolites and parent compounds were separated by reverse-phase HPLC, and peaks corresponding to the different products were quantified. The following HPLC conditions were used: 1) Ultrasphere ODS column, 4.6-mm × 25-cm, C18 (Beckman Coulter, Inc., Fullerton, CA,); 2) isocratic eluent MeOH/H2O 65:35 for paclitaxel and 10-deacetylpaclitaxel; linear gradient: 40 to 50% H2O/MeOH, 0 to 50 min for baccatin III and 10-deacetylbaccatin III; linear gradient 30 to 60% H2O/CH3CN, 0 to 55 min for docetaxel and acetyldocetaxel; 3) UV detection at 235 nm and a flow rate of 0.8 ml/min. The HPLC system included a Waters (Milford, MA) model equipped with a 600 MS pump and an U6K injector and were linked to a Waters M991 photodiode array for UV detection at 235 nm. To characterize the structure of taxoid metabolites in microsome or cell extracts, we used on-line HPLC/MS with an atmospheric pressure chemical ionization-mass spectrometry interface mode, as previously reported (Royer et al., 1995, 1996). Chromatographic conditions were the same as those described above; microsome and cell extracts were dissolved in 100 μl of a mixture of acetonitrile/water and introduced into the HPLC/MS system (TSQ 700; Thermo Finnigan MAT, San Diego, CA). Mass spectrometry data were acquired continuously in the full-scan mode (m/z 150-1200) at 2 ms/step.
Results
Metabolism of Paclitaxel.
The biotransformation of paclitaxel by human liver microsomes has been extensively studied by us and others, and we checked in a first set of experiments the formation of known metabolites in our conditions (Fig. 2 and3). As expected, two oxidation products were resolved by HPLC from incubation mixtures of liver microsomes with 50 μM paclitaxel and NADPH; the 6-hydroxy and the 3′-phenyl-hydroxy derivatives eluted at 9.5 and 16 min, respectively, in comparison with the parent molecule (20 min). These products were absent from incubations without NADPH. Their structures were explored by HPLC/MS and yielded fragments at m/z 286 (unchanged side chain at C13) and 569 (taxane ring) in the parent paclitaxel molecule shifting to either 302 or 585 after hydroxylation, which is in agreement with the structure already reported in the literature (Royer et al., 1995). The oxidation on the C2 benzoate ring could be excluded based on the shift of fragment from m/z 447 (−lateral chain − C2 benzoate) to 463 observed only in material eluting at 9.5 min.

Structure of paclitaxel, 10-deacetylpaclitaxel, docetaxel, and 10-acetyldocetaxel metabolites verified by HPLC/MS.

Chromatogram of incubation of paclitaxel with human liver microsomes.
Arrows refer to metabolites formed in the presence of NADPH eluting at 9.5 min (C3′-OH-paclitaxel) and 16 min (6-OH-paclitaxel).
When paclitaxel is incubated for 24 h with cells expressing or not expressing recombinant human P450, different HPLC profiles can be observed; in control Ad293 cells (expressing no P450), only the formation of 7-epipaclitaxel could be demonstrated and accounted for about 10% of the paclitaxel added to the culture medium. This peak of 7-epipaclitaxel was present in incubations performed with all cell lines and probably resulted from a non-P450-dependent reaction occurring during the 24-h incubation period. The HPLC profile obtained with Ad293-2C9, Ad293-1A1, and Ad293-1A2 cells was similar and confirmed that these P450 proteins play no role in the oxidation of paclitaxel. In contrast, a prominent peak of metabolite can be detected with Ad293-2C8 cells with a retention time corresponding to 6-hydroxypaclitaxel. In the same conditions, Ad293-3A4 cells produced only the 3′-phenyl-hydroxy derivative, whereas Ad293-3A5 and Ad293-3A7 cells were not capable of forming this oxidation product. Collectively, these data suggest that the metabolic pathway of paclitaxel is rather clear; 6-hydroxypaclitaxel is produced exclusively by CYP2C8 and 3′-phenyl-hydroxypaclitaxel by CYP3A4.
Metabolism of 10-Deacetylpaclitaxel.
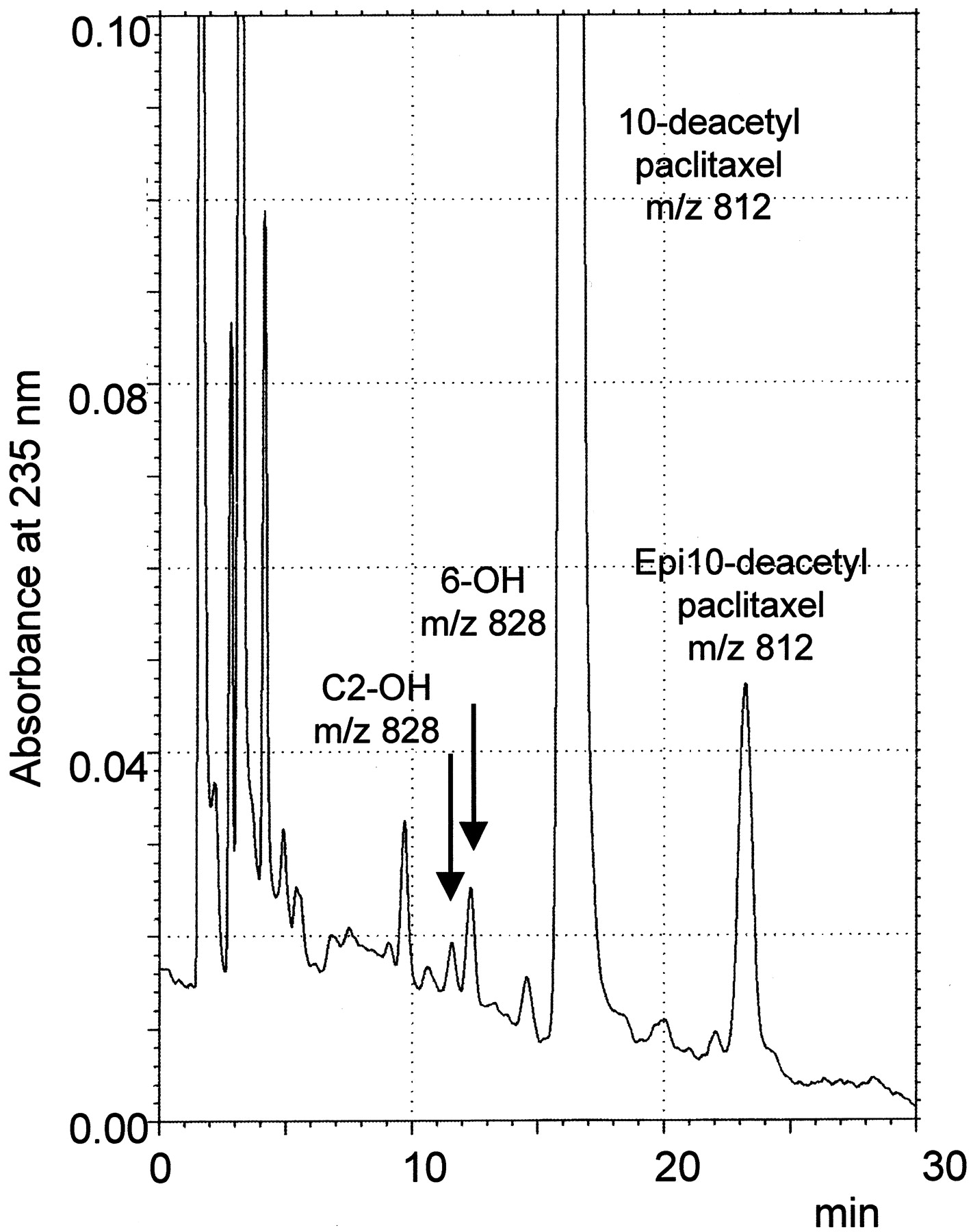
The HPLC profile of incubation mixture of 10-deacetylpaclitaxel with liver microsomes in the presence of NADPH showed two peaks eluting at 11.5 and 12.5 min, respectively. In addition, the unchanged 10-deacetylpaclitaxel had a retention time of 16 min (Fig.4). These two peaks were absent from incubations run in absence of NADPH and were identified as oxidation products. HPLC/MS analysis of 10-deacetylpaclitaxel generated a parent ion at m/z 812 and fragments atm/z 527 (intact taxane ring), 405 (taxane ring-C2 benzoate), and 286 (unchanged side chain at C13). The peak eluting at 11.5 min yielded a parent ion at m/z 828 and fragments compatible with an hydroxylation on the benzoate ring at C2, with fragments at m/z 543, 405, and 286, whereas the peak eluting at 12.5 min displayed a parent ion atm/z 828 and fragments atm/z 543, 421, and 286, indicating that oxidation occurred on the taxane ring, probably in position 6.

Chromatogram of incubation of 10-deacetylpaclitaxel with human liver microsomes.
Arrows refer to metabolites formed in the presence of NADPH eluting at 11.5 min (C2-OH-10-deacetylpaclitaxel) and 12.5 min (C6-OH-10-deacetylpaclitaxel).
Like paclitaxel, 10-deacetylpaclitaxel was steadily transformed by cells expressing P450 and by native Ad293 cells into its 7-epimer, having a retention time of 23 min; about 10 to 15% of the initial dose was recovered as the epimer after 24 h. In control Ad293 cells, no other product was identified. Similarly, Ad293-1A1 and Ad293-1A2 cells were not active toward 10-deacetylpaclitaxel. The peak eluted at 12.5 min was observed only when 10-deacetylpaclitaxel was incubated with Ad293-2C8 cells, whereas Ad293-2C9 cells were not capable to form this product. Conversely, the metabolite hydroxylated on the benzoate at C2 and eluted at 11.5 min was formed only by CYP3A proteins, although in variable amount; it is absent from incubates of Ad293-3A5 cells but present at a low content in Ad293-3A4 cells and at a higher concentration in Ad293-3A7 cell incubates.
Metabolism of Docetaxel.
The metabolism of docetaxel by human liver microsomes was consistent with those reported earlier; a single peak of product eluting at 23.5 min was observed and identified by mass spectrometry as the hydroxylated derivative on the tert-butyl of the lateral chain, shifting its mass from m/z 282 to 298, whereas a fragment at m/z 182 (lateral chain − tert-butyl − O-CO) was recovered in the product as in the native docetaxel molecule, indicating that oxidation occurred on the tert-butyl moiety.
In control and P450-expressing cells, two peaks can be monitored at 38.5 and 45 min and represented the unchanged docetaxel and its 7-epimer, respectively. In native Ad293 cells, as well as in Ad293-1A1, Ad293-1A2, Ad293-2C8, Ad293-2C9, and Ad293-3A7 cells, no other products can by detected in HPLC profiles, whereas the pattern of docetaxel metabolites generated by Ad293-3A4 and Ad293-3A5 cells was more complex (Fig. 5). The t-butyl hydroxylation was essentially mediated by Ad293-3A4 and to a lesser extent by Ad293-3A5 (0.110 and 0.034 nmol in 24 h, respectively). In addition to this oxidation product (marked with an asterisk) with a retention time of 24 min, two derivatives resulting from its cyclization (m/z 822; lateral chain 296) were also detected with retention times of 23 and 26 min. Epimers of the hydroxylated metabolite at t-butyl and its cyclized derivatives were also formed by Ad293-3A4 and Ad293-3A5 cells and were resolved with retention times of 32, 33, and 34 min, as previously identified (Shou et al., 1998).

Chromatogram of incubation of docetaxel with Ad293-3A4 cells.
Arrows refer to metabolites formed by Ad293-3A4 cells and absent from incubation with Ad293 cells. The asterisk indicates the C3′-OH-docetaxel produced by human liver microsomes.
Metabolism of 10-Acetyldocetaxel.
Metabolites generated during incubation of 10-acetyldocetaxel with liver microsomes in the presence of NADPH were resolved by HPLC and characterized by HPLC/MS (Fig. 6). The unchanged 10-acetyldocetaxel eluted at 49 min with a parent ionm/z 850 and yielded fragments atm/z 794 (−tert-butyl), 734 (−tert-butyl − acetyl at C10), 569 (−side chain at C13), 509 (−side chain at C13 − acetyl at C10), and 282 (side chain at C13). It was accompanied by several peaks; the oxidation product eluted at 37 min was hydroxylated on the tert-butyl of the lateral chain (t-butyl-OH-10-acetyldocetaxel), with a parent ion at m/z 866 and showed fragments atm/z 794, 569, 509, and 298. The oxidation on the taxane ring (presumably in position 6) led to a compound eluted at 44 min (parent ion m/z 866), having fragments atm/z 810, 750, 585, 525, and 282. Two other peaks were monitored at 40 and 42 min. The material present in these peaks cannot be distinguished by mass fragmentography and was believed to result from oxidation in two different positions on the phenyl ring of the lateral chain at C3′-parent ion m/z 866, with fragments at m/z 810, 750, 569, and 298 (phenyl-OH-10-acetyldocetaxel).

Chromatogram of incubation of 10-acetyldocetaxel with human liver microsomes.
Arrows refer to metabolites formed in the presence of NADPH eluting at 37 min (t-butyl-OH-10-acetyldocetaxel), 40 and 42 min (phenyl-OH-10-acetyldocetaxel), and 44 min (6-OH-10-acetyldocetaxel).
Like other taxoids, the epimerization of 10-acetyldocetaxel accounted for about 15% of the concentration of the parent drug in 24 h. No metabolite was produced during incubation with control Ad293, Ad293-1A1, Ad293-1A2, and Ad293-2C9 cells. In contrast, Ad293-2C8 cells were capable of forming a unique metabolite with a retention time of 44 min, which was identified as the hydroxylated derivative on the taxane ring. Conversely, only CYP3A4 was capable to actively carry out the hydroxylation of 10-acetyldocetaxel on the lateral chain at either thetert-butyl or the phenyl group but had no action on the taxane ring. These reactions were restricted to 3A4 since neither 3A5 nor 3A7 can support oxidation of 10-acetyldocetaxel.
Metabolism of Baccatin III.
When the lateral chain at C13 is absent from taxoid molecules, the biotransformation rate is severely reduced and the low amount of metabolites of baccatin III and 10-deacetylbaccatin III formed during incubation with human liver microsomes or P450-expressing cell lines made illustrating their structure difficult. In our conditions, baccatin III was eluted at 36 min after two peaks containing baccatin derivatives with retention times of 26 and 31 min. Fragments of baccatin III (parent ion m/z 587) indicated ions at m/z 527 (−acetyl), 405 (−acetyl − benzoate at C2), and 345 (−acetyl at C4 − benzoate at C2 − acetyl at C10). Fragments issued from the derivatives gave the same pattern with ions at m/z 543, 421, and 361 for a parent ion at m/z 603. This indicated that an oxygen atom was added to the taxane ring and was not compatible with an oxidation on the benzoate ring at C2, with expected fragments atm/z 603, 543, 405, and 345. These two hydroxylated metabolites on the taxane ring were produced only by Ad293-3A4 and Ad293-3A7 cells; all other P450-expressing cell lines were devoid of activity but retained the epimerization of baccatin. In addition to these two metabolites, a very low amount of hydroxylated metabolites of 7-epibaccatin can also be detected in HPLC but cannot be accurately analyzed in HPLC/MS.
Metabolism of 10-Deacetylbaccatin III.
Extracts prepared from incubations of NADPH with 10-deacetylbaccatin and liver microsomes contained a single peak considered a direct metabolite, with a retention time of 23 versus 24 min for 10-deacetylbaccatin. The structure of this metabolite was verified by HPLC/MS; the protonated molecular ion (m/z 545) and characteristic fragment ions were 16 mass units greater than 10-deacetylbaccatin, indicating that an oxygen atom was added to the taxane ring.
Neither control Ad293 cells nor Ad293-1A1, Ad293-1A2, Ad293-2C8, and Ad293-2C9 cells generated an oxidation product of 10-deacetylbaccatin. Only Ad293-3A4 and Ad293-3A7 cells produced the derivative eluted, in a very limited amount, at 23 min.
Quantitative Metabolism of Taxoids by Human Liver Microsomes.
The overall metabolism of taxoids and the balance between the different oxidative routes were investigated in liver microsomes and reflected the participation of individual P450 isoforms to the overall metabolism of taxoids by microsomes. The rate of biotransformation was quite variable, ranging from 0.36 to 4% of the substrate concentration transformed in 30 min by 1 mg of microsomal protein when taxoids were incubated at a concentration of 50 μM. A general feature is related to the presence of an acetyl group in position 10 of the taxane ring, which strikingly activates the biotransformation rate by liver microsomes. The rate of reaction is 2- to 5-fold higher with baccatin III, 10-acetyldocetaxel, and paclitaxel than with 10-deacetylbaccatin III, docetaxel, and 10-deacetylpaclitaxel, respectively (Table1).
Overall metabolism and kinetic parameters of individual biotransformation reactions of taxoids by human liver microsomes
Assuming that a single P450 protein is involved in individual reaction (Table 2), we ascribed biotransformation and its kinetic parameter calculated from data obtained with human liver microsomes to isoform 2C8 or 3A4. The hydroxylation on thetert-butyl of docetaxel could be attributed to both CYP3A4 and 3A5, and the value determined in liver microsomes should be the sum of the two contributions. As already reported, the formation of 6-hydroxypaclitaxel was higher than the formation of the 3′-phenyl-hydroxylated derivative (63 versus 37% of total metabolites) and was associated with a higher Vm, even if the Km value for the two reactions was similar. Using the ratioVm/Kmto assess the efficiency of P450 in paclitaxel metabolism, CYP2C8 was found to be more effective than CYP3A4 (130 versus 35). The same ratio between 6-hydroxylation and C2-hydroxylation was observed with 10-deacetylpaclitaxel. The hydroxylation on the taxane ring largely exceeded the hydroxylation on the benzoate ring at C2 (72 versus 28% at 50 μM 10-deacetylpaclitaxel), but the affinity of 10-deacetylpaclitaxel for both CYP3A4 and 2C8 was strikingly reduced. The Km value of 10-deacetylpaclitaxel and paclitaxel for CYP3A4 were 67 and 15 μM, respectively, whereas the Km values for CYP2C8 were 200 and 15 μM. Thus, the presence of the acetyl group in position 10 improved the affinity for P450 and would suggest that hydrophobic interactions existed between the substrate and the binding site of both P450 proteins.
Summary of the biotransformation reactions catalyzed by recombinant human PYSO proteins
With docetaxel a single primary metabolite resulting from the oxidation on the lateral chain was generated by CYP3A4 with an excellent affinity (2 μM). Consequently, the ratioVm/Kmwas the highest in the series. 10-Acetyldocetaxel retained the same affinity for the 3A4-dependent reaction (4 to 7 μM), but the hydroxylation on the taxane ring mediated by CYP2C8 exhibited a moderate affinity (50 μM). However, theVm of CYP2C8 largely exceeded theVm of CYP3A4. The higher affinity of 10-acetyldocetaxel for CYP3A4 than for CYP2C8 accounted for the preferential formation (74% of metabolites) of the 3′-phenyl-OH andt-butyl-OH derivatives when liver microsomes were incubated with 50 μM 10-acetyldocetaxel.
The affinity of baccatin III and 10-deacetylbaccatin III for liver microsomes ranged from 25 to 200 μM and the maximal velocities from 575 to 1920 pmol of metabolite formed in 1 min by 1 nmol of P450. These values were in the range of values calculated with other taxoids. The balance between the two unidentified metabolites of baccatin III was 4:1, the less abundant metabolite showing the lowest affinity and maximal velocity.
Collectively these data indicated that the affinity of taxoids for CYP3A4 was better than the affinity for CYP2C8, but this latter exhibited a higher maximal velocity than CYP3A4. Therefore, we can consider CYP3A4 as a high-affinity component involved in the biotransformation of taxoids compared with the low-affinity component CYP2C8.
Discussion
The rationale of this study was based on the observation that two changes in the structure of the taxoid molecule could shift the major hydroxylation site from the C6 of the taxane ring of paclitaxel to the side chain at C13 in docetaxel. Therefore, we intend to evaluate the impact of individual modification of the taxoid molecule on the site of hydroxylation and extent of the reaction by P450-dependent monooxygenases.
Data are summarized in Table 2. CYP3A4 and to a minor extent CYP3A7 were the only P450 isoforms tested in this study capable to actively hydroxylate taxoid molecules lacking the side chain at C13, such as in baccatin III and 10-deacetylbaccatin III. The reaction occurred on the taxane ring probably in position 6, whereas CYP2C8 responsible for this reaction in paclitaxel had no activity. The presence of atert-butyl group in the side chain at C3′, such as in docetaxel and 10-acetyldocetaxel, increased the overall oxidation of taxoids by CYP3A isoforms and orientated the metabolism toward the formation of the oxidation product on the tert-butyl group. A hydroxylation on a tert-butyl group by CYP3A4 has already been described for terfenadine, the tertiary butyl group being liable to oxidation because of its high number of available primary carbons (Yun et al., 1993). The hydroxylated derivative was the unique primary metabolite formed from docetaxel and accounted for 74% of the total metabolite formation from 10-acetyldocetaxel by human liver microsomes, with a similar affinity for docetaxel and 10-acetyldocetaxel. CYP3A4 was the major contributor to this reaction, whereas CYP3A5 only moderately catalyzed this reaction, as already reported by Royer et al. (1996) and Shou et al. (1998). Actually the affinity of CYP3A4 for docetaxel has been reported to be 10-fold higher than those of CYP3A5 and could explain why the participation of CYP3A5 to the biotransformation of taxoids at a dose of 50 μM might be very low in relation with the low affinity of CYP3A5 for taxoids. Conversely, the presence of a benzamide group in the side chain favored the hydroxylation in position 6 of the taxane ring, with more than 60% of the metabolite formed from either paclitaxel or 10-deacetylpaclitaxel by CYP2C8.
Collectively these results suggested that the side chain plays a crucial role in determining the orientation of the taxoid molecule into the active site of P450 proteins. The presence of the side chain is required for a correct orientation of the substrate into the active site of CYP2C8 and to place the putative hydroxylation site (e.g., carbon 6 of the taxane ring) in the vicinity of the heme-oxygen complex. Its absence completely prevents any reaction by CYP2C8. The replacement of the tert-butyl group of the side chain by a phenyl ring facilitates the positioning of the substrate into the CYP2C8-active site and would probably suggest that hydrophobic interactions exist between the phenyl group of the side chain and hydrophobic residues of the CYP2C8 peptide chain via π-π bonding. Conversely, the presence of a benzamide group instead of a boc-amino group in the side chain reduces the hydroxylation by CYP3A4 through a decreased affinity for the protein. This effect may be related to different conformations of the side chain of paclitaxel and docetaxel, as proposed earlier (Dubois et al., 1993).
Another feature is the 2- to 5-fold increase of the overall hydroxylation rate of 10-deacetylbaccatin III, docetaxel, and 10-deacetylpaclitaxel by human liver microsomes when an acetyl group is added in position 10 of the taxane ring. This higher biotransformation results from either a stimulation of the catalytic velocity for baccatin and 10-acetyldocetaxel or an increased affinity for paclitaxel without modification of the catalytic efficiency. Both CYP3A4- and CYP2C8-dependent activities are stimulated, indicating that the presence of a hydrophobic group in position 10 stimulates the activity, but without striking modification of the balance between CYP3A4 and 2C8. However the addition of the acetyl group in position 10 of docetaxel allowed the 6-hydroxylation of 10-acetyldocetaxel by CYP2C8 but with a weaker affinity than the oxidation on thetert-butyl group by CYP3A4.
These observations attribute a major role in the substrate recognition by P450 to hydrophobic groups and to interaction between these hydrophobic groups and hydrophobic amino acid residues. Several key residues have been hypothesized to play an active role in the binding of substrates to CYP3A4 based on sequence alignment or after site-directed mutagenesis. Thus, aromatic residues Phe215, Phe304, and Tyr307 (respectively Phe181, Phe263, and Tyr266 when aligned on BM3 or cyp102 fromBacillus megaterium) could interact with groups of 3A4 substrates but are also present in CYP3A5 and 3A7 (Lewis et al., 1996). Mutations in SRS4 within close proximity to the putative substrate-binding pocket have demonstrated altered oxidation products with progesterone but are similar in 3A4, 3A5, and 3A7 (Domanski et al., 1998). Similarly, the highly conserved residue Ser119 has been shown to play an important role in the active site topology of CYP3A4 but is also present in 3A5 and 3A7 (Roussel et al., 2000). To date no definitive clue has been provided regarding the identity of amino acids involved in substrate recognition and orientation, and the sequence homology between 3A4, 3A5, and 3A7 could not explain the difference observed in the transformation of taxoids. In this respect, the use of substituted taxoids allows exploring the orientation of substrates into the active site of CYP3A and constitutes a useful tool to probe the hydrophobic pocket of these enzymes.
The biological activity of taxoids tested upon microtubule assembly or in in vitro cytotoxic assays has demonstrated a partial relationship between the hydrophobicity of taxoids and their interaction with tubulin. Thus paclitaxel, 10-deacetylpaclitaxel, docetaxel, and 10-acetyldocetaxel showed a very similar potency to inhibit microtubule disassembly and modification of substituents at C10 has no or little effect on the activity except when very large groups are branched at C10 (Guénard et al., 2000). Furthermore, the addition of polar substituents has no effect in the tubulin assay. These data confirmed previous observations reporting that modifications at C10 (or C7) have generally no effect on the biological activity of taxoids (Parness et al., 1982). In contrast the potent biological activity of taxoids is generally reduced by modification on the lateral chain of paclitaxel and docetaxel (Lataste et al., 1984). Gueritte-Voegelein et al. (1991)and Guenard et al. (1993) reported that the configuration at position C2′ and C3′ of paclitaxel is important for the inhibition of microtubule assembly and postulated a positive correlation between the conformation of the side chain and the binding activity. These results differ from the striking difference reported here in the biotransformation of taxoids by P450 proteins and could be the basis for the design of molecule with high biological activity and low biotransformation.
Footnotes
-
This work was supported by Grant 9123 from Association de Recherche sur le Cancer and Grant 99001129 from Conseil régional Midi-Pyrénées.
- Abbreviations used are::
- P450
- cytochrome P450
- HPLC
- high-pressure liquid chromatography
- MS
- mass spectometry
- Received October 9, 2001.
- Accepted January 3, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}