


Abstract
Dehydroepiandrosterone (DHEA) is a steroid produced by the human adrenal gland. Administration of pharmacological doses of DHEA to rats changes expression of many genes, including the cytochrome P450 family members CYP4A1 and CYP3A23. It is known that induction of CYP4A expression by DHEA requires the peroxisome proliferator-activated receptor α (PPARα). In the current study, PPARα-null mice were used to examine the role of PPARα in expression of CYP3A. In wild-type mice, 150 mg/kg DHEA-sulfate induced Cyp4a and Cyp3a11 mRNAs by 5- and 2-fold, respectively. Induction of Cyp4a expression by DHEA-sulfate was not observed in PPARα-null mice, whereas induction of Cyp3a11 expression by DHEA-sulfate was similar between genotypes. This suggests that PPARα is not involved in induction of Cyp3a11 expression by DHEA. Because expression of CYP3A family members can be induced by activation of another member of the nuclear receptor superfamily, the pregnane X receptor (PXR), we examined the ability of DHEA to activate PXR. In transient transfection assays, DHEA and its metabolites androst-5-ene-3β,17β-diol (ADIOL), androst-5-ene-3,17-dione, and androst-4-ene-3,17-dione were activators of PXR. Maximal induction of a PXR-responsive reporter gene of approximately 3-fold was observed at concentrations of 50 to 100 μM, indicating that these steroids are relatively weak activators of PXR. Human and murine PXR exhibited different specificities for DHEA and its metabolites. ADIOL activated reporter gene expression in the presence of murine but not human PXR. Results of these studies suggest that the induction of rodent CYP3A expression upon treatment with high doses of DHEA occurs through activation of PXR.
Administration of dehydroepiandrosterone (DHEA2) to rodents results in several beneficial biological responses. DHEA acts as a chemopreventive agent in rodent cancer models (Schwartz, 1979; Schwartz and Tannen, 1981; Nyce et al., 1984; Pashko et al., 1984). Rodents with a predisposition toward obesity demonstrate decreased rates of weight gain without appetite suppression upon treatment with DHEA (Yen et al., 1977). Administration of DHEA ameliorated symptoms of diabetes and systemic lupus erythematosis in the appropriate rodent models (Coleman et al., 1982; Lucas et al., 1985). DHEA may act as a neurosteroid, and is thought to enhance memory function (Robel and Baulieu, 1995), as well as immune function (Morfin and Courchay, 1994).
DHEA is the most abundant circulating steroid in humans. It is secreted by the adrenal gland as the 3β-sulfate conjugate. DHEA-sulfate (DHEA-S) is taken up by target tissues (e.g., testis and ovary), and hydrolyzed by sulfatases back to DHEA (Kroboth et al., 1999). DHEA can then be further metabolized to active androgens and estrogens in steroidogenic tissues, as well as to several hydroxylated metabolites in liver (Fitzpatrick et al., 2001).
In rodents, DHEA is not produced by the adrenal, but limited amounts of DHEA are produced from progesterone in steroidogenic tissues (Pelletier et al., 1992; van Weerden et al., 1992). Although treatment of rodents with exogenous DHEA can produce the beneficial effects described above, high doses of DHEA induce peroxisome proliferation (Wu et al., 1989;Prough et al., 1994). Peroxisome proliferators, including DHEA, modulate expression of genes involved in fatty acid metabolism, including fatty acyl CoA oxidase, cytochrome P450 4A (CYP4A), and malic enzyme (Webb et al., 1996).3 DHEA and other peroxisome proliferators require the peroxisome proliferator-activated receptor α (PPARα) to modulate gene expression because these compounds are unable to induce expression ofCyp4a and other characteristic responses associated with peroxisome proliferation in PPARα-null mice (Peters et al., 1996). Peroxisome proliferators are typically able to activate PPARα in cell-based reporter assays; however, DHEA does not activate PPARα in these cell-based assays (Issemann and Green, 1990; Peters et al., 1996).
In addition to CYP4A, DHEA alters expression of other cytochromes P450 involved in metabolism of endogenous compounds and xenobiotics, including CYP3A23, the major glucocorticoid-inducible form of CYP3A in rat liver (Gonzalez et al., 1986; Singleton et al., 1999). The CYP3A subfamily of enzymes catalyzes the oxidation of endogenous steroids such as testosterone and cortisol, as well as the metabolism of a wide array of drugs. Several lines of evidence suggest that induction ofCYP3A23 expression by DHEA proceeds by a different mechanism than induction of CYP4A1 expression by DHEA.CYP3A23 expression is induced by DHEA treatment, but not by treatment with other peroxisome proliferators (Singleton et al., 1999). The degree of induction of CYP3A23 is much less than that ofCYP4A1 (i.e., hepatic CYP3A23 mRNA levels are induced by 2- to 3-fold in adult rats), whereas CYP4A1 mRNA levels are induced by approximately 30-fold. Finally, induction of CYP3A23expression is more marked in female animals than in male animals, whereas induction of CYP4A1 expression is more pronounced in males (Singleton et al., 1999). These observations suggest that DHEA's modulation of CYP3A23 and CYP4A1 expression proceeds by distinctly different mechanisms.
Recent studies have demonstrated that a member of the nuclear receptor superfamily, the pregnane X receptor (PXR), mediates induction of hepatic CYP3A23 expression in rat (Kliewer et al., 1998;Huss and Kasper, 2000). PXR also regulates the expression of other CYP3A family members in rodents, rabbits, and humans (Lehmann et al., 1998; Savas et al., 2000). Naturally occurring steroids that are known to activate PXR include some glucocorticoid and pregnane derivatives (Kliewer et al., 1998). PXR is also activated by potentially hepatotoxic bile acids. The activation of PXR by lithocholic acid leads to induction of CYP3A and the organic anion transporter 2, which then leads to increased metabolism and excretion of the bile acid. Therefore, activation of PXR is thought to provide liver protection from potentially toxic levels of bile acids (Staudinger et al., 2001). DHEA was tested for its ability to activate human and murine PXR in in vitro assays and was found to be a relatively weak activator of PXR up to 10 μM (Blumberg et al., 1998; Jones et al., 2000). However, a full concentration-response for PXR activation by DHEA or any of its metabolites has not been performed to date. The purpose of the current study was to examine the mechanism of induction of CYP3A expression by DHEA or its metabolites and determine potential participation of PPARα and PXR.
Materials and Methods
Chemicals.
Androst-5-ene-3β,17β-diol (ADIOL); androst-5-ene-3,17-dione (ADIONE); DHEA; DHEA-S; 3β,11β-dihydroxy-androst-5-en-17-one (11β-hydroxy-DHEA); 3β,16α-dihydroxy-androst-5-en-17-one (16α-hydroxy-DHEA); 3β-hydroxy-androst-5-ene-7,17-dione (7-oxo-DHEA); and 3β,7α-dihydroxy-androst-5-en-17-one (7α-hydroxy-DHEA) were purchased from Steraloids (Newport, RI). Dimethyl sulfoxide (DMSO) and ethyl acetate were purchased from Fisher Scientific (Pittsburgh, PA). Nafenopin was a gift from Novartis (Ardsley, NY).
In Vivo Treatments.
PPARα-null mice were generated as described (Lee et al., 1995). Male wild-type (+/+) and PPARα-null (−/−) mice (F3homozygotes or wild-type; hybrids of C57BL/6N × Sv/129 genetic background; 10–12 weeks of age) were given daily i.p. injections with either corn oil alone, or 150 mg/kg DHEA-S dissolved in corn oil for 4 days (Peters et al., 1996). Twenty-four hours after the final injection, mice were euthanized and livers removed and rapidly frozen in liquid nitrogen. Tissues were stored at −80°C until used for isolation of RNA.
Northern Blot Analyses.
Total RNA was isolated from mouse livers using guanidinium hydrochloride/phenol extraction with TRIzol reagent (Invitrogen, Carlsbad, CA). RNA (12 μg) was electrophoresed on a 1% agarose/10% formaldehyde gel and transferred to Zeta-probe nylon membranes (Bio-Rad, Hercules, CA). Plasmids containing cDNA for rat CYP4A1, mouse Cyp3a11, and rat glyceraldehydes-3-phosphate dehydrogenase (GAPDH) were generously provided by James Hardwick (Northeastern Ohio University, Rootstown, OH), Kazuo Nakayama (Hokkaido University, Hokkaido, Japan), and Jean-Marie Blanchard (University des Sciences et Techniques du Languedoc, Montpellier, France), respectively. cDNAs were cut from plasmids, purified, and labeled with [α-32P]dCTP using a random primed DNA labeling kit (Roche Applied Sciences, Indianapolis, IN). Hybridizations were carried out in UltraHyb solution (Ambion, Austin, TX) at 42°C overnight. Blots were washed twice in 2× standard saline citrate, 0.1% SDS for 15 min at 42°C, and once in 0.2× standard saline citrate, 0.1% SDS for 15 min at room temperature. Blots were exposed to phosphorscreens and scanned and quantitated using a PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Plasmids.
Expression plasmids containing the cDNA for murine PXR.1 (mPXR) or human PXR (hPXR) were generous gifts from Steve Kliewer (GlaxoSmithKline, Research Triangle Park, NC). The reporter plasmid PXRELUC was constructed by inserting two copies of a double-stranded oligonucleotide containing the CYP3A23 PXRE (5′-GATCAGACAGTTCATGAAGTTCATCTAGATC-3′) into the NdeI site of 0.164YaLUC (Falkner et al., 1998). 0.164YaLUC contains the minimal glutathione S-transferase A2 (GSTA2) promoter linked to the firefly luciferase reporter gene. The expression plasmid for β-galactosidase (pCMVβ) was purchased from CLONTECH (Palo Alto, CA). All plasmids were transformed into DH5αEscherichia coli bacteria, isolated, and prepared for use in transient transfections using QIAGEN plasmid prep kits (QIAGEN, Chatsworth, CA).
Transient Transfections.
CV-1 cells (ATCC no. CCL-70) were grown at 37°C in 5% carbon dioxide atmosphere. Cells were plated at 1.5 × 105cells/well in 12-well plates containing minimal essential medium supplemented with 5% charcoal-stripped fetal bovine serum. Twenty-four hours after plating, cells were transfected using 4 μg/ml LipofectAMINE (Invitrogen, Carlsbad, CA) with mPXR or hPXR expression plasmid (30 ng/ml), PXRELUC reporter plasmid (150 ng/ml), and pCMVβ (100 ng/ml) in serum-free medium. Each well was overlaid with 1 ml of transfection mixture and incubated overnight. After removal of the transfection mixture, cells were washed with phosphate-buffered saline and new medium supplemented with 5% charcoal-stripped serum was added. Transfected cells were treated with 500× concentrated stocks of DHEA and metabolites in DMSO, and harvested 24 h later with 100 μl of cell lysis buffer (Promega, Madison, WI). β-Galactosidase and luciferase activities were determined as described by Falkner et al. (1998). Data are expressed as luciferase activity relative to β-galactosidase activity to correct for transfection efficiency. All transient transfection experiments were performed in triplicate or quadruplicate, and experiments were repeated at least twice to confirm results.
Statistics.
Experiments were conducted in triplicate or quadruplicate and means ± standard deviations were determined. Statistical comparisons among treatment groups were determined using a two-tailedt test, with p < 0.05 as the criterion for significance. Where multiple comparisons were indicated, data were first analyzed by one-way analysis of variance at p < 0.05, followed by t tests adjusted using the Bonferroni procedure for multiple comparisons, with p < 0.05.
Results
Induction of Cyp3a11 Expression Is PPARα-Independent.
DHEA is known to induce expression of peroxisome proliferator-responsive genes, such as CYP4A, through activation of PPARα (Peters et al., 1996). Because CYP3A23 mRNA levels in rats were induced by DHEA, but not by the more potent peroxisome proliferator nafenopin (Singleton et al., 1999), we wanted to investigate whether PPARα was involved in CYP3Ainduction. PPARα-null mice were used for these studies. Wild-type or PPARα-null mice were treated with corn oil alone (control) or 150 mg/kg DHEA-S in corn oil for 4 days. Hepatic Cyp4a, Cyp3a11, and GAPDH mRNA levels were determined by Northern blot analysis (Fig.1). The probe used for Cyp4a was rat CYP4A1 cDNA sequence that has been shown to cross-react with a peroxisome proliferator-responsive mouse Cyp4a form (Peters et al., 1996). Cyp3a11 is the major glucocorticoid-inducible Cyp3a form in mice, and it is regulated in a similar manner toCYP3A23 in rats (Yanagimoto et al., 1997). Cyp4a mRNA levels were induced 5-fold by DHEA-S treatment and this induction was lost in PPARα-null mice, consistent with previous results (Peters et al., 1996). In contrast, Cyp3a11 mRNA levels were induced by approximately 2.0-fold in both wild-type and PPARα-null mice. The smaller degree of induction of Cyp3a11 expression by DHEA is consistent with previous studies in adult rats, which showed that induction ofCYP3A23 expression (approximately 2- to 3-fold) was much less pronounced than induction of CYP4A1 expression (approximately 30-fold). These results suggest that induction ofCyp4a and Cyp3a11 by DHEA proceed by distinct mechanisms, PPARα-dependent for Cyp4a and PPARα-independent for Cyp3a11.

Induction of Cyp3a11 expression is PPARα-independent.
Wild-type or PPARα-null mice were given daily i.p.injections of either corn oil alone (control) or 150 mg/kg DHEA-S dissolved in corn oil for 4 days. Hepatic mRNA was isolated, and Northern blot analyses was performed with probes to Cyp4a, Cyp3a11, and GAPDH (A). Levels of Cyp4a (B) and Cyp3a11 (C) mRNAs were quantified and normalized to GAPDH. Data are expressed as means ± S.D. for four separate animals. Northern analyses were repeated three times with nearly identical results. ★, significantly different from corn oil-treatment, p < 0.05; ■, corn oil; ▨, DHEA-S.
DHEA, ADIONE, and ADIOL Activate PXR in CV-1 Cells.
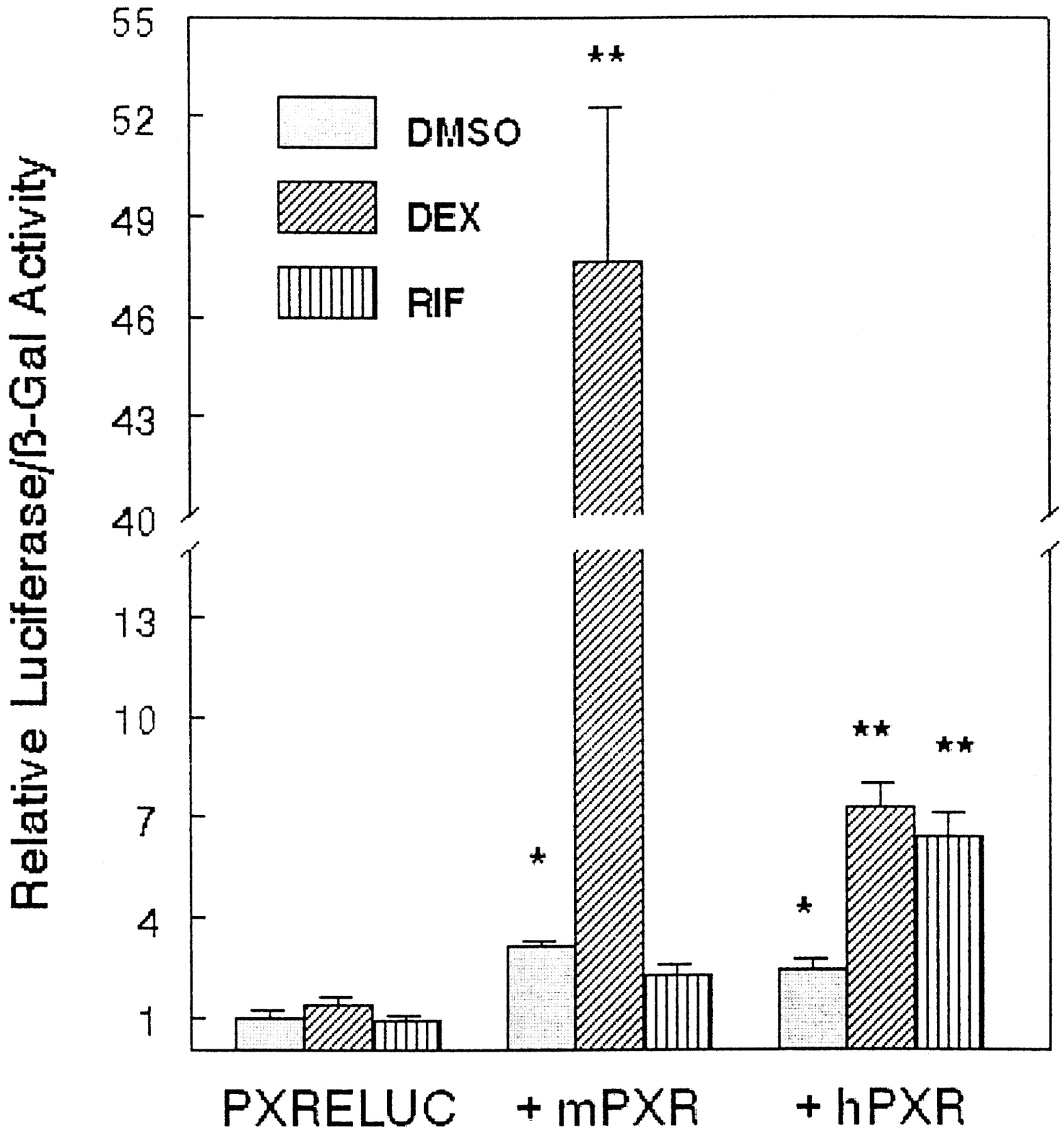
Because some steroid compounds are known to induce expression ofCYP3A23 through activation of the nuclear receptor PXR, we tested the ability of DHEA and its metabolites to activate gene transcription through PXR. A reporter plasmid was constructed that contained two copies of the PXR responsive element fromCYP3A23 located 5′ of a minimal promoter driving expression of a luciferase reporter gene (PXRELUC). Transient transfection assays were carried out in CV-1 cells cotransfected with the PXRELUC reporter construct and expression plasmids for either murine PXR or human PXR. The reporter construct was activated in response to the known PXR agonists dexamethasone t-butyl-acetate and rifampicin (Fig.2). Murine PXR was much more responsive to dexamethasone t-butyl-acetate than human PXR, whereas human PXR was more responsive to rifampicin than murine PXR. This is consistent with species differences characterized for these receptors by Lehmann et al. (1998).

PXR agonists increase expression of PXRELUC in the presence of cotransfected PXR.
CV-1 cells were transfected with PXRELUC reporter plasmid and expression vector for murine PXR or human PXR. Cells were treated for 24 h with vehicle (DMSO), 10 μM dexamethasonet-butyl-acetate (DEX), or 10 μM rifampicin (RIF). Cells were then harvested, and lysates were assayed for β-galactosidase and luciferase activities. Data represent the mean ± S.D. of four wells. Experiments were repeated twice with similar results. ★, significantly different from PXRELUC in the absence of coexpressed receptor, p < 0.05. ★★, significantly different from DMSO treatment, p < 0.05.
Next, DHEA and some of its known metabolites were tested for their ability to induce expression of PXRELUC through both human and murine PXR in CV-1 cells (Fig. 3). Initially, 100 μM concentrations of DHEA and metabolites were screened. DHEA induced expression of PXRELUC via both murine and human PXR, although the activation in the presence of the mouse receptor (2- to 3-fold) was slightly less than that seen in the presence of the human receptor (3- to 4-fold). Neither the sulfate conjugate of DHEA nor the oxidized derivatives 7α-hydroxy-DHEA, 7-oxo-DHEA, 16α-hydroxy-DHEA, and 11β-hydroxy-DHEA were active with either receptor. However, the cytosolic metabolites ADIONE and ADIOL were active. ADIONE activated both murine and human PXR to approximately the same degree, whereas ADIOL was active only with the mouse receptor, but not the human receptor.

Increased PXRELUC reporter activity in response to DHEA and metabolites in the presence of cotransfected hPXR or mPXR.
CV-1 cells were transfected with PXRELUC reporter plasmid and expression vector for either murine PXR or human PXR. Cells were treated for 24 h with vehicle (DMSO), 100 μM DHEA metabolites, or 10 μM dexamethasone t-butyl acetate. Cells were then harvested, and lysates were assayed for β-galactosidase and luciferase activities. Data represent the mean ± S.D. of four wells. Experiments were repeated three times with similar results. Statistical significance was determined using analysis of variance followed by t tests adjusted for multiple comparisons using the Bonferroni procedure. ★, significantly different from DMSO-treated cells, p < 0.05. Metabolites tested: DHEA, DHEA-S, ADIOL, ADIONE, 7α-hydroxy-DHEA (7-OH-DHEA), 7-oxo-DHEA, 11β-hydroxy-DHEA (11-OH-DHEA), 16α-hydroxy-DHEA (16-OH-DHEA), and dexamethasone t-butyl acetate (DEX).
Concentration-response studies were conducted to evaluate the potency of induction by DHEA, ADIONE, and ADIOL compared with dexamethasonet-butyl-acetate. Both human and mouse PXR are maximally responsive to dexamethasone t-butyl-acetate at a concentration of 10 μM (data not shown). Maximal activation of PXRELUC with the mouse receptor was approximately 50-fold, whereas maximal activation by the human receptor was approximately 10-fold. DHEA induced expression of PXRELUC by approximately 2- to 4-fold with either human or mouse PXR (Fig. 4). However, the human PXR showed significant induction at 50 μM DHEA, whereas mPXR did not show significant induction at concentrations below 100 μM. ADIONE produced a maximal induction of PXRELUC of approximately 3-fold with both hPXR and mPXR, and both androst-4-ene-3,17-dione and androst-5-ene-3,17-dione displayed nearly identical concentration dependence for activation of PXR (data not shown). ADIOL induced PXRELUC by approximately 3-fold only when mPXR was cotransfected, but not when hPXR was cotransfected. Activation of mPXR by ADIOL occurred at lower concentrations than those required for DHEA or ADIONE. DHEA, ADIONE, and ADIOL were less potent activators of PXR than dexamethasone t-butyl-acetate, requiring concentrations of approximately 100 μM for maximal activation, compared with the 10 μM required for dexamethasonet-butyl-acetate. The inhibitory effect of ADIONE and DHEA above 100 μM concentrations cannot be explained at this time, but similar effects of peroxisome proliferators were observed by Boie et al. (1993) with the murine PPARα. To exclude the possibility that the 7α-hydroxy-DHEA, 7-oxo-DHEA, 16α-hydroxy-DHEA, 11β-hydroxy-DHEA, and DHEA-S also exhibited a bell-shaped dose-response curve, these compounds were also tested at concentrations from 1 to 200 μM and did not activate reporter gene expression at any concentration (data not shown).

Concentration-dependent activation of PXR by DHEA, ADIONE, and ADIOL.
CV-1 cells were transfected with PXRELUC reporter plasmid and expression vector for either human PXR (A) or murine PXR (B). Cells were treated for 24 h with vehicle (DMSO) or varying concentrations of DHEA, ADIONE, or ADIOL. Cells were then harvested, and lysates were assayed for β-galactosidase and luciferase activities. Data represent the mean ± S.D. of four wells. Experiments were repeated at least twice with similar results. ★, significantly different from DMSO-treated cells, p< 0.05; ●, DHEA; ▴, ADIONE; ▪, ADIOL.
Discussion
Treatment of rats with pharmacological doses of DHEA results in modulation of expression of a number of genes typically responsive to peroxisome proliferators, including acyl CoA-oxidase, malic enzyme, and CYP4A family members (Wu et al., 1989; Webb et al., 1996). Treatment of rats with DHEA also induces expression of CYP3A23, a gene whose expression is not induced by other peroxisome proliferators (Singleton et al., 1999). Induction of expression of CYP4Afamily members by DHEA has previously been shown to be dependent on PPARα (Peters et al., 1996); however, the PPARα dependence ofCYP3A induction had not been determined. The studies described herein used PPARα-null mice to examine the role of PPARα in induction of Cyp3a11 (the mouse homolog to ratCYP3A23) by DHEA. Results indicate that induction ofCyp3a11 expression by DHEA is not dependent on PPARα. Moreover, DHEA and its metabolites ADIONE and ADIOL are activators of PXR in vitro. Although DHEA, ADIONE, and ADIOL are relatively weak activators of PXR, activation of PXR is the likely mechanism for induction of CYP3A forms in rats and mice fed high doses of DHEA.
Peters et al. (1996) have demonstrated that treatment with both DHEA and DHEA-S produce modulation of gene expression in vivo, with DHEA-S being slightly more potent. However, in our studies, only DHEA was able to activate PXR in vitro. The differential activities of DHEA and DHEA-S in vivo versus cultured cell lines may be due to differential abilities to transport DHEA-S. DHEA is expected to pass through the cell membrane by diffusion because it is a lipophilic compound. However, DHEA-S is taken up by active transport (Reuter and Mayer, 1995). The organic anion transport systems that transport DHEA-S may be lost in cultured cell lines. Once inside cells, due to the presence of steroid sulfotransferases and sulfatases, there is equilibrium between DHEA and DHEA-S (Yamada et al., 1994). Due to the interconversion of DHEA and DHEA-S in hepatocytes, it is unclear whether DHEA or DHEA-S is the active compound. Therefore, the apparent lack of activity of DHEA-S in the in vitro PXR activation assays may be due to lack of steroid transport and not lack of activity per se.
The PXR orthologs from rabbits, rats, mice, and humans exhibit high sequence similarity in their DNA-binding domains. However, they show considerable variability in ligand-binding domains, and therefore exhibit significant species-specific differences in response to ligands (Jones et al., 2000). For example, the antibiotic rifampicin is a good activator of human and rabbit PXR, but a very poor activator of rat and mouse PXR. In contrast, pregnenolone-16α-carbonitrile is a good activator of rabbit, rat, and mouse PXR, but a poor activator of human PXR. The studies described herein indicate that there are also species differences with respect to activation of PXR by DHEA and its metabolites. DHEA, ADIONE, and ADIOL were all able to activate expression of PXRELUC through mouse PXR; however, only DHEA and ADIONE were able to activate expression of PXRELUC through human PXR. This result suggests that ADIOL is a ligand for rodent PXR, but not for human PXR. With respect to DHEA, the human PXR had a slightly higher affinity than the mouse PXR.
The concentrations of DHEA, ADIONE, and ADIOL required to activate the PXR reporter system were approximately 50 to 100 μM. This is a similar concentration range required for activation of a PXR reporter system by other naturally occurring steroids, such as the pregnanes. However, it is an order of magnitude higher than the concentrations of synthetic steroids such as dexamethasone t-butyl acetate needed to activate the system (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998). Based on our results with the human PXR, we would not expect physiological concentrations of ADIONE and DHEA in humans to be high enough to significantly activate PXR. Physiological concentrations of ADIONE in blood are in the low nanomolar range, and even in individuals who supplement with over-the-counter ADIONE, levels do not reach the micromolar range (King et al., 1999). Physiological concentrations of DHEA in human plasma are approximately 1 to 8 μM (measured as the 3β-sulfate conjugate), with younger adults having higher levels than older adults (Nafziger et al., 1991). In addition, DHEA is available over-the-counter as a dietary supplement, and people who use DHEA supplements have higher concentrations of circulating DHEA and DHEA-S. For example, plasma DHEA-S levels in older adults taking 50 mg/day of DHEA per os for 6 months went from approximately 2 to 10 μM, with some individuals reaching significantly higher levels (Baulieu et al., 2000). Therefore, under normal physiological conditions or under conditions of modest DHEA supplementation, DHEA is not likely to significantly activate PXR. However, it is possible that individuals who supplement with high amounts of DHEA may reach concentrations high enough to activate PXR. Activation of PXR leading to increased expression of CYP3Aresults in enhanced drug metabolism; therefore, activation of PXR may be the basis for many important drug-drug interactions (Moore et al., 2000).
In summary, the studies described herein demonstrate that DHEA is able to induce expression of CYP3A family members in vivo in a PPARα-independent manner. Moreover, DHEA and its metabolites ADIONE and ADIOL are activators of PXR in vitro. Although DHEA, ADIONE, and ADIOL are relatively weak activators of PXR, this activation is the likely mechanism for the in vivo induction of CYP3A forms in rats and mice fed high doses of DHEA.
Acknowledgments
We thank Mary Pendleton for expert technical assistance with cell culture and transient transfection assays. We also are grateful to Cam Falkner and Steve Kliewer for providing plasmids and reagents for these experiments.
Footnotes
-
↵1 Present address: Department of Pharmacology, University of Washington School of Medicine, 1959 NE Pacific St., Box 356560, Seattle, WA 98195.
-
This study was supported by U.S. Public Health Service Grants DK54774 (to R.A.P.) and CA 89607 (to J.M.P.), National Research Service Award Fellowship F32 ES05927 (to S.L.R.), and Jewish Hospital Foundation Grant 9511-02 (to J.L.F.).
-
↵3 The major peroxisome proliferator-responsiveCYP4A form in the rat liver is CYP4A1. The mouse also expresses a hepatic Cyp4a form that is inducible by peroxisome proliferators and cross-reacts in Northern blot analysis with a cDNA probe to rat CYP4A1. In this article, this mouse gene will be designated Cyp4a, the rat gene will be referred to as CYP4A1, and when referring to both rat and mouse genes, CYP4A will be used. Similarly, the major glucocorticoid-inducibleCYP3A forms in rats and mice have different designations, CYP3A23 and Cyp3a11, respectively. However, when referring to both the rat and mouse genes, the more generic CYP3A is used.
- Abbreviations used are::
- DHEA
- dehydroepiandrosterone (3β-hydroxy-androst-5-en-17-one)
- DHEA-S
- dehydroepiandrosterone 3β-sulfate
- PPARα
- peroxisome proliferator-activated receptor α
- PXR
- pregnane X receptor
- ADIOL
- androst-5-ene-3β,17β-diol
- ADIONE
- androst-4-ene-3,17-dione
- 11β-hydroxy-DHEA
- 3β,11β-dihydroxy-androst-5-en-17-one
- 16α-hydroxy-DHEA
- 3β,16α-dihydroxy-androst-5-en-17-one
- 7-oxo-DHEA
- 3β-hydroxy-androst-5-ene-7,17-dione
- 7α-hydroxy-DHEA
- 3β,7α-dihydroxy-androst-5-en-17-one
- DMSO
- dimethyl sulfoxide
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- mPXR
- murine pregnane X receptor
- hPXR
- human pregnane X receptor
- PXRE
- pregnane X receptor responsive element
- LUC
- luciferase
- Received June 26, 2001.
- Accepted February 5, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}