


Abstract
Cytochrome P450 3A4 (CYP3A4) is the major cytochrome P450 present in adult human liver and is involved in the metabolism of over 50% of therapeutic compounds currently in use. Since expression levels of CYP3A4 are regulated by many of these compounds, this raises the potential for drug-drug interactions and subsequent altered efficacy or toxicity of the individual compounds at the dose prescribed. Hence, understanding the molecular mechanisms of CYP3A4 regulation is of key importance in predicting and understanding such interactions. To examine this we have used DNase I footprinting and bioinformatic analysis to identify putative transcription factor binding sites within the 250 base pairs of promoter proximal to the transcription start site. We identified several protected fragments within this region that corresponded to putative binding sites for Sp1, AP2, CCAAT/enhancer binding protein (C/EBPα), and hepatic nuclear factor-3 (HNF3), as well as confirming previously identified C/EBPα, pregnane X receptor (PXR), and HNF3 binding sites. Sequential site-directed mutagenesis of C/EBPα, Sp1, HNF3, and PXR binding sites was next used to examine the role of these sites in basal CYP3A4 expression. Disruption of the C/EBPα, HNF3, and PXR binding sites all affected basal expression. Finally, the role of these sites was examined in activation of CYP3A4 expression by rifampicin, metyrapone, clotrimazole, and phenobarbital. Disruption of any of these sites either led to an altered pattern of activation by the xenobiotic, as altered maximal activation, or altered the EC50 value of activation. Such effects were xenobiotic-specific, with each disrupted site playing a role in the activation of some of the xenobiotics.
The cytochrome P450 superfamily is a group of mixed-function oxidases present in both eukaryotes and prokaryotes, which carry out a large proportion of the initial metabolism of both endobiotics and xenobiotics (Nelson et al., 1996). In humans, the major site of metabolism is the liver, and as expected there are several distinct cytochrome P450 enzymes present within the liver possessing wide, overlapping, substrate specificities. The cytochrome P450 3A (CYP3A) family are the most abundant P450s1 present in human liver, comprising approximately 30% of the total P450 content (Watkins, 1994). In addition, approximately 60% of pharmaceutical drugs currently in use that are oxidized during metabolism are substrates for CYP3A enzymes (Cholerton et al., 1992), meaning that this family is of great clinical importance in xenobiotic metabolism in humans. Furthermore, of the four CYP3A enzymes present in man (CYP3As 4, 5, 7, and 43), CYP3A4 is the most prevalent in adults, being found in all but one adult liver sample so far screened (Aoyama et al., 1989). Hence, CYP3A4 probably contributes the major CYP3A-mediated metabolism in the population as a whole.
The molecular mechanisms underlying regulation of CYP3A4 gene expression have been studied by several groups, including ourselves. Initially, approximately 1 kilobase pair of genomic DNA proximal to the translation start site was isolated (Hashimoto et al., 1993), the transcription start site identified and in silico analysis of putative transcription factor binding sites was performed. Several putative binding sites for transcription factors were identified using this approach, including the estrogen receptor, glucocorticoid receptor, chicken ovalbumin upstream promoter transcription factor (COUPTF), HNF-4, HNF-5, p53, and Oct-1 (Hashimoto et al., 1993). However, no functional data were associated with these assignments, and further work has questioned the validity of these assignments. For example, binding of the glucocorticoid receptor to the putative progesterone response element/glucocorticoid response element binding site provided a possible explanation for the activation of CYP3A4 by glucocorticoids; however, no direct binding of GRα to the CYP3A4 promoter, let alone the existence of this site, has yet been demonstrated. Later experiments identified a novel steroid hormone receptor, the human pregnane X receptor (hPXR), which binds to both everted (ER6) and direct repeats of the consensus sequence AGGTCA, collectively referred to as a PXRE motif (Lehmann et al., 1998). In human CYP3A4, PXR binding site motifs are present at approximately -150 bp (as an ER6) (Lehmann et al., 1998) and also within an enhancer module some 8 kbp upstream of the transcription start site (as a direct three-strand repeat) (Goodwin et al., 1999). Indeed, recent evidence shows that glucocorticoid-mediated activation of CYP3A4 gene expression is more likely to occur via PXR activation and its interaction with the PXRE (Lehmann et al., 1998), rather than GR interaction with the putative progesterone response element/glucocorticoid response element site. To support such a hypothesis, Schuetz et al. (2000) demonstrated that CYP3A gene expression can be increased by glucocorticoids in GR-deficient mice. Further investigations into the protein-DNA interactions within the CYP3A4 promoter are now appearing, with elegant studies such as those carried out by Rodriguez-Antona et al. (2003) examining C/EBPα interactions at the CYP3A4 proximal promoter, and Tirona et al. (2003), who demonstrated the interaction of HNF4α within the distal enhancer.
Despite these recent advances, the molecular mechanisms underlying xenobiotic and endobiotic induction of CYP3A4 remain unclear. As a result, the current study reports a characterization of the CYP3A4 proximal promoter, identifying sites of protein-DNA interaction and examining their roles in both basal- and xenobiotic-mediated activation of CYP3A4 gene expression.
Materials and Methods
Chemicals. Phenobarbital, rifampicin, metyrapone, clotrimazole, and dimethyl sulfoxide were of cell culture grade and purchased from Sigma Chemical (Poole, Dorset, UK). Fugene-6 transfection reagent was purchased from Roche Diagnostics (Basel, Switzerland). pSG5-hPXRΔATG expression plasmid was a kind gift from Dr. S. Kliewer (University of Texas, Dallas, TX); pEVR2/Sp1 expression plasmid was a kind gift of Prof. G. Suske (University of Marbury, Marburg, Germany); pCDNA3.1/CEBPa expression plasmid was a kind gift of Dr. D. Heery (University of Leicester, Leicester, UK). Unless otherwise stated, all other chemicals were of molecular biology grade and obtained from Sigma Chemical.
Cell Culture. All cell culture medium and supplements were purchased from Invitrogen (Carlsbad, CA). HepG2 cells, a human hepatocyte carcinoma cell line, were obtained from the European Collection of Animal Cell Cultures (ECACC 85011430, Porton Down, UK). The HuH7 human hepatocellular carcinoma cell line (Nakabayashi et al., 1982) was a kind gift from Dr. Steve Hood (GlaxoSmithKline, Uxbridge, Middlesex, UK). All cells were routinely cultured in 75-cm2 vented tissue culture flasks (NUNC A/S, Roskilde, Denmark) using minimal essential medium with Earle's salts supplemented with 1% nonessential amino acids, 2 mM l-glutamine, 100 μg/ml gentamycin, and 10% Australasian fetal bovine serum. To maintain phenotypic consistency, HepG2 and HuH7 cells were only used up to passage 13 after receipt from ECACC.
Preparation of Nuclear Extracts. Nuclear protein extracts were isolated according to the protocol of Dignam (Dignam et al., 1983). Briefly, HepG2 cells were grown to approximately 90% confluence and then collected by trypsinization. Cells were pelleted by centrifugation (1300g for 5 min) and washed twice with phosphate-buffered saline. After the second wash, cells were resuspended in 5× packed cell volume of ice-cold phosphate-buffered saline. Cells were pelleted, resuspended in 2× packed cell volume of buffer A (10 mM Hepes-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCI, 0.5 mM DTT), and allowed to swell on ice for 10 min before disruption using a Dounce homogenizer. Nuclei were pelleted (2000g for 15 min) and resuspended in 0.5× packed nuclear volume (homogenate volume-supernatant volume) of buffer C (25% glycerol, 20 mM Hepes-KOH, pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, 20 mM NaCl, 0.5 mM DTT, 0.5 mM phenylmethylsulfonyl fluoride). Next, 0.5× packed nuclear volume of high salt buffer (buffer C containing 1.2 M NaCl) was added dropwise with swirling, and the suspension was homogenized with a Dounce homogenizer. The resulting homogenate was centrifuged at 16,000g for 30 min, and supernatant (nuclear protein) aliquots were stored at -80°C. Protein concentration was determined by a modification of the method of Stoscheck (1990), and samples analyzed by SDS-polyacrylamide gel electrophoresis to verify isolation of high-molecular-weight proteins, indicative of a good quality isolation. Each aliquot was taken through only three freeze/thaw cycles to maintain protein integrity.
DNase I Footprint Analysis. The radiolabeled probe was prepared by polymerase chain reaction amplification of the target region (-388 to -83 bp, -301 to +7 bp, and -240 to +69 bp) of the CYP3A4 promoter using appropriate primer sets. Dephosphorylated amplicons were 5′ labeled with [γ-32P]ATP (110 TBq/mmol; Amersham Biosciences UK, Ltd., Little Chalfont, Buckinghamshire, UK) for 60 min at 37°C using T4 PNK (Promega, Charbonnières, France), and restriction digestion was then carried out to remove one labeled end, creating either upper or lower strand-labeled probes. These probes were further purified by phenol-chloroform extraction and ethanol precipitation. DNase I footprinting was carried out using the Promega Core Footprinting System (Promega) according to the manufacturer's instructions. Samples were Cerenkov-counted to ensure equal loading, heat-denatured, and separated on a 6% denaturing polyacrylamide gel and opposed to X-Omat LS film (Eastman Kodak, Rochester, NY) for 48 h.
Electrophoretic Mobility Shift Assays. Double-stranded DNA probes representing either the consensus binding sites for PXR, C/EBPα, HNF3α, or Sp1, or the mutated binding sites generated in this study were generated by annealing complementary sense and anti-sense synthetic oligonucleotides (MWG Biotech, Milton Keynes, UK). The fragments used (sense strand only shown) are shown in Table 1.
Probe used in electromobility shift assay experiments Double-stranded probes were generated for EMSA experiments corresponding to the consensus-binding region for each factor, and the mutated sequence were generated via SDM. Sense strand only is shown for clarity with the putative binding sites underlined, and mutated bases are indicated in bold.
The protein binding reactions with 32P-end-labeled wild-type DNA probes and competing unlabeled wild-type or mutant fragments were performed as described previously (El-Sankary et al., 2002). After incubation, the reaction mixtures were electrophoresed in 4% nondenaturing polyacrylamide gels. The gels were exposed overnight to Kodak X-Omat film, and band intensity was quantified by computer-based densitometry. Control experiments, in which nuclear protein extracts were pretreated with proteinase K or heat-denatured before addition of probes, were also performed to ensure that the observed retarded bands were due to DNA-protein interactions.
Plasmid Construction. The secretory alkaline phosphatase reporter gene pSEAP basic (BD Biosciences, Franklin Lakes, NJ) was engineered to contain the CYP3A4 enhancer element (XREM; -7972 to -7673 bp), since this is required for maximal response to activators (Goodwin et al., 1999); this construct is hereafter termed pControl. Three hundred eight base pairs of the CYP3A4 5′ flanking region (-301bp→+7bp) were engineered into pControl by polymerase chain reaction cloning (hereafter termed p3A4). Both plasmid constructs were kindly provided by Dr. Hossein Hamzeiy (University of Surrey, Surrey, UK). Plasmids were grown in the Escherichia coli strain TOP10F′ (Invitrogen) and purified using Endo-free maxi preps (QIAGEN, Dorking, Surrey, UK) according to manufacturer's instructions. Removal of endotoxin contamination of DNA before transfection has previously been shown to be critical for optimal transfection of cell lines (Butash et al., 2000).
Site-Directed Mutagenesis (SDM). Mutated versions of the reporter plasmid construct were generated via a modification of the BD Biosciences Transformer system. Briefly, 2.5 μg of template DNA was incubated with 400 mM NaOH for 10 min at room temperature. The reaction was then neutralized by the addition of 3 M NaOAc (pH 4.8), and concentrated by ethanol precipitation. Annealing reactions consisted of buffer (20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, and 50 mM NaCl), 75 ng of selection primer, 1 μg of mutagenic primer, and 100 ng of denatured DNA; details of primers are presented in Table 2. This reaction was heated at 75°C for 5 min and cooled down to room temperature at the rate of 1°C/s using a thermal cycler. Strand synthesis was then carried out using ligase buffer (30 mM Tris-HCl, pH 7.8, 10 mM MgCl2, 10 mM DTT, and 1 mM ATP), 10 U T4 DNA polymerase, 3 U T4 DNA ligase, and 0.5 mM deoxynucleoside-5′-triphosphate in a final volume of 30 μl for 2 h at 37°C. Enzymes were denatured at 70°C for 5 min, digested using the appropriate selection restriction enzyme, and then 20 ng was transformed into mutS E. coli cells for selection. All SDM daughter constructs were sequenced for conformation of successful mutagenesis.
Site-directed mutagenesis primers and targets Primers used for mutagenesis of ER6, Sp1, HNF3, C/EBPα, and pSEAP-basic selection primer are indicated. Putative binding sites (restriction site for selection primer) to be disrupted are underlined. Mutated bases are indicated in bold, with the change described in the right column.
Transient Transfection. HuH7 cells were seeded into 96-well plates (NUNC) at a concentration of 24,000 cells/well and incubated at 37°C for 48 h in a humidified container for attachment. FuGENE 6-mediated DNA cotransfections, using either pControl or p3A4 derivatives containing wild-type or mutant CYP3A4 promoters and pSG5-hPXRΔATG plasmid DNA, were performed as described by Goodwin et al. (1999). Transfections were allowed to proceed for 24 h in medium from which the serum had been omitted. Cells were then cultured for an additional 60 h in fresh serum-containing medium in the presence or absence of classical CYP3A4 inducers (Goodwin et al., 1999).
Reporter Gene Assays and Data Analysis. Aliquots of cell culture medium (25 μl/well) were transferred into 96-well Optiplates (Canberra Industries, Meriden, CT). Endogenous alkaline phosphatase activity was deactivated by heat treatment of the medium at 65°C for 30 min. SEAP activity was then assayed using the AURORA system (ICN, Thame, UK), according to the manufacturer's protocol. Chemiluminescent output was measured using a LumiCount automated plate reader (Canberra Industries).
The relative change in SEAP activity before, and 60 h after, xenobiotic addition was calculated for p3A4 and pControl in the presence and absence of xenobiotic. These measurements allow for the control of variation in cell seeding, transfection efficiency, cytotoxicity, or cell proliferative effects of xenobiotics that might otherwise produce anomalous results.
A specific chemical effect was calculated using the measurements above and the statistical significance of this value over solvent control tested as described previously (Plant et al., 2000). Imax (representing the overall maximal induction produced, encompassing ligand-receptor, receptor-receptor, receptor translocation, and receptor-DNA interactions caused by the inducer) and EC50 (xenobiotic concentration required to achieve half-maximum induction) values were calculated using nonlinear regression analysis (GraphPad Prism v2.0; GraphPad Software Inc., San Diego, CA), and an indication of the overall effect of the xenobiotic, the overall inductive ability, was calculated by dividing Imax by EC50.
Results
Protein-DNA Interactions within theCYP3A4Minimal Promoter. DNase I footprinting was carried out over the first 250 bp of the CYP3A4 proximal promoter. This region has previously been shown to be the minimum required for xenobiotic-mediated transcriptional activation (Goodwin et al., 1999). Figure 1 shows occupancy of the Sp1 (-104 bp to -97 bp), C/EBPα (-132 bp to -121 bp), ER6 motif PXR binding site (PXRE, -169 bp to -152 bp), and HNF3: C/EBPα (-195 bp to -186 bp) targeted in subsequent site-directed mutagenesis experiments (Fig. 1, A—D, respectively). Nucleotide sequences representing the boundaries of the protected regions are indicated. To demonstrate that protected fragments were caused by specific protein-DNA interactions, all putative protein-DNA interaction sites were examined under increasing amounts of nuclear protein (e.g., PXRE shown in Fig. 1E). In addition, all putative protein-DNA interaction sites could be removed by pretreatment of the nuclear protein extract with proteinase K or heat denaturation (data not shown). To gain a complete picture of DNA-protein interactions within this region, DNase I assays were carried out on three overlapping fragments (-388 to -83 bp, -301 to +7 bp, and -240 to +69 bp), with interaction sites only being assigned if they were observed in at least two of the three fragments. Such an approach reduces potential errors caused by probe artifacts, where protein-DNA interactions toward the termini of probes may be disrupted. Figure 2 shows a composite picture of the DNA-protein interactions identified, and putative protein assignments gained through interrogation of the TransFac database with MatInspector Professional (Genomatix Software GmbH, Munich, Germany). In total, five protected areas were identified within the region -250 to -50 bp, and in silico analysis revealed these to contain putative transcription factor binding sites for Sp1, C/EBPα, HNF3, AP2, and PXR. In addition, a protected fragment corresponding to a putative TATAA box 26 bp upstream of the transcription start site was identified (-29 bp to -24 bp). However, because this was outside the overlapping regions of the probe set used and thus could not be confirmed by two probes, this classification has been left as preliminary. The TATAA box sequence identified herein, however, does show high homology to the consensus sequence, lending weight to its classification. Previous studies have indicated that approximately 25 bp upstream of the transcription start site is optimal for a TATAA box (Latchman, 2001), and hence the assignment presented herein agrees with such a paradigm.

DNase I protection of the PXR response element within the CYP3A4 proximal promoter.
Three hundred eight base pairs of the CYP3A4 proximal promoter (positions -301 to +7) were labeled with [32P]dATP and DNase I protection assay carried out as described under Materials and Methods, using HepG2 nuclear extract as the protein source. Reactions were compared with sequence ladders of the appropriate area, and solid bars represent areas protected from digestion by nuclear extract. Panels A–D represent protection with 20 μg of HepG2 nuclear protein (+) compared with control (0 μg; -). Protected regions encompassing the four sites used in subsequent site-directed mutagenesis experiments are indicated; Sp1 (-104 bp to -97 bp), C/EBPα (-132 bp to -121 bp), PXRE (-169 bp to -152 bp), and HNF3 (-188 bp). Boundaries of these protected regions are also indicated. Panel E represents the effect of increasing nuclear protein concentration on protection profile of the PXRE. Zero, 7, 14, 57, or 114 μg of HepG2 nuclear protein were used. Gels are representative of at least three independent protection experiments and were confirmed on both DNA strands.

Protein-DNA interactions within the CYP3A4 proximal promoter.
DNase I footprint analysis was carried out on the CYP3A4 proximal promoter, using HepG2 nuclear extract as the protein source, in both the sense and antisense strands. Open boxes represent protected areas, confirmed through triplicate repeat experiments and the use of overlapping probes. Bold nucleotides represent those mutated in site-directed mutagenesis experiments. Dashed box represents protected region confirmed through triplicate repeat experiments with a single probe. Putative transcription factor binding sites were assigned through interrogation of the TransFac database, as described under Materials and Methods.
To further examine the protein-DNA interaction sites and confirm the putative assignment of interacting factors, electromobility shift assays were next carried out. Figure 3 shows EMSA experiments on the four protein-DNA interactions sites further examined herein using site-directed mutagenesis. TransFac assignments for putative interacting factors were tested using expressed proteins for PXR, Sp1, HNF3α, and C/EBPα. Binding of these proteins to their respective putative target sequence in the CYP3A4 proximal promoter confirmed that these are the most likely interacting proteins; the retarded bands were also of the same mobility as those seen when HepG2 nuclear proteins were used, suggesting that these were indeed the factors interacting in the complex mixture as well. Competition of the binding of HepG2 nuclear protein to the sites was carried out, and in all cases a 10-fold excess of wild-type sequence competitor resulted in removal of the retarded band, demonstrating the specificity of the interaction (Fig. 3). Replacement of the wild-type sequence probe with one coding for the product of site-directed mutagenesis was carried out to examine how well the mutations disrupted binding at the sites. In all cases, binding to the mutant sequence was significantly less than to the wild-type sequence. For Sp1, HNF3, and C/EBPα interaction sites, binding was reduced to <10% of the wild type in the mutant, whereas disruption of binding was less successful for the PXR mutant, reducing the level of binding to approximately 40% of the wild-type sequence.

Binding of nuclear proteins to identified protein-DNA interaction sites is disrupted by consensus oligonucleotides.
Electromobility shift assay experiments were carried out using double-stranded oligomers corresponding either to the wild-type putative transcription factor binding sites or to the sequence resulting from site-directed mutagenesis (Table 1), as described under Materials and Methods. Wild-type oligomers for each binding site were exposed to recombinant transcription factor or 14 μg of HepG2 nuclear protein, and HepG2 nuclear binding was completed using a 10-fold excess of unlabeled wild-type oligomer. Binding of HepG2 nuclear protein to the mutated sequence was also examined. Dashed arrows represent excess probe, whereas solid arrows represent retarded fragments. Data are representative of at least three independent experiments.
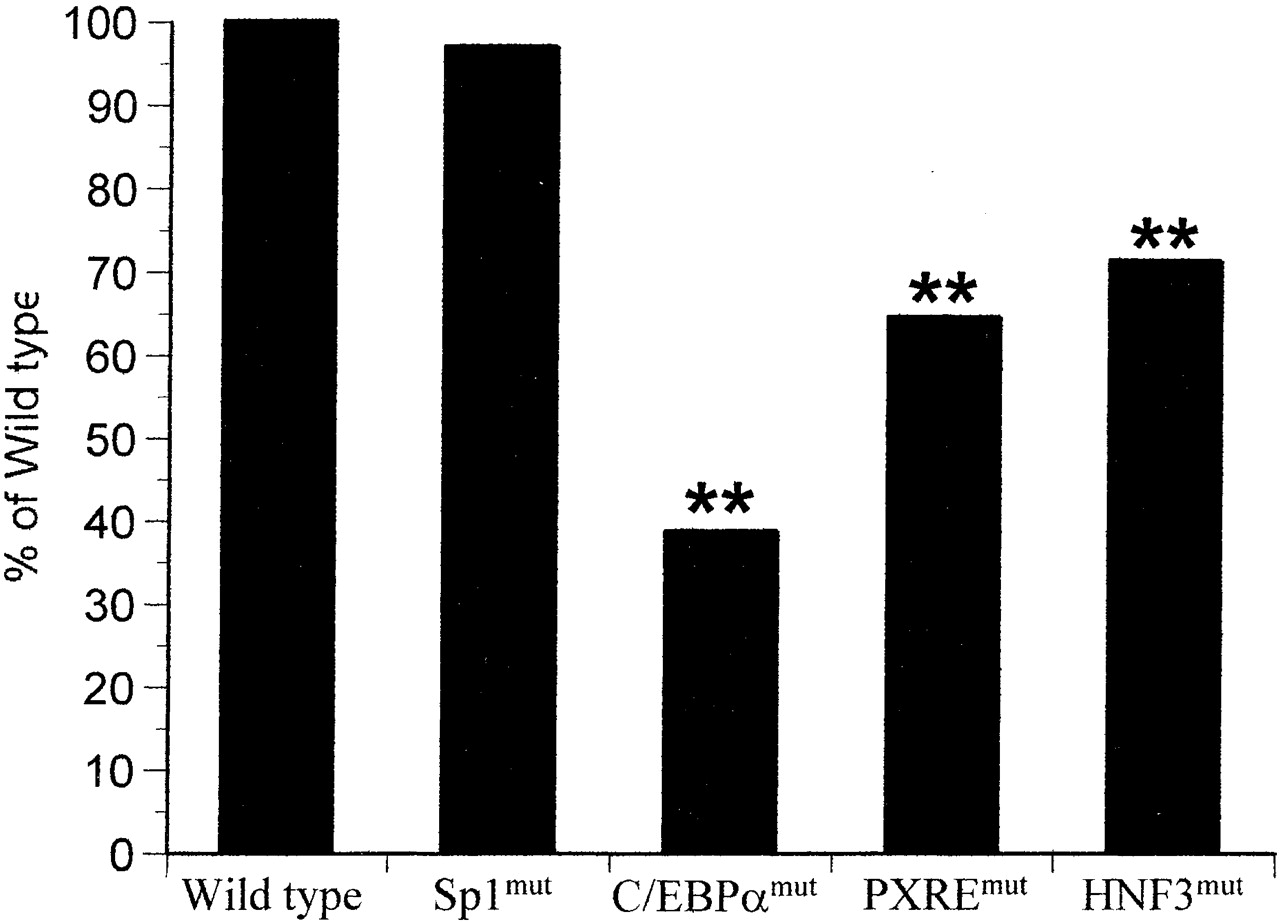
Disruption of Protein-DNA Interactions within theCYP3A4Proximal Promoter Reduces Basal Gene Expression. To examine the importance of the identified protein-DNA interaction sites in basal expression of CYP3A4, we used site-directed mutagenesis to disrupt four of these sites. We then examined the effect of these mutations on the expression of a CYP3A4 reporter gene, comprising the CYP3A4 XREM (-7972 to -7673 bp), 308 bp of proximal promoter linked to a secretory alkaline phosphatase reporter. Reporter gene studies carried out without addition of the XREM within the reporter gene produce very low expression values (data not shown), and hence are not appropriate for studying the potentially small variations in expression caused by disruption of single response elements. Figure 4 shows that disruption of either the C/EBPα (-132 bp to -121 bp), PXRE (-169 bp to -152 bp), or HNF3 (-195 bp to -186 bp) binding sites results in a significant decrease in basal expression, with the most marked effect being caused by disruption of the C/EBPα site (39% of control). In contrast, disruption of the putative Sp1 (-104 bp to -97 bp) site had no discernible effect on basal expression of the CYP3A4 reporter gene (96% of control).

Effect on basal gene expression of transcription factor binding sites within the CYP3A4 proximal promoter.
Site-directed mutagenesis of the wild-type CYP3A4 secretory alkaline phosphatase reporter gene construct (termed wild type) was carried out to disrupt PXRE (-169 bp to -152 bp), Sp1 (-104 bp to -97 bp), C/EBPα (-132 bp to -121 bp), and HNF3 (-195 bp to -186 bp) binding sites as described under Materials and Methods (termed PXREmut, Sp1mut, C/EBPαmut, and HNF3mut, respectively). These five constructs were transiently transfected separately into HuH7 cells and alkaline phosphatase activity measured 24 h later. Bars are calculated from eight wells per experimental point, and statistical significance was calculated as described under Materials and Methods (**, p < 0.01). Data are representative of experiments undertaken on two separate occasions.
Disruption of Protein-DNA Interactions within theCYP3A4Proximal Promoter Alters Xenobiotic-Mediated Activation of Gene Expression. To further examine the roles of the observed protein-DNA interactions in regulating CYP3A4 gene expression, we next examined their effects on the activation reporter gene expression by four known CYP3A4 transcriptional activators: rifampicin, phenobarbital, clotrimazole, and metyrapone (El-Sankary et al., 2001). The CYP3A4 reporter gene comprised the CYP3A4 XREM (-7972 to -7673 bp) and 308 bp of proximal promoter linked to a secretory alkaline phosphatase reporter. As might be expected, disruption of the PXRE motif had a dramatic effect on the activation caused by all four compounds (Fig. 5). In the case of rifampicin, metyrapone, and phenobarbital, disruption of the PXRE resulted in an approximate 50% decrease in the maximal induction observed (Imax), with no change in EC50. However, in the case of clotrimazole, a right-shift in EC50 from 1.6 to 4.6 μM with no significant change in Imax was observed.
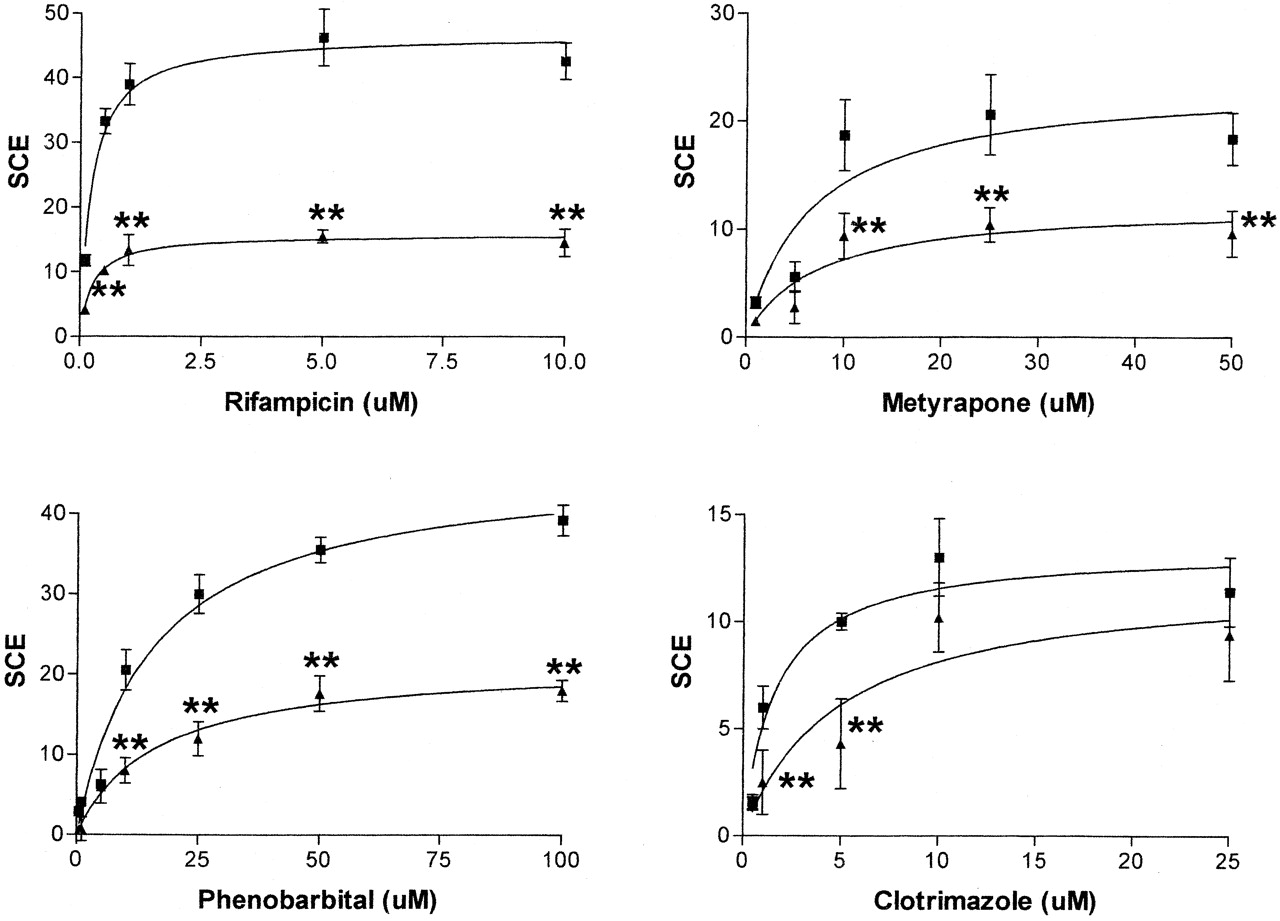
Effect of disruption of the CYP3A4 proximal promoter PXRE (-169 bp to -152 bp) on xenobiotic-mediated gene activation.
Either the wild-type CYP3A4 secretory alkaline phosphatase reporter gene containing both XREM and CYP3A4 proximal promoter (3A4), PXREmut derived from this or control plasmid lacking the CYP3A4 proximal promoter (Control) were transiently transfected into HuH7 cells and exposed to a range of concentrations of rifampicin, phenobarbital, metyrapone, and clotrimazole for 48 h. Alkaline phosphatase activity was measured before and after dosing, and the specific chemical effect was calculated as described under Materials and Methods. Values were then plotted on a lin-log graph; maximal effect, EC50, and inductive ability (IA, Imax/EC50) were calculated using GraphPad Prism PC software. Closed squares represent induction of wild-type reporter plasmid, and closed triangles represent induction of PXREmut reporter plasmid. Error bars show compound variation from eight wells per experimental point and statistical significance was calculated as described under Materials and Methods (*, p < 0.05; **, p < 0.01). Data are representative of experiments undertaken on at least two separate occasions.
Figure 6 shows the effect of disruption of the Sp1 (-104 bp to -97 bp) site on xenobiotic-mediated CYP3A4 transcriptional activation. Unlike the lack of effect observed on basal expression, disruption of the Sp1 (-104 bp to -97 bp) site had statistically significant effects on xenobiotic induction of the CYP3A4 reporter gene. Whereas neither rifampicin nor clotrimazole induction was affected, Imax values for phenobarbital and metyrapone were markedly reduced (47% and 50%, respectively).

Effect of disruption of the CYP3A4 proximal promoter Sp1 (-104 bp to -97 bp) element on xenobiotic-mediated gene activation.
Either the wild-type CYP3A4 secretory alkaline phosphatase reporter gene containing both XREM and CYP3A4 proximal promoter (3A4) and Sp1mut derived from this or control plasmid lacking the CYP3A4 proximal promoter (Control) were transiently transfected into HuH7 cells and exposed to a range of concentrations of rifampicin, phenobarbital, metyrapone, and clotrimazole for 48 h. Alkaline phosphatase activity was measured before and after dosing, and the specific chemical effect was calculated as described under Materials and Methods. Values were then plotted on a lin-log graph; maximal effect, EC50, and inductive ability (IA, Imax/EC50) were calculated using GraphPad Prism PC software. Closed squares represent induction of wild-type reporter plasmid, and closed triangles represent induction of Sp1mut reporter plasmid. Error bars show compound variation from eight wells per experimental point, and statistical significance was calculated as described under Materials and Methods (*, p < 0.05; **, p < 0.01). Data are representative of experiments undertaken on at least two separate occasions.
Disruption of the C/EBPα site identified herein and by Rodriguez-Antona et al. (2003) resulted in significant effects on the transcriptional activation of rifampicin, metyrapone, and phenobarbital, whereas no effect was observed on clotrimazole-mediated transcriptional activation (Fig. 7). In the cases of rifampicin and metyrapone, Imax values were decreased by 52% and 54%, respectively, whereas maximal induction by phenobarbital was decreased to only 19% of wild-type values. In addition, the EC50 value for phenobarbital induction was decreased from 15.5 to 8.8 μM, potentially suggesting a switch to an alternate receptor target and/or DNA binding site.

Effect of disruption of the CYP3A4 proximal promoter C/EBPα (-132 bp to -121 bp) element on xenobiotic-mediated gene activation.
Either the wild-type CYP3A4 secretory alkaline phosphatase reporter gene containing both XREM and CYP3A4 proximal promoter (3A4) and C/EBPαmut derived from this or control plasmid lacking the CYP3A4 proximal promoter (Control) were transiently transfected into HuH7 cells and exposed to a range of concentrations of rifampicin, phenobarbital, metyrapone, and clotrimazole for 48 h. Alkaline phosphatase activity was measured before and after dosing, and the specific chemical effect was calculated as described under Materials and Methods. Values were then plotted on a lin-log graph; maximal effect, EC50, and inductive ability (IA, Imax/EC50) were calculated using GraphPad Prism PC software. Closed squares represent induction of wild-type reporter plasmid, and closed triangles represent induction of C/EBPαmut reporter plasmid. Error bars show compound variation from eight wells per experimental point, and statistical significance was calculated as described under Materials and Methods (**, p < 0.01). Data are representative of experiments undertaken on at least two separate occasions.
Finally, an HNF3 (-195 bp to -186 bp) binding site was disrupted, and its effect on xenobiotic induction was examined (Fig. 8). As previously observed, disruption of this HNF3 site had no effect on rifampicin induction (El-Sankary et al., 2002). However, disruption of the HNF3 resulted in statistically significant reductions in Imax values for phenobarbital and clotrimazole. Interestingly, in contrast to all previous experiments, CYP3A4 activation was increased (132% of wild type) when the HNF3 mutant was challenged with metyrapone, suggesting that this site may be involved in both positive and negative regulation of CYP3A4 gene expression.

Effect of disruption of the CYP3A4 proximal promoter HNF3 (-195 bp to -186 bp) element on xenobiotic-mediated gene activation.
Either the wild-type CYP3A4 secretory alkaline phosphatase reporter gene containing both XREM and CYP3A4 proximal promoter (3A4) and HNF3mut derived from this or control plasmid lacking the CYP3A4 proximal promoter (Control) were transiently transfected into HuH7 cells and exposed to a range of concentrations of rifampicin, phenobarbital, metyrapone, and clotrimazole for 48 h. Alkaline phosphatase activity was measured before and after dosing, and the specific chemical effect was calculated as described under Materials and Methods. Values were then plotted on a lin-log graph; maximal effect, EC50, and inductive ability (IA, Imax/EC50) were calculated using GraphPad Prism PC software. Closed squares represent induction of wild-type reporter plasmid, and closed triangles represent induction of HNF3mut reporter plasmid. Error bars show compound variation from eight wells per experimental point, and statistical significance was calculated as described under Materials and Methods (*, p < 0.05; **, p < 0.01). Data are representative of experiments undertaken on at least two separate occasions.
Discussion
Understanding the molecular mechanisms underlying transcriptional regulation of drug-metabolizing enzymes is a field of emerging importance. Rather than merely being able to test for transcriptional activation by a compound or even place this in the context of other activating compounds (El-Sankary et al., 2001), interest has extended to studying the transcription factors that underlie such phenomena. With the identification of PXR, many of the mysteries for CYP3A transcriptional regulation appear to have been solved (Kliewer et al., 1998). Human PXR has a wide ligand binding profile and is activated by many of the compounds that activate CYP3A4 transcription (Lehmann et al., 1998). However, despite the apparent all-encompassing role of PXR in CYP3A4 gene expression, several questions remain unanswered. First, it is known that PXR competes for ligands and DNA binding sites with other receptors such as constitutive androstane receptor (Xie et al., 2000); the role of this competitive interaction in controlling gene expression of CYP3A4, and indeed the host of other genes that constitutive androstane receptor and PXR regulate (Maglich et al., 2002), remains to be fully elucidated. Second, interactions of ligand-activated transcription factors with other transcription factors may modulate their effects. It is clear that other transcription factors, including members of the C/EBP and HNF families, play a vital role in modulating gene activation, both through direct interaction with ligand-activated transcription factors and indirectly through chromatin remodeling. Finally, the role of epigenetics in the control of CYP3A4 expression is a field that, at the present time, is largely untouched.
We have used a variety of techniques to study the second of these questions, examining the protein-DNA interactions within the CYP3A4 proximal promoter, both at the level of physical mapping and also functional consequences of disruption of these protein-DNA interaction sites. DNase I footprint analysis revealed a complex set of protein-DNA interactions within the CYP3A4 proximal promoter, and in silico analysis identified putative transcription factor binding sites for C/EBPα, HNF3, Sp1, and AP2. Further analysis using EMSA demonstrated the validity of these assignments, with recombinantly expressed proteins showing specific interactions with the target sequences.
Both HepG2 and Huh7 hepatoma cell lines are used within this study. Transcriptome analysis of these cell lines demonstrates that both the expression of CYP3As and the transcription factors under study herein are comparable between the two cell lines (A. Phillips, personal communication). In addition, CYP3A4 reporter gene constructs show similar basal- and xenobiotic-mediated expression when transfected into either HepG2 or Huh7 cells (data not shown), demonstrating the comparability of these two hepatoma cell lines when used in studies such as this one. The use of a reporter gene system encompassing the CYP3A4 enhancer (XREM) raises the possibility of interactions within the XREM affecting the binding sites within the proximal promoter and hence altering their functioning. Inclusion of the XREM within a reporter gene is common for two reasons: first, experiments carried out without one produce significantly lower expression levels which are often difficult to quantify accurately (El-Sankary et al., 2000); and second, experiments using the native enhancer will, in all probability, more accurately reflect the in vivo situation.
It is of interest that several of these sites exist as overlapping complexes; the resulting competition for DNA binding sites may be important in the process of gene expression (Schaufele et al., 1990; Roux et al., 1995). Although it is important to recognize that such an in vitro analysis, as compared with in vivo footprinting, does not take into account the potential effects of chromatin structure in organizing these interactions, it does provide the first complete analysis of the potential interactions. Furthermore, in vivo, analysis will be required to understand which interactions occur in vivo in response to stimuli.
As expected, protein-DNA interactions were observed encompassing the PXRE within the CYP3A4 proximal promoter, the binding site for PXR. Disruption of this site via site-directed mutagenesis led to an approximate 30% decrease in basal CYP3A4 reporter gene expression and alterations in the induction profiles for all the drugs tested, again confirming the importance of this site in regulation of CYP3A4 gene expression. The fact that complete loss of basal activation was not observed may suggest one of two scenarios. First, binding to the PXRE may not have been completely ablated, with the observed activations representative of this residual binding. Lehmann et al. (1998) have previously shown that disruption of only one half of the site of the PXRE results in only partial disruption of PXR binding, and the EMSA experiments carried out herein show that binding to the mutated PXRE did occur, although at a reduced level compared with the wild-type sequence. An intriguing alternative explanation is that the residual activation could represent gene activation occurring through alternate transcription factors and/or protein-DNA interaction sites, although no direct evidence exists to support such a hypothesis at this time.
Whereas disruption of the proximal PXRE significantly decreased the Imax for rifampicin, metyrapone, and phenobarbital, no such effect was observed when cells were exposed to clotrimazole, with an alteration in EC50 being observed instead. This unexpected result may suggest that clotrimazole may act via two equally efficacious activation pathways, one PXR-dependent and one PXR-independent. In the presence of an active PXRE, activation occurs via PXR with an EC50 of 1.6 μM. However, if the PXRE is disrupted, a second activation pathway with lower affinity (EC50 = 4.6 μM) is used to mediate the transcriptional activation. Since the EC50 value represents both activation of the receptor by ligand and the kinetics of any resulting protein-DNA interaction, such a shift may represent binding of activated PXR to a second lower-affinity response element or activation of a second receptor pathway by clotrimazole. The latter scenario seems more probable, since it is unlikely that PXR could bind to an alternate binding site when activated by clotrimazole but not after activation by the other chemicals.
For disruption of Sp1, C/EBPα, and HNF3 binding sites by SDM, the nucleotides mutated were those most highly conserved in the consensus binding site or shown to be important in the mechanisms of binding. Using such an approach, it would be expected that the majority of binding at these sites has been removed, and EMSA analysis has shown significantly decreased HepG2 nuclear extract binding at these sites (Fig. 2).
The transcription factor Sp1 is a ubiquitous transcription factor that binds to glucocorticoid-rich elements in the promoters of many genes and thus controls their expression (Kaczynski et al., 2003). Hence, disruption of a putative Sp1 (-104 bp to -97 bp) site in the proximal promoter of the CYP3A4 gene might be expected to have major effects on the transcriptional regulation of this gene. However, disruption of this site does not alter the basal transcription of the reporter gene, possibly due to the presence of a second putative Sp1 binding site adjacent to the one disrupted in this study. In the basal situation it is possible that both or only the distal unit are required for basal gene expression. Disruption of the proximal Sp1 binding site significantly affected activation of the reporter construct by metyrapone and phenobarbital, however, reducing the Imax values by approximately 50%. Such a finding is in keeping with those of Muangmoonchai et al. (2001), who demonstrated that Sp1 bound to two sites in the CYP2B1 promoter and that phenobarbital induction in primary rat hepatocytes was enhanced by cotransfection with an Sp1 expression plasmid.
The C/EBP family of proteins comprises ubiquitous transcription factors involved in a number of biological processes including differentiation, inflammation, and liver regeneration (Ramji and Foka, 2002). Recent work by Rodriguez-Antona et al. (2003) demonstrated the presence of active C/EBPα binding sites in the CYP3A4 promoter, one of which is in agreement with the site identified and mutated in the present study. The latter authors demonstrated that overexpression of C/EBPα resulted in increased levels of CYP3A4 mRNA. The data in the present study are in agreement with such an observation, with disruption of the C/EBPα (-132 bp to -121 bp) binding site resulting in a 60% decrease in basal expression. In addition, we now demonstrate for the first time that this site is also responsible for the regulation of xenobiotic-mediated activation of CYP3A4 gene expression. Disruption of this site results in a decrease in the Imax calculated for rifampicin-, phenobarbital-, and metyrapone-mediated activation, whereas clotrimazole-mediated activation is unaffected.
The final site of protein-DNA interaction to be disrupted in this study is a putative HNF3 binding site positioned -195 bp to -186 bp upstream of the transcription start site. Whereas three isoforms of HNF3 exist, HNF3α, β, and γ, the present study does not distinguish between them, labeling the site “HNF3” only. Since all three isoforms share highly similar DNA binding sites, only in vivo footprinting would be able to truly confirm which isoform was bound at this site and, as such, is beyond the current investigation. In the present study, a weak down-regulation of basal expression was seen upon disruption of the HNF3 binding site, although this was markedly less than was seen in the case of the C/EBPαmut construct. Xenobiotic-mediated activation of the CYP3A4 gene was affected by disruption of the HNF3 binding site, although the pattern observed was the most complicated. Rifampicin activation was unaffected, as previously demonstrated through an alternate mutation that disrupted this overlapping HNF3-C/EBPα site (El-Sankary et al., 2002). Both phenobarbital- and clotrimazole-mediated activation were decreased, with reductions in Imax values observed; in contrast, metyrapone-mediated activation was seen to be increased. The complexity of the response observed may reflect the role that HNF molecules play in gene expression in general. HNF transcription factors are often associated with chromatin remodeling functions, thus improving access for other transcription factors (Roux et al., 1995). As can be seen from Fig. 3, both HNF3 binding sites are adjacent to C/EBPα binding sites, and it is possible that cooperativity exists between them, as is seen in the tyrosine aminotransferase gene (Grange et al., 1989; Rigaud et al., 1991). Indeed, Rodriguez-Antona et al. (2003) observed that HNF3γ did not affect the expression of CYP3A4 in HepG2 when transfected alone; however, it augmented the effect of C/EBPα cotransfection. Chromatin remodeling by liver-specific factors such as the HNFs may underlie the tissue-specific expression and activation of CYP3A4.
It should be noted that for all the DNA-protein interactions examined, the effects observed, although significant, do not represent a complete ablation of transcriptional activity. This suggests that none of the sites studied are obligated to respond, but rather play supportive roles in the transcriptional regulation of CYP3A4. Such a concept is not inconsistent with the data generated from PXR-knockout mice; such mice show no alteration in basal expression of CYP3A4, whereas xenobiotic-mediated transcriptional activation is ablated. Indeed, it is perhaps not unsurprising that the transcriptional control of an important enzyme such as CYP3A4 is under the net control of several transcription factors, as opposed to the gross control of a single factor.
In summary, we have conducted an analysis of protein-DNA interactions within the proximal promoter of the CYP3A4 gene. This region is relatively complex, and in addition to the interaction site for PXR, contains interaction sites for HNF3, C/EBPα, Sp1, and AP2, as well as a consensus TATAA box. We have examined the functionality of these sites and shown that they are capable of affecting both the basal- and xenobiotic-mediated activation of gene expression.
Footnotes
-
↵1 Abbreviations used are: P450, cytochrome P450; bp, base pair; PXR; pregnane X receptor; GR, glucocorticoid receptor; hPXR, human pregnane X receptor; ER6, everted six-strand repeat; PXRE, PXR element; C/EBP, CCAAT/enhancer binding protein; HNF, hepatic nuclear factor; DTT, dithiothreitol; XREM, xenobiotic-responsive enhancer module; SDM, site-directed mutagenesis; EMSA, electrophoretic mobility shift assay.
- Received November 7, 2003.
- Accepted February 4, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}