


Abstract
The cytochrome P450 (P450) CYP2E1 enzyme metabolizes and activates a wide array of toxicological substrates, including alcohols, the widely used analgesic acetaminophen, acetone, benzene, halothane, and carcinogens such as azoxymethane and dimethylhydrazine. Most studies on the biochemical and pharmacological actions of CYP2E1 are derived from studies with rodents, rabbits, and cultured hepatocytes; therefore, extrapolation of the results to humans can be difficult. Creating “humanized” mice by introducing the human CYP2E1 gene into Cyp2e1-null mice can circumvent this disadvantage. A transgenic mouse line expressing the human CYP2E1 gene was established. Western blot and high-performance liquid chromatography/mass spectrometry analyses revealed human CYP2E1 protein expression and enzymatic activity in the liver of CYP2E1-humanized mice. Treatment of mice with the CYP2E1 inducer acetone demonstrated that human CYP2E1 was inducible in this transgenic model. The response to the CYP2E1 substrate acetaminophen was explored in the CYP2E1-humanized mice. Hepatotoxicity, resulting from the CYP2E1-mediated activation of acetaminophen, was demonstrated in the livers of CYP2E1-humanized mice by elevated serum alanine aminotransferase levels, increased hepatocyte necrosis, and decreased P450 levels. These data establish that in this humanized mouse model, human CYP2E1 is functional and can metabolize and activate different CYP2E1 substrates such as chlorzoxazone, p-nitrophenol, acetaminophen, and acetone. CYP2E1-humanized mice will be of great value for delineating the role of human CYP2E1 in ethanol-induced oxidative stress and alcoholic liver damage. They will also function as an important in vivo tool for predicting drug metabolism and disposition and drug-drug interactions of chemicals that are substrates for human CYP2E1.
The cytochrome P450 (P450) family of enzymes play an important role in the metabolic activation of chemicals to cytotoxic or carcinogenic products, as well as the oxidation of therapeutically used drugs and endogenous steroids (Guengerich et al., 1991; Lieber, 1997). In particular, CYP2E1 is of interest since it metabolizes and activates a wide array of toxicologically important substrates, including acetaminophen (APAP), acetone, benzene, halothane, ethanol, carbon tetrachloride, and carcinogens such as the low molecular weight nitrosamines (Kessova and Cederbaum, 2003). CYP2E1 metabolizes a small number of clinically employed drugs such as disulfiram and the muscle relaxant chlorzoxazone, which undergoes 6-hydroxylation (Peter et al., 1990; Carriere et al., 1993). CYP2E1 also oxidizes a significant number of important occupational and industrial chemicals, including alkanes, alkenes, solvents, and aromatic and halogenated hydrocarbons (Bolt et al., 2003).
APAP is a widely used analgesic and is considered safe at therapeutic doses; hepatotoxicity occurs at low frequency. Metabolism of APAP was shown to be primarily contributed by CYP2E1 (Raucy et al., 1989; Snawder et al., 1994a; Kostrubsky et al., 1997; Zhang et al., 2004) However, studies have shown that CYP1A2 and CYP3A are also involved in the metabolism of APAP, although CYP2E1 has a lower Km than CYP1A2 (Raucy et al., 1989; Snawder et al., 1994a; Kostrubsky et al., 1997; Zhang et al., 2004). APAP is metabolized to N-acetyl-p-benzoquinone imine (NAPQI), a metabolite that is capable of reacting with cellular nucleophiles such as glutathione (Forte et al., 1984). NAPQI is rapidly metabolized to nontoxic cysteine and mercapturic acid conjugates; however, in an overdose of APAP, NAPQI accumulates as hepatic glutathione becomes depleted, and the formation of reactive intermediate metabolites outstrips their detoxification.
The expression of CYP2E1 is highest in the liver; however, it is also present in the kidney cortex, with low levels detected in nasal mucosa, lung, testis, ovaries, small intestine, colon, and brain (Lieber, 1997). The human CYP2E1 gene is located on chromosome 10 and encodes an mRNA that is 1.6 kb in length. The human CYP2E1 cDNA shares 79% similarity over the coding region with mouse CYP2E1 (Freeman et al., 1992). The molecular mechanism responsible for the induction of human CYP2E1 are not as well understood as those described for other P450s that involve the pregnane X receptor or constitutive active receptor (Goodwin et al., 1999, 2002). The CYP2E1 gene is under transcriptional control during development. In rats, immediately after birth, CYP2E1 expression is activated, and maximal transcription occurs within the first week (Kessova and Cederbaum, 2003). CYP2E1 is clearly expressed in human fetal liver, and postnatal data suggest that infants less than 90 days old would have decreased clearance of CYP2E1 substrates compared with older infants and adults (Johnsrud et al., 2003). Levels of CYP2E1 mRNA are induced under a variety of pathophysiological conditions, such as fasting, obesity, or induced diabetes (Hong et al., 1987; Raucy et al., 1991; Woodcroft et al., 2002). This increase in CYP2E1 mRNA was shown to be due to post-transcriptional mRNA stabilization. Regulation of CYP2E1 by exogenous chemicals such as ethanol, acetone, or pyrazole is less clear, since CYP2E1 mRNA levels were reportedly not increased after administration of these chemicals to rats (Song et al., 1986). Indeed, CYP2E1 is not transcriptionally activated by an acute bolus dose or chronic administration of ethanol, acetone, or other exogenous-inducing agents, but levels of CYP2E1 were increased as a result of stabilization of the protein by inhibiting the rapid phase of its biphasic degradation (Song et al., 1989; Kessova and Cederbaum, 2003). Consequently, increased half-lives of the enzyme or its mRNA result in higher steady state enzyme levels.
HepG2 cell lines that constitutively overexpress human CYP2E1 (Dai et al., 1993; Chen and Cederbaum, 1998) and primary hepatocyte cultures from rats treated with pyrazole to elevate levels of CYP2E1 have been used to understand basic effects and actions of CYP2E1 (Wu and Cederbaum, 2000, 2001). Since most studies on the biochemical and pharmacological actions of CYP2E1 are derived from studies with rodents, rabbits, and cultured hepatocytes, extrapolation of the results to humans can be difficult. Creating humanized mice by introducing the human CYP2E1 gene into Cyp2e1-null mice can circumvent this disadvantage. A transgenic mouse line was described that expresses the human CYP2E1 cDNA under the control of the mouse albumin enhancer promoter (Morgan et al., 2002). However, this transgenic mouse line contains endogenous mouse CYP2E1 and is therefore not a true humanized model.
In this study, the development of a CYP2E1-humanized transgenic mouse model that expresses functional and inducible human CYP2E1 is described. Comparisons between CYP2E1-humanized mice with mouse Cyp2e1-null and wild-type mice allow immediate functional differences to be examined. The generation of such a model can contribute to understanding these functional differences between human and mouse CYP2E1.
Materials and Methods
Chemicals. Chlorzoxazone, 6-hydroxychlorzoxazone, p-nitrophenol, p-nitrocatechol, and acetaminophen were purchased from Sigma-Aldrich (St. Louis, MO). High-performance liquid chromatography grade solvents were purchased from Fisher Scientific Co. (Pittsburgh, PA).
Animals and Treatments. Mice were maintained under a standard 12-h light/dark cycle with water and chow provided ad libitum. Handling was in accordance with animal study protocols approved by the National Cancer Institute Animal Care and Use Committee. At least five male mice were used for each treatment group. For the APAP toxicity study, mice were given one dose of APAP dissolved in saline (0–400 mg/kg b.wt.) by intraperitoneal injection. Serum was collected from the mice by retro-orbital bleeding 24 h after the APAP injection, following which the mice were sacrificed and the tissues were collected. To examine CYP2E1 induction, mice were administered 2% acetone (v/v) in the drinking water for 7 days. For histology, liver samples were placed into 10% neutral buffered formalin and embedded in paraffin; sections were stained with hematoxylin and eosin. Otherwise, tissues were snap-frozen in liquid nitrogen and stored at –80°C until further analysis. Alanine aminotransferase (ALT) levels were measured in serum using a commercial kit (Sigma-Aldrich).
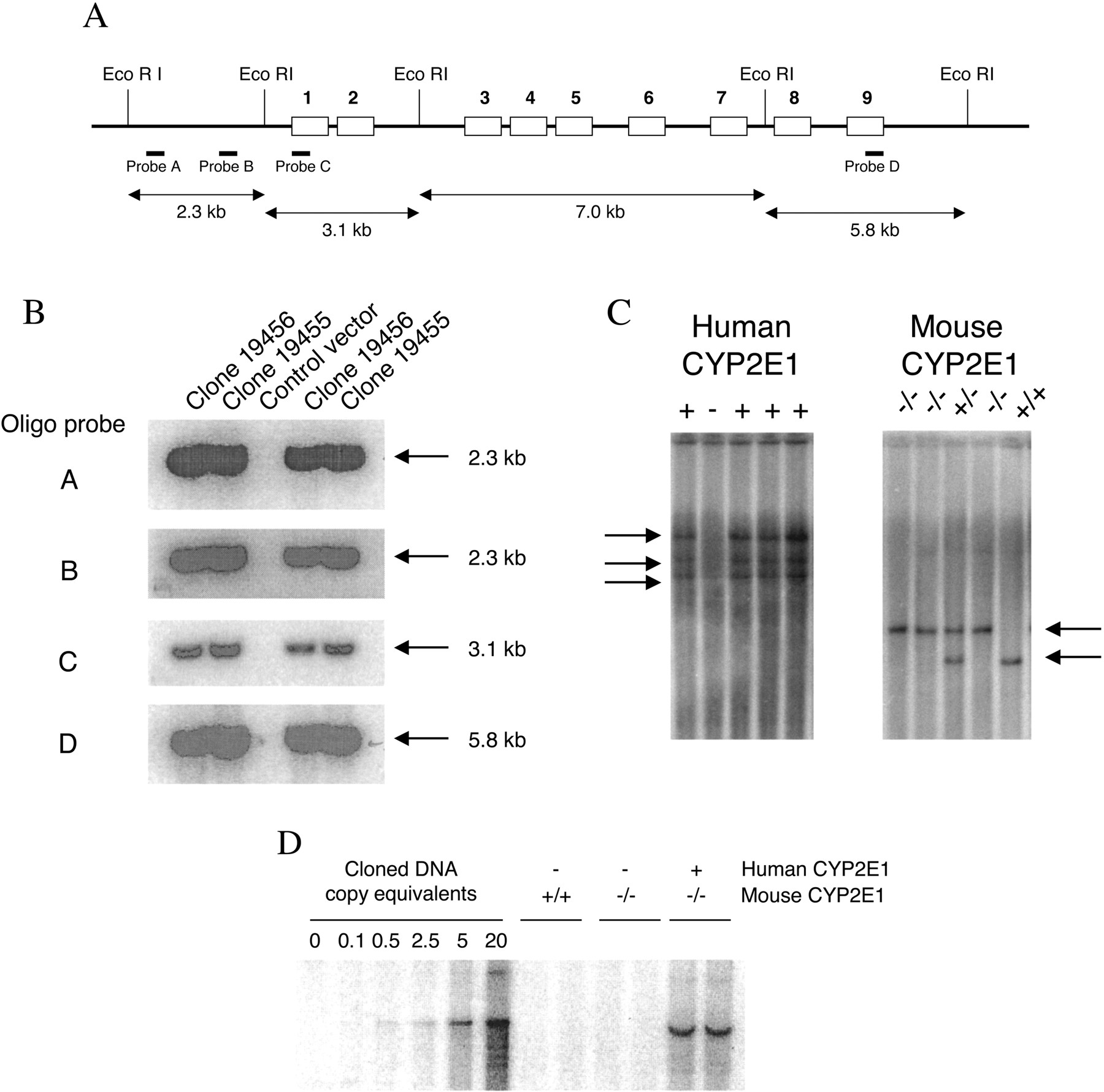
Generation ofCYP2E1-Humanized Mice. A bacterial artificial chromosome (BAC) genomic library (Genome Systems, St. Louis, MO) was screened using CYP2E1 cDNA (Umeno et al., 1988). Positive BAC clones were identified and verified by Southern blot analysis using 32P end-labeled DNA oligonucleotide probes A to D, recognizing specific regions (–2650 and –539 bp, exon 1 and 9, respectively) of the human CYP2E1 gene (Fig. 1A) (Umeno et al., 1988). A BAC clone containing the human CYP2E1 gene was linearized using restriction enzyme digestion (NotI) and purified before microinjection into fertilized FVB/N mouse eggs. Transgenic founders were screened by Southern blot analysis and bred with Cyp2e1-null mice (Lee et al., 1996). From this breeding, mice positive for the human CYP2E1 transgene were identified by Southern blotting and further mated to Cyp2e1-null mice. Litters from this breeding were again screened to identify which were positive for the human CYP2E1 transgene and the mouse Cyp2e1-null allele. Mice containing the human CYP2E1 transgene in a mouse Cyp2e1-null background were bred together to generate homozygous mice, and these mice were designated CYP2E1-humanized mice.

Generation of CYP2E1-humanized mice. A, diagrammatic representation of the human CYP2E1 gene showing EcoRI digestion sites. B, Southern blot analysis of the BAC clone DNA using end-labeled oligo probes recognizing specific regions of the gene (probe A, –2650-bp region; probe B, –539-bp region; probe C, exon 1 region; and probe D, exon 9 region). C, typical genotyping results by Southern blot hybridization for the human CYP2E1 transgene and mouse Cyp2e1-null allele. D, transgene copy number determination using purified genomic clone DNA diluted with mouse DNA as a standard to give copy equivalents for 5 μg of genomic DNA. Genomic DNA (5 μg) from wild-type, Cyp2e1-null, and CYP2E1-humanized mice were compared with the standard.
Southern Blot Analysis. For analysis of the BAC clone, DNA was digested with EcoRI and SalI (Umeno et al., 1988), whereas for genotyping mice, tail genomic DNA was digested with BamHI or SpeI. Electrophoresis and Southern hybridization conditions were as described previously (Granvil et al., 2003). Random primer 32P-labeled DNA probes were used for hybridizations; probes A to D (Fig. 1A) were used in the analysis of the BAC clone, a NotI fragment of the BAC clone was used for screening mice containing the human CYP2E1 transgene, and the probe P5 was used to screen for the mouse Cyp2e1-null allele (Lee et al., 1996). Transgene copy number was determined as described previously, with some minor alterations (Granvil et al., 2003). Cloned DNA was diluted with mouse DNA to yield the equivalent of 0.1, 0.5, 2.5, 5, and 20 copies of the gene per diploid genome (based on 7.8 × 105 base pairs per diploid genome). DNA was digested with EcoRI and subjected to Southern blot analysis with genomic DNA isolated from wild-type, Cyp2e1-null, and CYP2E1-humanized mice.
Microsome Preparation. Tissues were homogenized in ice-cold buffer [50 mM Tris-HCl, 150 mM KCl, 1 mM EDTA, 20% glycerol, and 0.5 M (4-amidino-phenyl)-methanesulfonyl fluoride], and microsomes were prepared by differential centrifugation as described previously (Cheung et al., 2003).
Western Blot Analysis. Microsomal protein (5–30 μg) from each sample was subjected to SDS-polyacrylamide gel electrophoresis in 10% polyacrylamide gels, electrophoretically transferred to Immobilon-P membranes (Millipore Corporation, Bedford, MA), and probed using anti-human CYP2E1 (clone 1-156-3) (Gelboin et al., 1996), anti-rat CYP2E1 (1-98-1) (Ko et al., 1987), anti-rat CYP3A1/2 (clone 2-13-1) (Park et al., 1986), and anti-rat CYP1A2 (clone 22-341) (Park et al., 1982).
In Vitro Metabolism of Chlorzoxazone andp-Nitrophenol. Incubation reactions were carried out in a final volume of 200 μl containing 100 mM potassium phosphate, pH 7.4, pooled liver microsomes, chlorzoxazone, NADPH, and the monoclonal antibody, if necessary. For each reaction, the final concentration was 0.2 μg/μl for liver microsomal proteins and 2.5 μM for chlorzoxazone. For immunoinhibition analysis, 15 μl of an anti-CYP2E1 monoclonal antibody (clone 11-73-18) (Gelboin et al., 1996) was used. Antilysozyme (HyHel) was used as a control for nonspecific binding. Reactions were initiated by the addition of 20 μl of 10 mM NADPH after 5 min of preincubation at 37°C. Incubations were terminated by the addition of 200 μl of ice-cold acetonitrile, followed by 20 μl of 10 μM 7-hydroxycoumarin (internal standard, in methanol). The mixture was vortexed for 20 s, cooled on ice for 10 min, and centrifuged at 14,000g for 10 min. The supernatant was transferred to a glass tube and extracted with a mixture of ethyl acetate (1 ml) and methyl t-butyl ether (2 ml). The organic phase was separated by centrifugation at 3300g for 15 min, transferred to a new glass tube, and evaporated to dryness under a stream of nitrogen gas. The sample was reconstituted with 50% methanol containing 0.2% formic acid for LC/MS/MS analysis. All reactions were performed in triplicate. p-Nitrophenol O-hydroxylation to p-nitrocatechol was measured using a previously published method (Sai et al., 1999).
Identification and Quantitation of Metabolites by LC/MS/MS. LC/MS/MS analysis was performed on a PE Sciex API2000 electrospray ionization triple-quadrupole mass spectrometer (PerkinElmer Life and Analytical Sciences, Boston, MA) controlled by Analyst software. Separation of chlorzoxazone and its metabolite was performed on an Aquasil C18 column (5 μm, 50 × 2.1-mm i.d.) (Thermo Hypersil, Keystone Scientific Operations, Bellefonte, PA). The high-performance liquid chromatography system was operated isocratically at a flow rate of 0.2 ml/min with the mobile phase [water/methanol = 20:80 (v/v)] containing 0.1% formic acid. The mass spectrometer was equipped with a turbo ion spray source and run in the negative ion mode. The turbo ion spray temperature was maintained at 350°C, and a voltage of 4.8 kV was applied to the sprayer needle. Nitrogen was used as the turbo ion spray and nebulizing gas. The detection and quantification of analytes were accomplished by multiple reactions monitoring with the transitions of m/z 183.8/119.8 for 6-hydroxychlorzoxazone, 167.8/131.8 for chlorzoxazone, and 160.8/132.9 for the internal standard 7-hydroxycoumarin. Each assay was completed in 3 min, and all raw data were processed using Analyst software. Chlorzoxazone and 6-hydroxychlorzoxazone were eluted at 1.09 and 0.90 min, respectively. Calibration curves were linear for the concentrations of 6-hydroxychlorzoxazone ranging from 0.1 to 5 μM and chlorzoxazone from 1.0 to 50 μM. The coefficient of variation was less than 20%.
Northern Blot Analysis. Total RNA was extracted from tissues using TRIzol reagent (Invitrogen, Carlsbad, CA) and quantitated by optical densitometry at 260 nm. Total RNA (0.1–20 μg) was subjected to electrophoresis in a 0.22 M formaldehyde containing 1.0% agarose gel and transferred to Gene Screen Plus hybridization transfer membranes (PerkinElmer Life and Analytical Sciences). Membranes were hybridized using random primer 32P-labeled cDNA probes as described previously (Cheung et al., 2003). For a loading control, 36B4 was used.
Statistical Analysis. All values are expressed as the means ± S.D. or means ± S.E.M. All data were analyzed by paired or unpaired Student's t test for significant differences between the mean values of each group.
Results
Generation and Expression Analysis ofCYP2E1-Humanized Mice. From the initial screening of the human BAC genomic library with CYP2E1 cDNA, two positive BAC clones (19456 and 19455) were identified. The BAC clones were analyzed by Southern blotting to verify the presence of the human CYP2E1 gene, and both were shown to contain at least 2.6 kb of DNA upstream of the transcriptional start site and the last exon 9 (Fig. 1, A and B). One clone was selected and used to make a transgenic mouse expressing the human CYP2E1 gene. Humanized transgenic mice containing the human CYP2E1 gene in the mouse Cyp2e1-null background were generated (see Materials and Methods), with the presence of transgenes confirmed by Southern blotting at each breeding step (Fig. 1C). CYP2E1-humanized mice seem normal and have litter sizes comparable to wild-type mice. The copy number of the transgene in the CYP2E1-humanized mice was approximately five, as estimated by densitometric analysis of the intensity of hybridization signal to that obtained with known amounts of human DNA (Fig. 1D).
Detection of microsomal human CYP2E1 protein was demonstrated in the livers of CYP2E1-humanized mice (Fig. 2A). Human CYP2E1 protein was undetectable in the microsomes of extrahepatic tissues examined (data not shown). However, human CYP2E1 mRNA was detectable in extrahepatic tissues such as heart, kidney, lung, small intestine, and spleen (Fig. 2B). Human CYP2E1 activity in the liver microsomal fractions of the CYP2E1-humanized mice was confirmed by significant increases in both chlorzoxazone 6-hydroxylation and p-nitrocatechol formation compared with Cyp2e1-null mice (Fig. 2C). There was no significant difference between the chlorzoxazone 6-hydroxylation activity in the liver microsomal fractions of the wild-type and CYP2E1-humanized mice. However, using p-nitrophenol as a substrate, the formation of p-nitrocatechol, formed as a result of p-nitrophenol O-hydroxylation, was just significantly higher (p = 0.047) in the CYP2E1-humanized mice compared with wild-type mice (Fig. 2C). Using an inhibitory monoclonal antibody to human CYP2E1, chlorzoxazone 6-hydroxylation was reduced by approximately 30% in both wild-type and CYP2E1-humanized mice. The same inhibitory monoclonal antibody to human CYP2E1 significantly reduced the p-nitrophenol O-hydroxylation in the CYP2E1-humanized mice by approximately 75%, whereas this inhibitory effect was not as pronounced in the wild-type mice (∼36% reduction, p = 0.15).

Expression analysis of CYP2E1-humanized mice. A, Western blot analysis of liver microsomes from wild type, Cyp2e1-null, and CYP2E1-humanized mice detected using monoclonal antibodies specific to human or rat CYP2E1. HLM, human liver microsomes. B, Northern blot analysis of RNA isolated from liver and extrahepatic tissues of CYP2E1-humanized mice. Membranes were probed with CYP2E1 or 36B4 (loading control). C, CYP2E1 enzymatic activity as measured by the relative percentage of chlorzoxazone 6-hydroxylation or p-nitrophenol O-hydroxylation in the liver microsomal fractions of untreated wild-type, Cyp2e1-null, and CYP2E1-humanized mice. The relative activities were calculated based on the mean value of the untreated wild-type liver sample (the mean absolute values for this sample were 129.9 pmol/mg protein/min for chlorzoxazone 6-hydroxylation and 0.85 nmol/mg protein/min for p-nitrocatechol formation and were defined as 100%). Inhibition of activity was demonstrated using a monoclonal anti-inhibitory antibody to human CYP2E1 (hCYP2E1 MAb). Results shown are the means ± S.E.M. of pooled liver microsomes (n = 5).
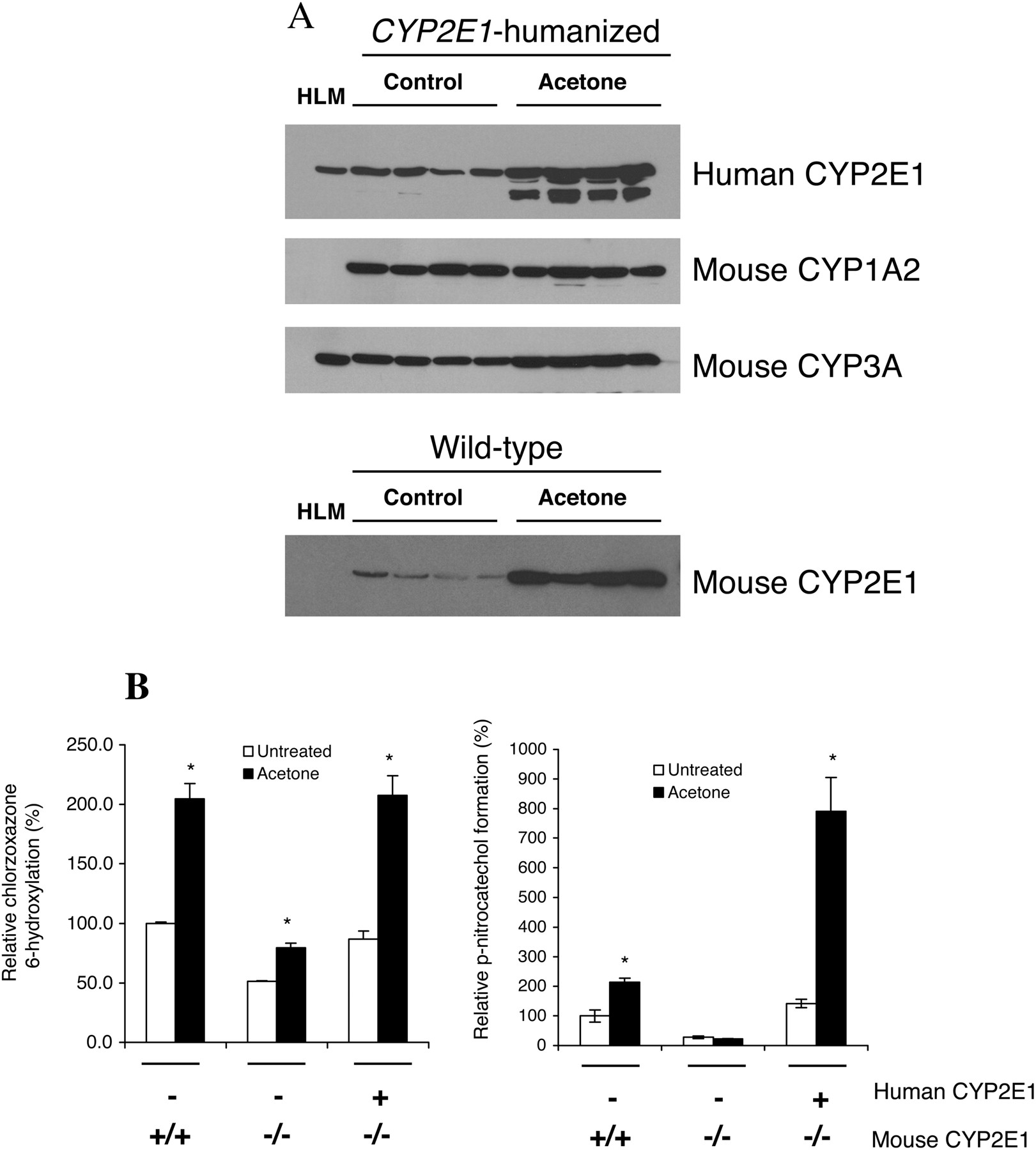
Induction of CYP2E1 Protein and Activity. Acetone induction of CYP2E1 is well characterized in rodent models with the mechanism established as stabilization of the CYP2E1 protein as opposed to direct gene transcriptional regulation (Song et al., 1986; Hong et al., 1987). To demonstrate the inducibility of human CYP2E1 in this CYP2E1-humanized mouse model, both wild-type and CYP2E1-humanized mice were treated with acetone. Using Western blot analysis, human CYP2E1 microsomal protein was induced by 2- to 3-fold, with an additional lower molecular weight protein also induced (Fig. 3A). Mouse CYP3A immunoreactive proteins were additionally induced by acetone; however, mouse CYP1A2 remained unchanged (Fig. 3A). In wild-type mice, CYP2E1 protein was induced by ∼5- to 6-fold following acetone treatment (Fig. 3A). Microsomal CYP2E1 activity measured by chlorzoxazone 6-hydroxylation was also correspondingly induced by 2- to 3-fold in both the CYP2E1-humanized and wild-type mice treated with acetone compared with no treatment (Fig. 3B). Using p-nitrophenol as a substrate to measure CYP2E1 activity, the formation of p-nitrocatechol was induced 2-fold in the wild-type mice; however, a 5- to 6-fold induction was observed in the CYP2E1-humanized mice (Fig. 3B).

Induction of CYP2E1 protein and activity in CYP2E1-humanized mice. A, Western blot analysis of liver microsomes from CYP2E1-humanized mice detected using monoclonal antibodies specific to human CYP2E1, rat CYP2E1, CYP1A2, and CYP3A. HLM, human liver microsomes. B, CYP2E1 enzymatic activity as measured by the relative percentage of chlorzoxazone 6-hydroxylation or p-nitrophenol O-hydroxylation in the liver microsomal fractions of untreated and acetone-treated wild-type, Cyp2e1-null, and CYP2E1-humanized mice. The relative activities were calculated based on the mean value of the untreated wild-type liver sample (the mean absolute values for this sample were 101.7 pmol/mg protein/min for chlorzoxazone 6-hydroxylation and 0.85 nmol/mg protein/min for p-nitrocatechol formation and were defined as 100%). Mice were administered 2% acetone in the drinking water for 7 days. Results shown are the means ± S.E.M. of pooled liver microsomes (n = 5). *, p < 0.05 compared with control.
Acetaminophen Toxicity. To determine whether any functional differences in response to APAP in the CYP2E1-humanized mice compared with wild-type mice exists, the mice were treated with increasing doses of APAP, and the liver toxicity was examined. Hepatocyte necrosis was centrilobular and marked according to the degree of necrosis (Table 1). Representative H&E-stained liver sections from these mice are shown in Fig. 4. At the lower APAP dose of 200 mg/kg, two of five wild-type mice exhibited mild-to-moderate degrees of centrilobular hepatocyte necrosis, whereas both the Cyp2e1-null and CYP2E1-humanized mice had no hepatic necrosis lesions detected histologically (Table 1). At the higher APAP dose of 400 mg/kg, all wild-type mice exhibited a moderate degree of centrilobular hepatocyte necrosis, whereas the Cyp2e1-null mice had no hepatic necrosis lesions detected histologically (Table 1). In the livers of CYP2E1-humanized mice treated with 400 mg/kg APAP, there was variation in the response from mild-to-severe degrees of centrilobular hepatocyte necrosis in the 10 mice examined at this dose (Table 1). No Cyp2e1-null mice were found dead 24 h following saline or APAP treatment; however, with the 400-mg/kg APAP dose, one CYP2E1-humanized mouse and two wild-type mice were found dead, presumably due to very severe hepatocyte necrosis leading to mortality.
Degree of centrilobular hepatic necrosis in mice treated with saline or APAP
The livers were examined histologically and classed according to the degree of centrilobular hepatocyte necrosis, marked as none, mild, moderate, or severe. Mice found dead 24 h after the APAP treatment were also noted. The numbers indicate how many mice were classed with which degree of hepatocyte necrosis per total number of mice examined in each group (total numbers of mice in each group, n = 5–10).

Histological analysis of livers of mice treated with acetaminophen. Representative H&E-stained liver sections are shown from wild-type, Cyp2e1-null, and CYP2E1-humanized mice treated with saline or 400 mg/kg APAP 24 h following injection. Liver necrosis is centrilobular; magnification, 400×.
Further confirmation of liver toxicity was demonstrated by measuring serum ALT, elevated levels of which serve as a marker for liver damage, cirrhosis, carcinoma, viral or toxic hepatitis, and obstructive jaundice. In wild-type mice 24 h following APAP administration, ALT was increased by approximately 8- and 12-fold in mice treated with 200 and 400 mg/kg, respectively, compared with saline. The elevated ALT levels after 200 and 400 mg/kg APAP were statistically significant (p = 0.026 and p = 0.005, respectively) (Fig. 5). There was great variability in the response to the 200-mg/kg APAP dosing in these wild-type mice, as shown by both the serum ALT levels (Fig. 5) and histological examinations (Table 1). In the CYP2E1-humanized mice, the levels of ALT were unchanged following 200 mg/kg APAP dosing but significantly elevated by approximately 12-fold following 400 mg/kg APAP (p = 0.002) (Fig. 5). In the Cyp2e1-null mice, there was no significant difference in levels of ALT between the APAP- and saline-treated mice (Fig. 5). In addition, there was no significant difference between the ALT levels in saline or 200 mg/kg APAP-treated wild-type and CYP2E1-humanized mice, with Student's t test values of p = 0.08 and p = 0.06, respectively (Fig. 5).

Serum alanine aminotransferase activity in mice treated with acetaminophen. Wild-type, Cyp2e1-null, and CYP2E1-humanized mice were treated with saline or 200 or 400 mg/kg APAP, and serum was collected after 24 h. The serum was analyzed for ALT activity, and the results are shown as mean ± S.D. of the -fold change compared with saline (n = 5). *, p < 0.05 compared with control.
To determine the P450 levels in the livers following APAP treatment, both protein and mRNA levels were examined (Fig. 6). In both the wild-type and CYP2E1-humanized mice, the levels of CYP2E1, CYP1A2, and CYP3A, all of which are known to contribute to APAP metabolism (Snawder et al., 1994b; Kostrubsky et al., 1997), were decreased with increasing APAP dosing. Levels of CYP1A2 protein and mRNA were unchanged by APAP dosing in the Cyp2e1-null mice (Fig. 6). CYP3A protein seemed unchanged by APAP dosing in the Cyp2e1-null mice (Fig. 6A); however, CYP3A11 mRNA was decreased with increasing APAP dosing (Fig. 6B).

Effect of acetaminophen on the cytochrome P450 levels in the liver. A, Western blot analysis of liver microsomes prepared from wild-type, Cyp2e1-null, and CYP2E1-humanized mice treated for 24 h with saline or 200 or 400 mg/kg APAP. HLM, human liver microsomes. Membranes were probed with monoclonal antibodies specific for human CYP2E1, rat CYP2E1, CYP1A2, and CYP3A. B, Northern blot analysis of RNA isolated from the livers of wild-type, Cyp2e1-null, and CYP2E1-humanized mice treated for 24 h with saline or 200 or 400 mg/kg APAP. Membranes were probed with CYP2E1, CYP1A2, CYP3A11, glutathione S-transferase-π, and 36B4.
Discussion
Transgenic mice expressing the human CYP2E1 gene were generated and bred into a mouse Cyp2e1-null background, thus creating humanized mice. Using this mouse model, the functional differences between human and mouse CYP2E1 can be directly compared. Human CYP2E1 protein and mRNA expression were expressed at high levels in the liver of CYP2E1-humanized mice, with the human CYP2E1 mRNA additionally detected in some extrahepatic tissues such as kidney, small intestine, and lung. The distribution pattern is similar to that reported in humans—a high level of CYP2E1 in the liver and low levels in other extrahepatic organs (Lieber, 1997). Human CYP2E1 activity was demonstrated in the livers of CYP2E1-humanized mice by the increased chlorzoxazone 6-hydroxylation and increased p-nitrocatechol formation compared with the level in Cyp2e1-null mice. Chlorzoxazone 6-hydroxylation has been commonly used to measure human CYP2E1 activity (Lucas et al., 1996, 1999; Haufroid et al., 2002). Measurement of chlorzoxazone and trimethadione metabolism by CYP2E1 has also been used as an indicator for liver diseases in vivo and in vitro (Dilger et al., 1997; Burckart et al., 1998; Mishin et al., 1998; Dupont et al., 2000). An alternative method for measuring CYP2E1 activity has been to analyze p-nitrophenol hydroxylation (p-nitrocatechol formation) (Zerilli et al., 1997). In this study, complete inhibition of chlorzoxazone 6-hydroxylation was not achieved due to the contribution of other P450s such as CYP1A2 to the metabolism of chlorzoxazone (Carriere et al., 1993; Ono et al., 1995). Similarly, the chlorzoxazone 6-hydroxylation activity detected in the Cyp2e1-null mice is contributed by other mouse P450s such as CYP1A2 (Carriere et al., 1993; Ono et al., 1995). The lack of complete inhibition of chlorzoxazone 6-hydroxylation demonstrated in this study is consistent with previous reports also using an anti-human CYP2E1 antibody (Peter et al., 1990; Ono et al., 1995). Using p-nitrophenol as substrate to measure CYP2E1 activity, a greater level of inhibition was achieved with the anti-human CYP2E1 antibody compared with inhibition of chlorzoxazone 6-hydroxylation activity in the CYP2E1-humanized mice. This suggests that p-nitrophenol may be a better substrate for assessing CYP2E1 activity in the CYP2E1-humanized mice.
Inducible human CYP2E1 was demonstrated in the CYP2E1-humanized mice using the known CYP2E1 inducer acetone. Chlorzoxazone 6-hydroxylation activity was induced by approximately 2-fold with acetone in both wild-type and CYP2E1-humanized mice, a finding comparable to the 1.3-fold increase in chlorzoxazone 6-hydroxylation reported for wild-type mice given acetone by the same treatment (Rosenberg and Mankowski, 1994). p-Nitrocatechol formation was similarly induced 2-fold in the wild-type mice treated with acetone; however, a significant 5- to 6-fold increase in p-nitrocatechol formation was seen in the CYP2E1-humanized mice treated with acetone, a result that could be explained by a species difference in response to acetone, although chlorzoxazone 6-hydroxylation activity was similar in these mice. In this study, the induction of CYP2E1 protein was shown to be ∼5- to 6-fold after acetone treatment in wild-type mice, which was consistent with the ∼5-fold increase seen in a previous study in wild-type mice by Western blot analysis (Forkert et al., 1994). In comparison, a lower induction of human CYP2E1 protein was seen (∼2- to 3-fold) in the CYP2E1-humanized mice treated with acetone. However, some lower molecular weight bands were observed to be additionally induced in these livers, suggesting that other proteins similar to CYP2E1 (and therefore recognized by the anti-human CYP2E1 antibody) were affected by acetone treatment; the identity of these other proteins are not yet known. Since acetone stabilizes the CYP2E1 enzyme against degradation, these lower weight bands are not likely to be degradation products.
The effect of APAP on the wild-type, Cyp2e1-null, and CYP2E1-humanized mice were compared. At a dose of 400 mg/kg APAP, liver necrosis was detected in wild-type but not Cyp2e1-null mice, as previously reported (Lee et al., 1996). The 400-mg/kg dose in the CYP2E1-humanized mice resulted in liver necrosis, which was marked as moderate-to-severe, whereas in wild-type mice moderate liver necrosis was identified. However, the serum ALT levels were elevated to approximately the same levels in both wild-type and CYP2E1-humanized mice treated with 400 mg/kg APAP, indicating a similar toxic response. There was variability in the liver necrosis seen in wild-type mice given 200 mg/kg APAP with normal-to-moderate liver damage, as reflected in the serum ALT levels. This result was consistent with previous reports showing significantly elevated serum ALT levels in wild-type mice given 200 mg/kg APAP (Lee et al., 1996). In contrast, the same 200-mg/kg dose given to the CYP2E1-humanized mice did not result in liver necrosis or elevated serum ALT. This functional difference in APAP response with 200 mg/kg could indicate a species difference between mouse and human CYP2E1, a possibility that remains to be examined. In humans, an APAP dose lower than 125 mg/kg results in rare liver toxicity, whereas a dose greater than 250 mg/kg is considered the minimum hepatotoxic dose. A dose of 350 mg/kg invariably causes severe liver damage in humans (Lewis and Paloucek, 1991). Consistent with previous reports describing how hepatotoxic doses of APAP decrease the levels of hepatic P450s (Thorgeirsson et al., 1976; Snawder et al., 1994b), levels of CYP2E1, CYP1A2, and CYP3A were shown to be decreased with increasing APAP dose in wild-type and CYP2E1-humanized mice in this study.
CYP2E1 is an effective generator of reactive oxygen species; therefore, CYP2E1-induced oxidative stress has been suggested to play a central role in alcohol-induced liver damage (Caro and Cederbaum, 2004). Most ethanol is metabolized by alcohol dehydrogenase; however, at elevated concentrations of ethanol and after chronic consumption of ethanol, CYP2E1 assumes a more important role (Caro and Cederbaum, 2004). Alcohol-induced liver injury is a multifactorial process involving several mechanisms, and the contribution of CYP2E1 to this injury requires further study (Caro and Cederbaum, 2004); however, some studies have suggested that CYP2E1 may not play a role in alcohol liver injury (Koop et al., 1997; Kono et al., 1999). A previous study demonstrated that in a transgenic mouse model expressing human CYP2E1 specifically in the liver, early alcohol-induced liver injury was evident following feeding with a nutritionally complete alcohol diet (Morgan et al., 2002). However, since this mouse line also contained endogenous mouse CYP2E1, the role of human CYP2E1 alone cannot explain this finding (Morgan et al., 2002). To clearly elucidate the role of human CYP2E1 in alcohol-induced liver injury, the CYP2E1-humanized model described herein would provide an invaluable model to investigate such questions.
Species differences between rodent and human CYP2E1 have been reported. Differences in the rate of trichloroethylene oxidation, a reaction catalyzed by CYP2E1, were observed between rats and humans (Lipscomb et al., 1998). Species differences in the rate of metabolism and distribution of 1,3-butadiene, a volatile organic compound used in the production of resins and plastics that is also oxidized by CYP2E1, was reported between human and rodents (Jackson et al., 2000). However, these reports of species differences in metabolic rates for CYP2E1 substrates depend on the CYP2E1 content of the preparation or system under investigation and therefore cannot be based on differences in the intrinsic activity of the enzyme between these species (Lipscomb et al., 2004). In this study, we have shown a difference in the hydroxylation of p-nitrophenol to p-nitrocatechol between wild-type and CYP2E1-humanized mice, suggesting a potential species difference in substrate metabolism, although chlorzoxazone metabolism was similar. In the CYP2E1-humanized mice, p-nitrocatechol formation from p-nitrophenol seems to be more effective in assessing CYP2E1 activity.
In contrast to experimental animal models, humans tend to show interindividual variations in P450-catalyzed reactions of drugs and chemicals, which have been associated with different susceptibilities of humans to toxicological and pharmacological effectors (Guengerich, 1989; Bolt et al., 2003). A number of genetic polymorphisms have been described for the human CYP2E1 gene, with consistent ethnic differences in CYP2E1 expression, mostly demonstrated between European and Japanese populations (Bolt et al., 2003).
By establishing a CYP2E1-humanized mouse model, where the human CYP2E1 gene is expressed in a mouse Cyp2e1-null background, functional differences in APAP metabolism have been shown. Transgenic animal models expressing human P450 genes were previously described that have been used to investigate the contribution of regulatory factors that control gene expression in vivo (Corchero et al., 2001; Granvil et al., 2003; Galijatovic et al., 2004). Such transgenic mouse models provide valuable tools for predicting drug metabolism, disposition, and drug-drug interactions of chemicals that are human CYP2E1 substrates.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.002402.
-
ABBREVIATIONS: P450, cytochrome P450; APAP, acetaminophen; NAPQI, N-acetyl-p-benzoquinone imine; kb, kilobase(s); ALT, alanine aminotransferase; BAC, bacterial artificial chromosome; bp, base pair(s); LC/MS/MS, liquid chromatography/tandem mass spectometry.
-
↵1 Current address: Department of Pharmaceutical Sciences, School of Pharmacy and Pharmaceutical Sciences, The State University of New York at Buffalo, Buffalo, NY.
-
↵2 Current address: Metabolic Disorders, Merck Research Labs, Rahway, NJ.
- Received September 16, 2004.
- Accepted November 30, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}