


Abstract
The purpose of this study is to investigate reliable prediction methods for in vivo pharmacokinetics and the likelihood of drug interactions with several cytochrome P450 inhibitors in humans for (S,S)-3-[3-(methylsulfonyl)phenyl]-1-propylpiperidine (PNU-96391). By allometric scaling of in vivo animal data, clearance of PNU-96391 in humans was over-predicted by 4-fold, half-life was under-predicted by 3-fold, and volume of distribution was accurately predicted. High correlation coefficients (>0.99) were observed for these parameters. Neither the in vitro-in vivo correlation approach nor the modified allometric scaling with maximum life span potential or brain weight accurately provided the predicted clearance value. Using an alternative method, based on normalization of in vitro human data with the ratio of in vivo to in vitro animal data, the in vivo clearance in humans was predicted to be 0.39 l/h/kg. This value correlated well with the in vivo value (0.43 l/h/kg). Regarding the interactions of PNU-96391 with cytochrome P450 inhibitors, only quinidine, haloperidol, and ketoconazole showed significant inhibition on the metabolic clearance of PNU-96391 in human hepatocytes. By comparing in vitro Ki values with in vivo maximum unbound concentrations of the inhibitor, the increases in systemic exposure of PNU-96391 by coadministration of the inhibitors were estimated to be less than 1.5-fold. A preliminary comparison of pharmacokinetics of PNU-96391 between CYP2D6 extensive and poor metabolizers in the clinical study showed only a slight increase in systemic exposure in poor metabolizers (approximately 1.4-fold as area under the concentration-time curve). Therefore, clinically significant drug-drug interactions of PNU-96391 would be unlikely to occur with coadministration of CYP2D6 inhibitors.
Drugs are most frequently administered orally, and the majority of these are intended to act systemically. A number of important factors limit the systemic availability of orally administered drugs. In particular, its removal as it passes through the liver is one of the most critical steps that can dramatically reduce the systemic availability. Thus, predicting pharmacokinetics in humans, especially metabolic clearance in the liver, is important at several stages of the drug development process for the selection of compounds with predicted suitable pharmacokinetics properties as well as for design of the first clinical trial. Initial attention was focused on the use of allometric scaling as a technique to predict pharmacokinetic parameters in human based on the use of empirical relationships observed between mammalian body weight and physiological parameters (Boxenbaum, 1980; Mordenti, 1986). Considerations of the relationship between drug elimination and physiological parameters such as hepatic or renal blood flow reasonably led to the application of allometric scaling in correlating pharmacokinetics in humans and animal species (Boxenbaum, 1980; Ings, 1990; Ritschel, 1992). Accurate predictions of pharmacokinetics in human by allometric scaling were demonstrated for renally excreted antibiotics (Sawada et al., 1984; Mordenti, 1986) and proteins (Mordenti et al., 1991) as well as for a number of drugs displaying high hepatic extraction (Boxenbaum and D'Souza, 1990). However, for compounds characterized by low and intermediate hepatic extraction, elimination strongly depends on biochemical parameters such as intrinsic clearance and protein binding, which often are highly species specific. Allometric scaling generally fails to predict in vivo clearance in humans in such cases (Ings, 1990; McNamara, 1991; Suzuki et al., 1995). More recently, with the increased availability of human liver samples for the generation of microsomes, hepatocytes, or liver slices, approaches that utilize in vitro metabolic data in animals and humans have demonstrated an increased accuracy in the prediction of metabolic clearance for compounds with medium or low hepatic extraction (Lave et al., 1999). A number of investigators have reported hepatocytes to be a superior system to microsomes and liver slices for accurate prediction of in vivo clearance in humans (Houston, 1994; Li et al., 1999; Naritomi et al., 2003).
(S,S)-3-[3-(Methylsulfonyl)phenyl]-1-propylpiperidine [PNU-963912 or (-)-OSU6162; Fig. 1] is a substituted (S)-3-phenylpiperidine derivative that exhibits some affinity to the dopamine D2 receptor family and is an orally active modulator of central dopaminergic function that has good activity in accepted models of parkinsonian dyskinesias and schizophrenia. PNU-96391 displays a unique normalizing profile on psychomotor activity by an intriguing mixture of stimulatory and inhibitory properties (Ekesbo et al., 1997; Tedroff et al., 1998). As a consequence, PNU-96391 (hydrochloride salt) is being developed for the treatment of l-DOPA-induced dyskinesias in Parkinson's disease patients. In the present study, prediction of pharmacokinetics of PNU-96391 in humans was investigated by the allometric scaling method for in vivo animal data and the normalization method of in vitro human data with the ratio of in vivo to in vitro animal data. Furthermore, the effects of a number of known cytochrome P450 inhibitors on clearance of PNU-96391 were studied in human hepatocytes to evaluate the likelihood of in vivo interactions of PNU-96391 with cytochrome P450 inhibitors.

Chemical structure of (S,S)-3-[3-(methylsulfonyl)phenyl]-1-propylpiperidine (PNU-96391)
Materials and Methods
Chemicals. PNU-96391 (hydrochloride salt: chemical purity >99%) and [13C,2H3]PNU-96391 (hydrochloride salt: >99%) were obtained from Pharmacia Corporation (Kalamazoo, MI). Dextromethorphan was provided by F. Hoffman-La Roche (Nutley, NJ), and ketoconazole was provided by Sigma/RBI (Natick, MA). (S)-Mephenytoin was obtained from Dr. W. F. Trager (University of Washington, Seattle, WA). 7-Ethoxyresorufin, haloperidol, quinidine, and trypan blue were obtained from Sigma-Aldrich (St Louis, MO). Other reagents were of reagent grade.
Hepatocytes. Freshly isolated hepatocytes from male Sprague-Dawley rats, beagle dogs, and cynomolgus monkeys (n = 4 per species) were obtained from CEDRA Co. (Austin, TX). Cryopreserved dog hepatocytes were also used after in-house preparation from four male beagle dogs (Marshall Farms USA, Inc., North Rose, NY) according to the procedure of Hengstler et al. (2000). Cell viability was routinely checked by the trypan blue (0.4%, w/v) exclusion test and preparations in excess of 95% viable were used. Cryopreserved human hepatocytes (n = 8; Caucasian; 43-, 43-, 57-, and 59-year-old males and 44-, 47-, 55-, and 61-year-old females) were obtained from In Vitro Technologies (Baltimore, MD), and cell viability, assessed by trypan blue, was approximately 70% in each case.
In Vitro Protein Binding. The binding of PNU-96391 to plasma protein was determined by incubating the compound with pooled plasma from Sprague-Dawley rats, beagle dogs, cynomolgus monkeys and humans (Caucasian) in a final drug concentration of 0.05 to 25 μM. PNU-96391, dissolved in saline (1% v/v), was added to plasma samples and incubated for 15 min. The samples were transferred to Centrifree ultrafiltration units (Amicon; Millipore, Bedford, MA), and, after centrifugation of the samples (2000g, 37°C), the ultrafiltrates were removed and analyzed by liquid chromatography-tandem mass spectrometry (LC/MS-MS). The free fraction of PNU-96391 was determined by dividing the amount of drug in the ultrafiltrate by the amount in the original plasma sample. Nonspecific binding of PNU-96391 to the ultrafiltration membrane was negligible (<1%) since the drug concentrations in saline solution were not significantly different between both sides of the ultrafiltration device. Therefore, the data were not corrected for nonspecific binding.
Blood-to-Plasma Ratio. The partitioning of PNU-96391 into blood cells was determined by incubating the compound with whole blood from Sprague-Dawley rats, beagle dogs, cynomolgus monkeys, and humans (Caucasian). PNU-96391, dissolved in saline (1%, v/v), was added to blood to give a final drug concentration of 0.1 to 5 μM, and the samples were incubated at 37°C for 15 min. Plasma was separated by centrifugation, and aliquots were taken for LC/MS-MS.
In Vivo Pharmacokinetic Study. For intravenous administration to the animals, PNU-96391 (hydrochloride salt) was dissolved in saline. The concentration of the formulation was approximately 2.5 mg/ml. Dose levels of PNU-96391 were expressed as free base equivalents. Pharmacokinetics in Sprague-Dawley rats and beagle dogs have been previously reported (Shobe et al., 2000). Male cynomolgus monkeys (3.1–3.6 kg; Pharmacia monkey colony, Kalamazoo, MI) were given 5.8 mg/kg PNU-96391 intravenously via the saphenous vein. Blood samples (approximately 0.5 ml) were collected from the jugular vein at 0 (predose), 2, 10, 20, and 30 min and 1, 2, 4, 6, 8, 12, and 24 h after administration. All blood samples were collected with K2EDTA as the anticoagulant and were then centrifuged. The resulting plasma samples were stored at approximately -20°C until analysis. The studies were conducted in compliance with the Animal Welfare Act Regulations (9 CFR Parts 1, 2, and 3) and the Guide for the Care and Use of Laboratory Animals (the Institute of Laboratory Animal Resources), as well as with internal company policies and guidelines.
Incubation Conditions. All incubations with hepatocytes were carried out at a cell density of approximately 1 × 106 cells/ml in Krebs-Henseleit buffer containing 10 mM HEPES (pH 7.4) at 37°C. A reaction was started by the addition of PNU-96391 (final concentration 0.5 μM) after a 2-min preincubation. For the inhibition experiments, the different inhibitors were dissolved in acetonitrile, and 10 μl (final concentration of acetonitrile 0.8%, v/v) were added to the incubation medium just before preincubation. Incubations were terminated at 20-min intervals over 120 min by the addition of 10% trichloroacetic acid. Hepatocyte protein was precipitated by centrifugation at 10,000 rpm for 5 min. The resulting supernatants were transferred into autosampler vials and stored at approximately 4°C to await analysis. The apparent rates of PNU-96391 disappearance were linear with respect to cell density (0.5–2 × 106 cells/ml) in animal and human hepatocytes.
Assay of PNU-96391. Concentrations of PNU-96391 in plasma and hepatocyte samples were quantitated using LC/MS-MS. [13C,2H3]PNU-96391 was used as an internal standard. The separation of PNU-96391 was achieved using a Waters Alliance 2790 Chromatography system (Waters, Milford, MA) with an Ace 5 Phenyl, 5 cm × 2.1 mm i.d. column (Advanced Chromatography Technologies, Aberdeen, Scotland, UK). Mass spectrometric analyses were performed on a Micromass Quattro Ultima (Waters) using electrospray ionization. A gradient mobile phase of 10 mM ammonium acetate buffer, pH 4, and acetonitrile was maintained at a constant flow rate of 0.25 ml/min for a total run time of 6 min. The gradient started with 90% buffer for 0.5 min and then changed to 80% acetonitrile over a 1-min period. After 0.5 min of flow, the gradient proceeded back to 90% buffer over a 0.1-min time period and was held for the remaining run time. The injection volume was 5 μl. The retention time of PNU-96391 was approximately 4 min. Sample analysis was performed in the positive ionization multiple reaction monitoring mode with unit resolution for the transitions 282.15 to 129.25 and 286.15 to 129.25 for PNU-96391 and [13C,2H3]PNU-96391, respectively. The calibration range was 0.009 to 9 μM. The back-calculated calibration standard concentrations of PNU-96391 were within ±10% of their theoretical concentrations with CVs of less than 15%. The precision and accuracy of the quality control samples were within ±15%.
Pharmacokinetic Analysis. For the in vivo studies, plasma concentration-time data for each species were analyzed by model-independent methods (Gibaldi and Perrier, 1982). The area under the plasma concentration-time curve from time 0 to the last time point with a quantifiable concentration (Ct), AUC0-t, was calculated using the linear trapezoidal rule. The area was extrapolated to infinity time (AUC0-∞) by the following equations:  where kel was the elimination rate constant determined by linear regression of the last three or four quantifiable data points in the apparent terminal phase of the log concentration-time curve. The apparent terminal half-life (t1/2,z) was calculated as:
where kel was the elimination rate constant determined by linear regression of the last three or four quantifiable data points in the apparent terminal phase of the log concentration-time curve. The apparent terminal half-life (t1/2,z) was calculated as:  Plasma clearance (CLplasma) was calculated by use of the relationship:
Plasma clearance (CLplasma) was calculated by use of the relationship:  The volume of distribution at steady-state (Vss) was calculated as:
The volume of distribution at steady-state (Vss) was calculated as:  where AUMC0-∞ was the area under the first moment of the plasma concentration-time curve from time 0 to infinity:
where AUMC0-∞ was the area under the first moment of the plasma concentration-time curve from time 0 to infinity: 
For the in vitro studies, in vitro intrinsic clearance (CLint) was calculated from the ratio of the initial amount of PNU-96391 in the hepatocytes and the corresponding AUC0-∞ based on the assumption of monoexponential decline over the time course of incubation. Values of CLint were scaled to in vivo units using scaling factors such as the number of hepatocytes present in the whole liver, and the liver weights and body weights (Bayliss et al., 1990; Davies and Morris, 1993; Iwatsubo et al., 1996). Hepatic clearance (CLhep) was thereafter calculated from intrinsic clearance, the unbound fraction in plasma (fu), the blood-to-plasma concentration ratio (Rb), and the hepatic blood flow (Qh) using the dispersion model analysis (Roberts and Rowland, 1986; Iwatsubo, 1996):  where a = (1 + 4 · RN · DN)1/2 and RN = (fu/Rb) × (CLint/Qh). In these equations, dispersion number (DN) was assumed to be 0.17 (Roberts and Rowland, 1986). Finally, the CLhep in humans was normalized by the ratio of in vivo blood clearance (CLblood) to in vitro CLhep in animal species, to predict systemic clearance in humans:
where a = (1 + 4 · RN · DN)1/2 and RN = (fu/Rb) × (CLint/Qh). In these equations, dispersion number (DN) was assumed to be 0.17 (Roberts and Rowland, 1986). Finally, the CLhep in humans was normalized by the ratio of in vivo blood clearance (CLblood) to in vitro CLhep in animal species, to predict systemic clearance in humans:  For many drugs, extrahepatic clearance in part contributes to total body clearance. Therefore, this normalization method could improve the predictive performance for CLblood in humans since the method takes account of the correction of in vitro to in vivo scaling, including extrahepatic clearance.
For many drugs, extrahepatic clearance in part contributes to total body clearance. Therefore, this normalization method could improve the predictive performance for CLblood in humans since the method takes account of the correction of in vitro to in vivo scaling, including extrahepatic clearance.
Allometric Scaling. For allometric scaling, pharmacokinetic parameters (y) for CLblood, Vss, and t1/2,z of PNU-96391 in animals were correlated with their corresponding mean body weights (W), using the allometric equation: y = aWx (Boxenbaum, 1980; Mordenti, 1986). The values of the allometric coefficients (a) and exponent (x) were estimated by linear least squares regression of the log-transformed allometric equation (log y = log a + x log W). The pharmacokinetic parameters observed in humans in vivo were compared with the values determined by conventional allometric scaling using the body weight of 70 kg for humans.
Prediction of Increase in AUC from in Vitro Data. As a prediction of the likelihood of in vivo interactions of PNU-96391 with P450 inhibitors, the AUC ratios in the presence and absence of P450 inhibitors were estimated from the following equation assuming that the protein binding is not altered by the inhibitor (Ito et al., 1998):  where fm is the fraction of PNU-96391 eliminated by hepatic metabolism, Iin,max,u is maximum unbound concentration of the inhibitor in the portal vein, and Ki is the inhibition constant of the inhibitor determined from in vitro inhibition studies. The value of fm for PNU-96391 was assumed to be 0.3 to 0.4. To avoid false-negative predictions, Iin,max,u was calculated as the sum of maximum unbound concentrations in circulating blood and those coming from gastrointestinal absorption after oral administration, assuming that the unbound concentration in liver equals that in blood (Ito et al., 1998):
where fm is the fraction of PNU-96391 eliminated by hepatic metabolism, Iin,max,u is maximum unbound concentration of the inhibitor in the portal vein, and Ki is the inhibition constant of the inhibitor determined from in vitro inhibition studies. The value of fm for PNU-96391 was assumed to be 0.3 to 0.4. To avoid false-negative predictions, Iin,max,u was calculated as the sum of maximum unbound concentrations in circulating blood and those coming from gastrointestinal absorption after oral administration, assuming that the unbound concentration in liver equals that in blood (Ito et al., 1998):  where Imax is the maximum concentrations of the inhibitor in circulating blood, ka is the absorption rate constant, Fa is the fraction absorbed, and Dose is the amount of inhibitor administered. Fa values were assumed to be 0.8, 0.6, and 1.0 for quinidine (Greenblatt et al., 1997), haloperidol (Schaffer et al., 1982), and ketoconazole, respectively. The value of ka can be calculated from tmax and kel:tmax = ln(ka/kel)/(ka - kel). In general, the ka value of an orally administered drug is maximal when gastrointestinal absorption of the drug is so rapid that the limiting step is the gastric emptying rate. Therefore, the theoretical maximal gastric emptying rate of 0.1 ml-1 was used for ka in the present study to avoid false-negative prediction (Oberle et al., 1990).
where Imax is the maximum concentrations of the inhibitor in circulating blood, ka is the absorption rate constant, Fa is the fraction absorbed, and Dose is the amount of inhibitor administered. Fa values were assumed to be 0.8, 0.6, and 1.0 for quinidine (Greenblatt et al., 1997), haloperidol (Schaffer et al., 1982), and ketoconazole, respectively. The value of ka can be calculated from tmax and kel:tmax = ln(ka/kel)/(ka - kel). In general, the ka value of an orally administered drug is maximal when gastrointestinal absorption of the drug is so rapid that the limiting step is the gastric emptying rate. Therefore, the theoretical maximal gastric emptying rate of 0.1 ml-1 was used for ka in the present study to avoid false-negative prediction (Oberle et al., 1990).
Results
Allometric Scaling of Clearance, Volume of Distribution, and Half-life across Animal Species and Humans. The pharmacokinetic parameters obtained for PNU-96391 from Sprague-Dawley rats, beagle dogs, and cynomolgus monkeys after single intravenous administration are shown in Table 1. Values of CLplasma were estimated to be 2.8, 2.2, and 2.0 l/h/kg in rats, dogs, and monkeys, respectively. The unbound fractions in plasma (fu) were 0.91, 0.72, and 0.81 in rats, dogs, and monkeys, respectively. The fu values were concentration-independent in these animals. Blood-to-plasma concentration ratio (Rb) ranged from 1.1 to 1.2 across animal species, and the values were concentration-independent. Using the values of fu and Rb for PNU-96391, CLblood values were estimated to be 2.6, 1.9, and 1.8 l/h/kg in rats, dogs, and monkeys, respectively. The values of CLblood represented 70 to 100% of hepatic blood flow in these species (3.3, 1.9, and 2.6 l/h/kg, respectively) (Davies and Morris, 1993). Accordingly, PNU-96391 is a high hepatic extraction ratio compound in animal species. Values of Vss in rats, dogs, and monkeys (1.5, 2.5, and 1.4 l/kg, respectively) exceeded total body water (0.6–0.7 l/kg) (Davies and Morris, 1993), indicating extensive distribution of drug into tissues. Half-life of PNU-96391 was short, ranging from 0.6 to 1.1 h in these species.
Pharmacokinetic parameters of PNU-96391 in Sprague-Dawley rats, beagle dogs, and cynomolgus monkeys after a single intravenous administration
Animals were administered intravenously a single bolus dose of PNU-96391 (mean ± S.D.; n = 4 in rats and dogs, n = 3 in monkeys). Plasma concentrations were determined by LC/MS-MS following protein precipitation, and pharmacokinetic parameters were calculated by the noncompartmental method. Data for rats and dogs are cited from Shobe et al. (2000).
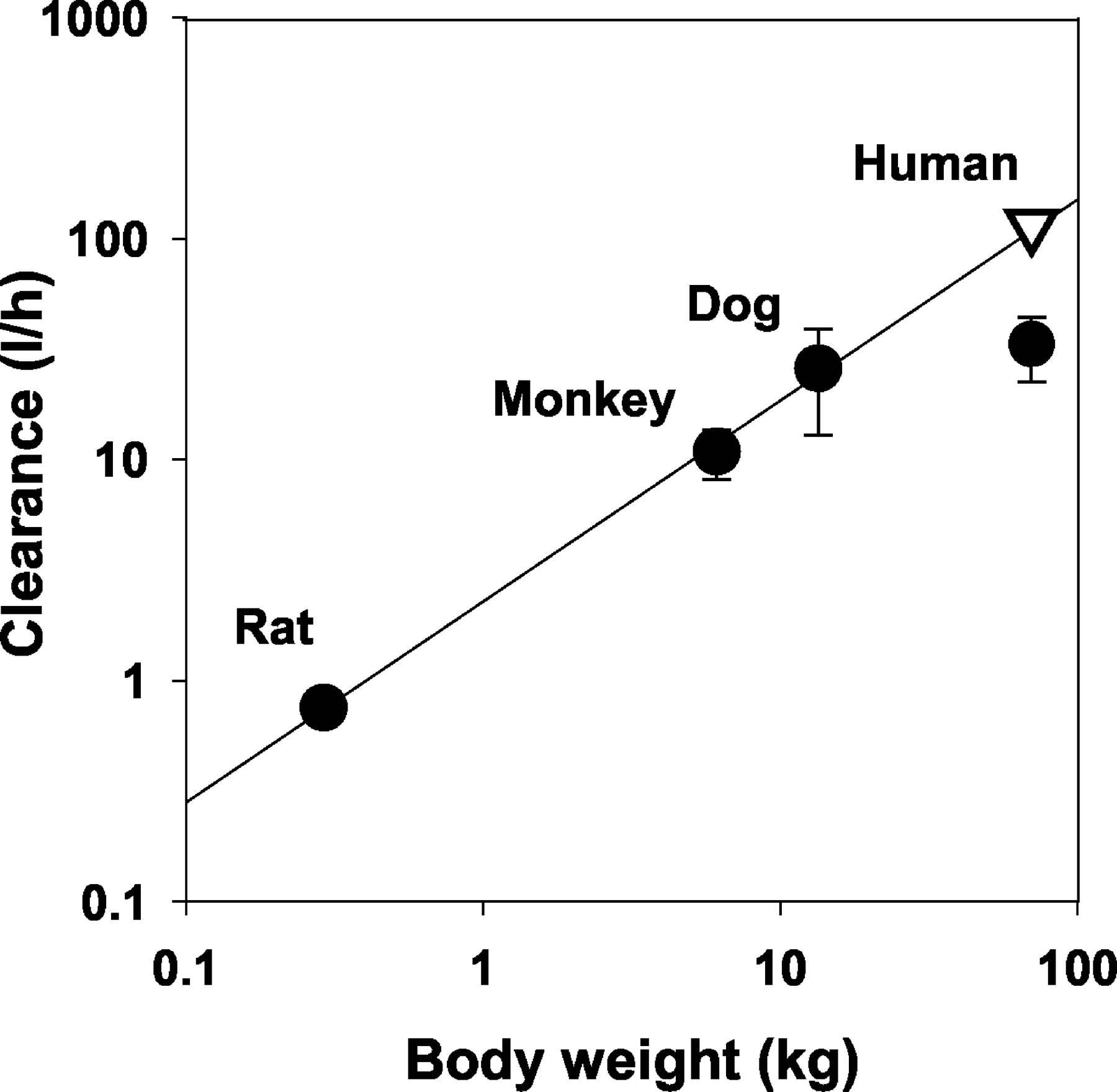
Allometric scaling results for CLblood, Vss, and t1/2,z across animal species are illustrated in Figs. 2, 3, and 4, respectively. Table 2 shows the results of least-squares fitting of log CLblood, log Vss, and log t1/2,z against log W. The pharmacokinetic parameters corresponding to a 70-kg man were then estimated by use of allometric scaling equations. The observed values in the clinical study of healthy volunteers were also presented in Figs. 2, 3, and 4 for comparison with the predictions of allometric scaling. Human data (n = 39) were obtained after a single oral administration of PNU-96391 (3–200 mg) to healthy volunteers (n = 35) (C. A. Rodriguez, N. E. Azie, G. Adams, K. Donaldson, S. F. Francom, B. A. Staton, and P. A. Bombardt, manuscript in preparation) and after a single intravenous and oral administration at 0.1 mg/kg to healthy volunteers (n = 4) (Pharmacia, unpublished data). The values of oral clearance and volume of distribution obtained from the former study were corrected with the oral bioavailability (F = 0.6) that was obtained from the latter study. The lower limit of quantitation for plasma concentrations of PNU-96391 in the clinical studies was 0.008 μM, which was similar to that of preclinical pharmacokinetic studies. Dose-proportionality of clearance was observed over the whole dose range. The values of fu and Rb in humans were 0.73 and 1.1, respectively. Despite high correlation coefficients for the allometric regression of CLblood (0.998) and t1/2,z (0.994) across animal species, these parameters were not predicted adequately for humans (Table 2; Figs. 2, 3, 4). Compared with the observed values in humans (0.43 l/h/kg), CLblood was over-predicted by approximately 4-fold, whereas t1/2,z was under-predicted by approximately 3-fold. Only Vss was accurately predicted (2.4 versus 2.2 l/kg, correlation coefficient = 0.987).

Allometric scaling of blood clearance of PNU-96391 across animal species and humans.
Predicted value (▿) in humans by allometric scaling was determined by regression analysis (—) using in vivo data across animal species (•).

Allometric scaling of volume of distribution at steady state of PNU-96391 across animal species and humans.
Predicted value (▿) in humans by allometric scaling was determined by regression analysis (—) using in vivo data across animal species (•).

Allometric scaling of terminal elimination half-life of PNU-96391 across animal species and humans.
Predicted value (▿) in humans by allometric scaling was determined by regression analysis (—) using in vivo data across animal species (•).
Allometric scaling of the pharmacokinetic parameters of PNU-96391 across animal species and comparison of estimated and observed values in humans
Prediction of Systemic Clearance of PNU-96391 Using in Vitro and in Vivo Data. In hepatocytes from all species, PNU-96391 concentrations demonstrated monoexponential decline over the time course of incubation as shown in Fig. 5. Apparent half-lives calculated for PNU-96391 ranged from 0.34 h to 1.2 h in animal species but was much longer (4.2 h) in human hepatocytes (Table 3). Values of CLint (l/h/106 cells) for PNU-96391 were in the order of rat > dog ≈ monkey > human (Table 3). Values of CLhep calculated by the dispersion model were in the order of rat > monkey > dog > human (Table 3). The value of CLhep (0.32 l/h/kg) in humans was approximately 0.7-fold lower than the observed CLblood (0.43 l/h/kg). Finally, the normalized CLblood in vivo in humans calculated from in vivo/in vitro data from animal species showed good agreement with the observed value in human in the order of rat > monkey > dog (Table 4). Rat data provided the most accurate prediction for humans with a deviation from the observed value of only 8% (0.39 l/h/kg compared with the in vivo observed value of 0.43 l/h/kg). On the other hand, the values predicted from dog and monkey data (0.65 and 0.49 l/h/kg, respectively) were also acceptable for prediction of CLblood in vivo because these values were over-estimated by only 50% and 20%, respectively.
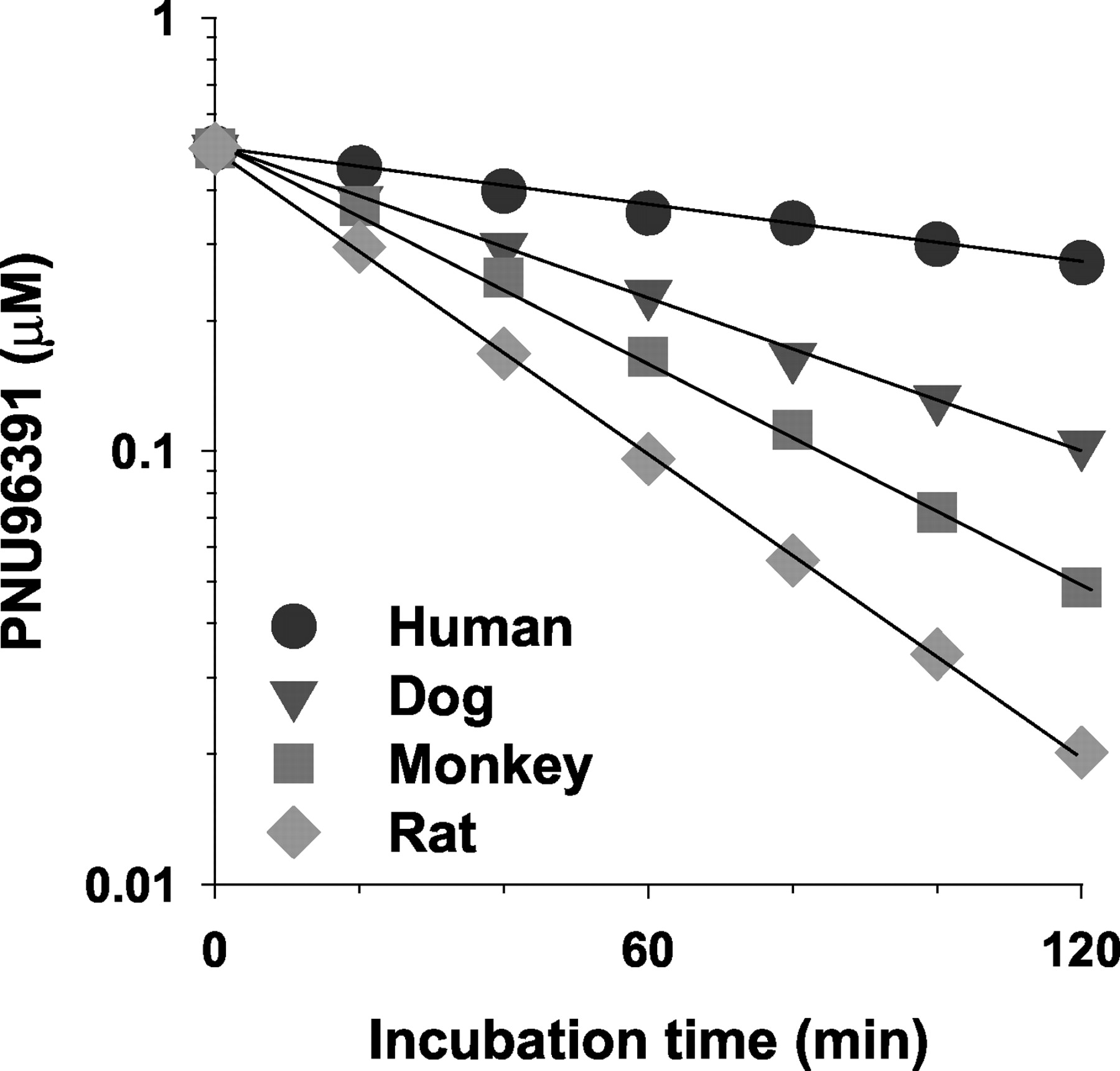
Time course for the disappearance of PNU-96391 in typical hepatocytes of rats (♦), dogs ( ▾), monkeys (▪), and humans (•).
Hepatocytes (approximately 1 × 106 viable cells/ml) in Krebs-Henseleit buffer containing 10 mM HEPES (pH 7.4) were preincubated for 2 min at 37°C, and reactions were started by the addition of PNU-96391 (final concentration 0.5 μM). Incubations were terminated at 20-min intervals over 120 min by the addition of 10% trichloroacetic acid. PNU-96391 concentrations were determined by LC/MS-MS after protein precipitation. Data are the mean of triplicate determinations.
Pharmacokinetic parameters of PNU-96391 in rat, dog, monkey, and human hepatocytes PNU-96391 at 0.5 μM was incubated in hepatocytes (ca. 1 × 106 viable cells/ml) in Krebs-Henseleit buffer containing 10 mM HEPES at 37°C up to 120 min. PNU-96391 concentrations were determined by LC/MS-MS after protein precipitation, and pharmacokinetic parameters were calculated using scaling factors to in vivo basis followed by dispersion model analysis (mean ± S.D.; n = 4 in rats and monkeys, n = 8 in dogs and humans).
Predicted in vivo clearance of PNU-96391 in humans by in vivo and in vitro data of rats, dogs, and monkeys
Interaction of PNU-96391 with Cytochrome P450 Inhibitors in Hepatocytes. Interactions of PNU-96391 with various cytochrome P450 substrates/inhibitors (Guengerich, 1995; Parkinson, 1996) were investigated in human hepatocytes. The effects of different inhibitor concentrations on the disappearance of PNU-96391 in pooled human hepatocytes are shown in Fig. 6. Vehicle (acetonitrile, final concentration of 0.8%, v/v) used for the inhibitor solution inhibited the disappearance of PNU-96391 by approximately 10%. Ethoxyresorufin (50 μM), a substrate of CYP1A isozymes, and (S)-mephenytoin (100 μM), a substrate of CYP2C19, did not significantly inhibit the disappearance of PNU-96391. Dextromethorphan (0.5–50 μM), a substrate of CYP2D6, and midazolam (0.5–50 μM), a substrate of CYP3A isozymes, also did not show significant effects. Quinidine, which is regarded as a specific inhibitor of CYP2D6, inhibited metabolism of PNU-96391 by 50% at 0.5 μM and 80% at 5 μM. Haloperidol, an inhibitor of CYP2D6, also inhibited the disappearance at 0.5 to 50 μM in a concentration-dependent manner. Strong inhibition of the disappearance of PNU-96391 was observed with ketoconazole, an inhibitor of CYP3A isozymes, which demonstrated 50% inhibition at 0.5 μM, increasing to 90% at 5 μM. Inhibition constants (Ki) were determined for quinidine, haloperidol, and ketoconazole under the same incubation condition using PNU-96391 concentrations of 0.25, 0.5, and 1 μM; Ki values were estimated to be 1.4, 36, and 0.78 μM for quinidine, haloperidol, and ketoconazole, respectively.

Effects of various cytochrome P450 inhibitors on the disappearance of PNU-96391 in human hepatocytes.
The P450 inhibitors were dissolved in acetonitrile, and 0.01 ml (final 0.8%, v/v) was added to the incubation mixture just before the preincubation: blank (no vehicle), quinidine (0.05–5 μM), haloperidol (0.5–50 μM), dextromethorphan (0.5–50 μM), ketoconazole (0.05–5 μM), and midazolam (0.5–50 μM). The incubation conditions and the measurement of PNU-96391 were the same as those described in Fig. 2. Data are the mean of duplicate determinations and expressed as percentage of vehicle control (acetonitrile, final 0.8%, v/v).
Discussion
In the present study, allometric scaling provided an accurate prediction of Vss in human, whereas CLblood and t1/2,z were not predicted adequately (Table 2). The allometric exponents for equations relating CLblood (0.910) and Vss (1.10) to body weight in animal species were slightly higher than the values (0.75 and 1.0) expected for physiological process and small organic molecules (Mordenti, 1986). In addition, high correlation coefficients (>0.987) were observed for these pharmacokinetic parameters, indicating that reasonable exponents and high correlation coefficients are not necessarily associated with successful extrapolation to humans. In the case of PNU-96391, where hepatic metabolism is the major determinant of clearance across species, it was anticipated that allometric scaling would be a useful prediction method across species since the method relies on the fact that physiological processes (in this case liver blood flow) show an empirical relationship with body weight across species (Boxenbaum, 1980; Mordenti, 1986). The predicted CLblood value for PNU-96391 in humans was, however, approximately 4-fold higher than the observed value. The values of CLblood for animal species (1.8–2.6 l/h/kg) were near hepatic blood flow (1.9–3.3 l/h/kg), whereas that for humans (0.43 l/h/kg) was approximately 40% of hepatic blood flow (1.2 l/h/kg) (Davies and Morris, 1993).
To improve the predictive performance of allometric scaling, Mahmood and Balian (1996) evaluated three different allometric scaling methods: 1) clearance versus body weight (simple allometry, CL = aWb), 2) product of clearance and maximum life span potential (MLP) versus body weights (CL × MLP = aWb), and 3) product of clearance and brain weights (BW) versus body weights (CL× BW = aWb). They proposed the selection of one of the methods based upon the exponents of simple allometric scaling: 1) if the exponent of the simple allometry lies between 0.55 and 0.70, simple allometry will predict CLblood more accurately than CL × MLP or CL × BW; 2) if the exponent of the simple allometry lies between 0.71 and 1.0, the CL × MLP approach will predict CLblood better than simple allometry or CL × BW; and 3) if the exponent of the simple allometry is ≥1.0, the CL × BW approach is suitable to predict CLblood in humans compared with the other two methods. Using the CL × MLP method based on the exponent (0.910) of simple allometry, predicted CLblood value for PNU-96391 was over-predicted by approximately 7-fold, despite a high regression coefficient (0.992) across animal species. Alternatively, the CL × BW method provided an under-prediction (0.4-fold) with a regression coefficient of 0.864. Recently, Lave et al. (1999) proposed a normalized allometric scaling approach for compounds of low or intermediate hepatic extraction ratios. When in vitro clearances were used as correction factors, the in vivo clearance in each animal species was normalized using the ratio: CLblood,animal × (CLhep,human/CLhep,animal). The normalized values were then extrapolated to humans using allometric scaling. Using this approach, the CLblood value for PNU-96391 was over-predicted by approximately 3-fold, with a regression coefficient of 0.782. As another prediction method, an in vitro-in vivo correlation approach was provided with felodipine (Bäärnhielm et al., 1986) and bosentan (Ubeaud et al., 1995), using in vitro clearance data in animals and humans. This approach is based upon a regression analysis between in vitro and in vivo clearances in several animal species, and then the corresponding in vitro clearance in humans is used to predict the in vivo clearance. In the case of PNU-96391, the predicted CLblood value was approximately 2-fold higher than the observed value with high regression coefficient of 0.999 across rats, dogs, and monkeys. Thus, the in vitro-in vivo correlation approach could predict CLblood value more accurately than the simple allometry or the modified allometric methods. Finally, the results of the present study show that CLblood for PNU-96391 is most accurately predicted by the correction of in vitro human data with the ratio of in vivo to in vitro animal data. This could be due to the improvement of the predictive performance by the normalization of in vitro human data with the ratio of in vivo to in vitro animal data, namely in vitro to in vivo scaling including extrahepatic clearance. Urinary excretion of PNU-96391 ranged from 10 to 20% of dose in rats and dogs, indicating that renal clearance in part contributes to total body clearance. The urinary excretion data therefore support the hypothesis that the prediction of in vivo clearance from in vitro data should take extrahepatic clearance into consideration. In this normalization approach, choice of the animal species for correction by in vivo and in vitro animal data are key, as demonstrated by the results of the present study, which show that the rat was a suitable species for PNU-96391 in contrast to dog and monkey (Table 4). However, the values predicted from dog and monkey data were also acceptable because the predicted values were less than 2-fold. Although further investigation will be necessary, it can be considered that an approach using only in vivo and in vitro data in one animal species, such as the rat, and in vitro data in humans could be as predictive as approaches using in vivo and in vitro data in at least three animal species and in vitro human data.
The metabolism of PNU-96391 has been reported in vivo and in vitro with particular emphasis placed on characterizing the importance of the N-depropylation pathway (Sood et al., 1999; Wienkers and Wynalda, 2002). The N-depropylation of PNU-96391 is principally mediated by CYP2D6 (Wienkers and Wynalda, 2002). The involvement of CYP2D6 is well supported by 1) a good correlation between the formation rate of the N-despropyl-metabolite and dextromethorphan-O-demethylation activity in a panel of human liver microsomes, 2) extensive inhibition of PNU-96391 metabolism by quinidine, a selective inhibitor of CYP2D6, and 3) a high turnover of PNU-96391 during incubation with cloned CYP2D6 (Km = 4 μM). Additionally, CYP1A1/2, CYP2C19, and CYP3A4 are reported to contribute a small fraction of metabolite formation (Wienkers and Wynalda, 2002).
Li et al. (1999) have suggested that human hepatocytes, with the complete enzyme pathways and cofactors, should represent a more accurate system for the evaluation of drug-drug interactions than microsomes. The influence of selective substrates/inhibitors of CYP1A1/2, CYP2C19, CYP2D6, and CYP3A4 on the metabolism of PNU-96391 was therefore studied during incubations performed with human hepatocytes in the present study. CYP2D6 inhibitors, quinidine and haloperidol, inhibited the apparent disappearance rates of PNU-96391 in human hepatocytes up to 80% and 60%, respectively. The CYP3A4 inhibitor ketoconazole also strongly inhibited the metabolism of PNU-96391. Previous data indicated that ketoconazole (5 μM) slightly inhibited the N-despropyl-metabolite formation (approximately 25%) in human liver microsomes (Wienkers and Wynalda, 2002). Li et al. (1999) demonstrated that the apparent Ki value for the inhibition of terfenadine metabolism by ketoconazole was significantly lower for human hepatocytes than that for human liver microsomes. This could be due to the bioaccumulation of ketoconazole in the hepatocytes, thereby leading to a lower apparent Ki value, and/or the nonspecific binding of the drugs to microsomes, leading to a higher apparent Ki value. This inhibition mechanism by ketoconazole is being investigated further.
The AUC ratios in the presence and absence of P450 inhibitors, quinidine, haloperidol, and ketoconazole, were estimated as a prediction of the likelihood of in vivo interactions of PNU-96391 with P450 inhibitors. At first, values of Iin,max,u for quinidine, haloperidol, and ketoconazole were estimated to be approximately 10, 0.20, and 0.35 μM, respectively, based on the literature data for Imax, Fa, Dose, and fu for quinidine (Greenblatt et al., 1997), haloperidol (Schaffer et al., 1982), and ketoconazole (Stockly et al., 1986). Consequently, the AUC ratios were estimated to be approximately 1.3, 1.0 and 1.1 for quinidine, haloperidol, and ketoconazole, respectively. Because quinidine and haloperidol are often administered intravenously, the AUC ratios were also estimated to be approximately 1.3 and 1.0, respectively, using Imax,u values for intravenous doses. Therefore, systemic exposure for PNU-96391 in humans in vivo would not significantly increase with coadministration of these P450 inhibitors. A preliminary comparison of pharmacokinetic parameters of PNU-96391 between extensive and poor metabolizers for CYP2D6 indicated an approximate 1.4-fold increase in AUC in the poor metabolizers, with a corresponding similar reduction in the clearance (0.7-fold) and increase in the half-life (1.3-fold) (C. A. Rodriguez, N. E. Azie, G. Adams, K. Donaldson, S. F. Francom, B. A. Staton, and P. A. Bombardt, manuscript in preparation). Clinical safety profiles of PNU-96391 were confirmed at a wide range of plasma concentrations in healthy volunteers, although large intersubject variances of plasma clearance were observed. These findings make it less likely that clinically significant drug-drug interactions would occur with coadministration of P450 inhibitors.
Acknowledgments
We greatly acknowledge the expert technical assistance of members of the Bioanalytical Resource Laboratory, including Tracy L. Chinigo, Joe Palandra, and Brian A. Staton, in the PNU-96391 analyses. We thank the members of the Preclinical Resource Laboratory Group for expert technical assistance; Brian W. Jones, Jeri L. Nederhoed, Lori R. Norris, Renae J. Ouding, Kenneth E. Rousch, and Terry L. VandeGiessen for assistance with the PNU-96391 animal experiments. We thank Mark P. Grillo and Fengmei Hua for preparing cryopreserved dog hepatocytes, and Carlos A. Rodriguez and Nkechi E Azie for providing the clinical pharmacokinetic data.
Footnotes
-
↵2 Abbreviations used are: PNU-96391, (S,S)-3-[3-(methylsulfonyl)phenyl]-1-propylpiperidine [(-)-OSU6162]; AUC, area under the concentration-time curve; AUMC, area under the first moment curve; BW, brain weight(s); CLblood, blood clearance estimated from in vivo study; CLint, intrinsic clearance estimated from in vitro hepatocyte study (intrinsic hepatic clearance); CLhep, hepatic clearance estimated from in vitro hepatocyte study; CLplasma, plasma clearance estimated from in vivo study; Fa, fraction absorbed; fm, fraction eliminated by hepatic metabolism; fu, unbound fraction in plasma; Iin,max,u, maximum unbound concentration of the inhibitor in the portal vein; Imax, maximum concentration of the inhibitor in circulating blood; ka, absorption rate constant; kel, elimination rate constant; Ki, inhibition constant; LC/MS-MS, liquid chromatography/tandem mass spectrometry; MLP, maximum life span potential; Qh, hepatic blood flow; Rb, blood to plasma concentration ratio; t1/2,z, terminal elimination half-life; Vss, volume of distribution at steady state; W, body weight; P450, cytochrome P450; PK, pharmacokinetic.
-
↵1 Current address: Pharmacokinetics, Dynamics and Metabolism, La Jolla Laboratories, Pfizer Inc., 10724 Science Center Drive, San Diego, CA 92121.
- Received July 24, 2003.
- Accepted December 19, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}