


Abstract
Caspofungin (CANCIDAS, a registered trademark of Merck & Co., Inc.) is a novel echinocandin antifungal agent used in the treatment of esophageal and invasive candidiases, invasive aspergillosis, and neutropenia. Available data suggest that the liver is a key organ responsible for caspofungin elimination in rodents and humans. Caspofungin is primarily eliminated by metabolic transformation; however, the rate of metabolism is slow. Accordingly, it was hypothesized that drug uptake transporters expressed on the basolateral domain of hepatocytes could significantly influence the extent of caspofungin uptake and subsequent elimination. In this study, experiments ranging from perfused rat livers to heterologous expression of individual hepatic uptake transporters were utilized to identify the transporter(s) responsible for the observed liver-specific uptake of this compound. Data from perfused rat liver studies were consistent with the presence of carrier-mediated caspofungin hepatic uptake, although this process appeared to be slow. To identify a relevant hepatic uptake transporter, we developed novel Tet-on HeLa cells expressing OATP1B1 (OATP-C, SLC21A6) and OATP1B3 (OATP8, SLC21A8), whose target gene can be overexpressed by the addition of doxycycline. A modest but statistically significant uptake of caspofungin was observed in cells overexpressing OATP1B1, but not OATP1B3. Taken together, these findings suggest that OATP1B1-mediated hepatic uptake may contribute to the overall elimination of this drug from the body.
Caspofungin is a semisynthetic, lipopeptide antifungal agent of the novel echinocandin class of compounds (Letscher-Bru and Herbrecht, 2003). Its antifungal activity is mediated by inhibition of synthesis of β-(1,3) glucan, an essential component in the cell wall of target organisms. Caspofungin has demonstrated potent antifungal activity in vitro against Candida and Aspergillus isolates (Bartizal et al., 1997) and in vivo efficacy in mouse models of disseminated aspergillosis and candidiasis (Abruzzo et al., 1997). Caspofungin has been successfully used in the clinical arena for the treatment of esophageal candidiases, candidemia, and invasive aspergillosis (Arathoon et al., 2002; Koss et al., 2002; Mora-Duarte et al., 2002). More recently, caspofungin has also been used as an empirical antifungal therapy for patients with persistent fever and neutropenia (Walsh et al., 2004). Due to its high molecular weight (1093; Fig. 1) and unfavorable lipophilicity, oral bioavailability was extremely low in animals. This drug is, thus, used parenterally in humans. Following intravenous administration to healthy human volunteers, hepatic elimination appeared to be the main route of removal for the parent drug in that only 2% of unchanged drug was recovered in human urine (Stone et al., 2002). The drug does not appear to be subject to metabolism by the cytochrome P450 system or other enzyme systems studied, and part of the metabolism is thought to be nonenzymatic hydrolysis (Sandhu et al., 2004; Stone et al., 2004b). Overall, the metabolism of caspofungin is slow and most of the plasma area under the curve is associated with distributional phases. These physiochemical and observed pharmacokinetic properties associated with caspofungin suggest a role of hepatic transporter(s) as a key determinant of its elimination.
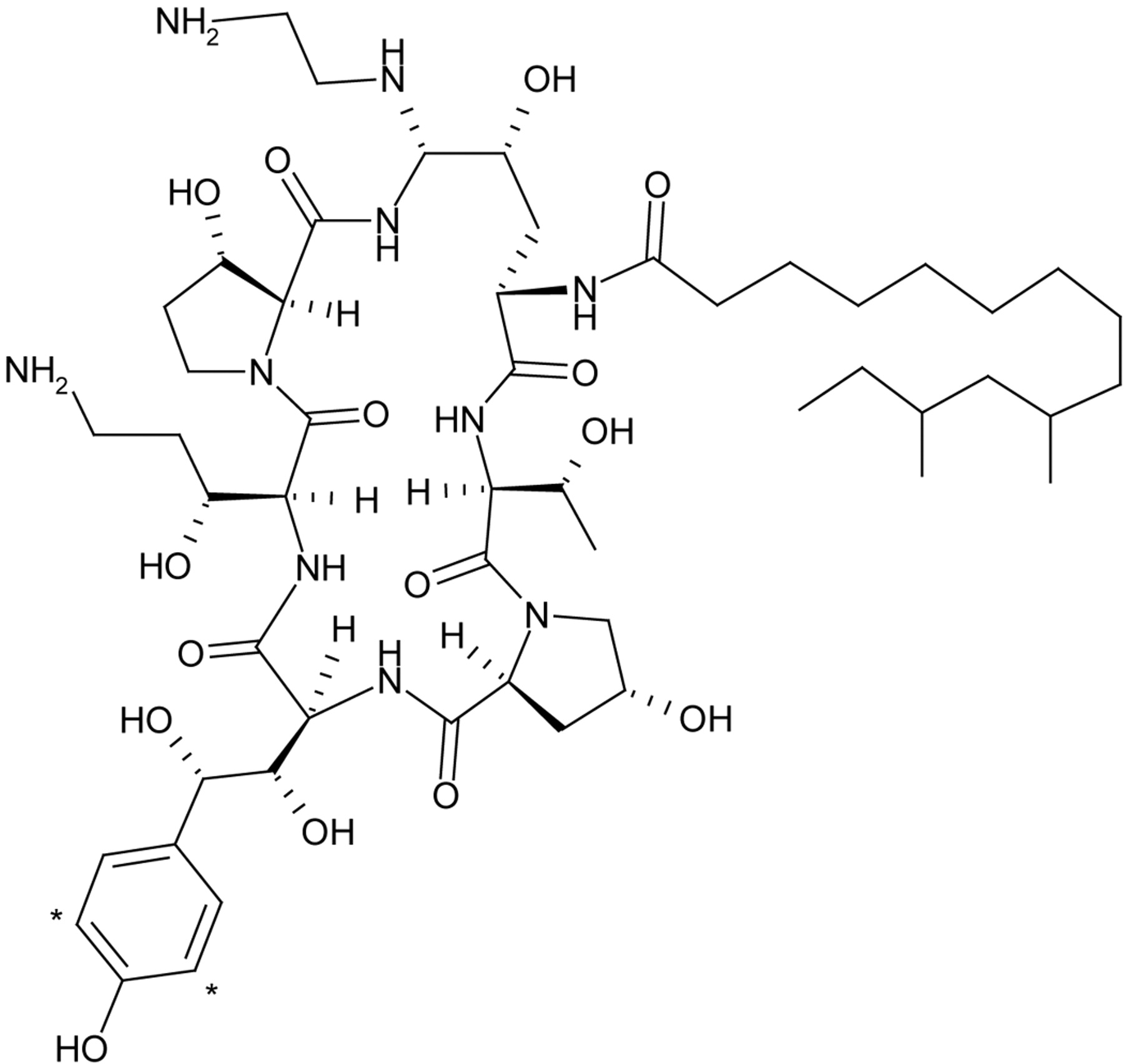
Structure of [3H]caspofungin. The symbol * denotes position of tritium label.
Since caspofungin is often utilized in the setting of severe life-threatening illnesses, a better understanding of the processes governing the disposition of this novel antifungal agent will enhance our knowledge of the potential caspofungin-drug interactions. In this study, a combination of in vivo animal models, in situ rat perfusion and in vitro heterologous transporter expression experiments, was carried out to determine the relative extent and contribution of hepatic caspofungin uptake and to show that the liver-specific transporter OATP1B1 (SLC21A6) may be a relevant transporter responsible for this uptake.
Materials and Methods
Chemicals. [3H]Caspofungin tris-trifluoroacetate salt in ethanol was synthesized by Labeled Compound Synthesis (Merck Research Laboratories, Rahway, NJ). The specific activities of the radiolabeled lots of caspofungin were ∼5 and 17 Ci/mmol and the radiochemical purities were ≥96%. Unlabeled caspofungin diacetate salt was synthesized by the Department of Process Research (Merck Research Laboratories). [3H]Taurocholate (2.0 Ci/mmol, >98% purity), [14C]tetraethylammonium (5.0 mCi/mmol, >99% purity), [3H]para-aminohippurate (3.95 Ci/mmol, >98% purity), and [3H]estradiol-17β-d-glucuronide (44 Ci/mmol, >97% purity) were obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). [G-3H]Vinblastine sulfate (15.5 Ci/mmol, >94% purity) and [3H]cholecystokinin 8 (84 Ci/mmol, >91% purity) were purchased from Amersham Biosciences UK, Ltd. (Little Chalfont, Buckinghamshire, UK). The molecular weight of the free base was used for the preparation of all stock solutions. All other solvents and reagents were of either high-performance liquid chromatography or analytical grade and, unless otherwise stated, were obtained from Sigma-Aldrich (St. Louis, MO).
Time Course of [3H]Caspofungin in the Liver. To establish the time course of radioactivity in hepatic tissue, rat livers were collected at 0.5, 1, 2, 6, 12, 24, 48, 120, and 288 h after i.v. administration of 2 mg/kg [3H]caspofungin (1 ml/kg). Male Sprague-Dawley rats from Taconic Farms (Germantown, NY), weighing approximately 200 to 350 g, were used for the study. A cannula was implanted in the right jugular vein for dose administration. The surgical procedure was performed under pentobarbital anesthesia (40–50 mg/kg, intraperitoneally) 1 day before the experiment. [3H]Caspofungin was administered as a bolus intravenously via the jugular vein. Liver samples were homogenized in water (2- to 5-fold dilution). Total radioactivity in the liver was determined by adding aliquots of the homogenate (1.0 ml) for combustion on Combusto-cones. The cones were allowed to dry overnight in a fume hood. The samples were combusted in a PerkinElmer Oximate 80 model 307 sample oxidizer. The resulting 3H2O was trapped and radioactivity was measured by liquid scintillation spectrometry.
Hepatic Uptake of [3H]Caspofungin in Perfused Rat Liver. An in situ perfused rat liver system was used to investigate the mechanism underlying the hepatic uptake of caspofungin. The surgical procedure and the method used were as described by Pang (1984). A commercially available Two/Ten Perfuser (MX International, Aurora, CO), equipped with two reservoir units, was used for perfusions. The perfusate consisted of 0.3% glucose and bovine serum albumin (Sigma-Aldrich) at various concentrations (0.1–4.0%) in Krebs-Henseleit bicarbonate buffer (pH 7.4). A peristaltic pump was used to deliver the perfusate into the liver at a flow rate of 30 ml/min/10 g of liver. The bile duct was also cannulated and bile samples were collected over 10-min intervals.
To assess the hepatic uptake of [3H]caspofungin, rat livers were first perfused with drug-free perfusate in a single-pass mode to stabilize the system. The perfusate (100 ml) from a second reservoir containing 10 μg/ml [3H]caspofungin was subsequently recirculated through the liver for 60 min. Perfusate samples were collected directly from the reservoir. At the end of the perfusion, livers were removed and homogenized as described previously. Radioactivity in the perfusate, bile, and liver homogenates was determined by liquid scintillation spectrometry.
Adsorption of [3H]Caspofungin on the Surface of Liver Cells. To enable determination of the extent of adsorption of [3H]caspofungin on the liver cell surface, rat livers, taken from a 1-h in situ perfusion study or at 0.5 and 24 h after i.v. administration of [3H]caspofungin at 2 mg/kg, were reperfused with a drug-free 4% albumin solution for 1 h to wash out the radioactivity associated with nonspecific binding on cell membranes. At the end of the wash, the remaining radioactivity in each liver was measured as described previously.
Effect of Protein Binding on Hepatic Uptake of [3H]Caspofungin. Due to the high nonspecific binding properties of [3H]caspofungin (the free fraction was approximately 4% and independent of drug concentrations from 0.1 to 100 μg/ml), plasma protein binding was determined by an ultracentrifugation method as described by Legg and Rowland (1987). The effect of plasma protein binding on the hepatic uptake of [3H]caspofungin was investigated with the perfusate containing various albumin concentrations (0.1–4.0%), whereas the drug concentration was maintained at 10 μg/ml. The unbound fraction of [3H]caspofungin in the perfusate was determined by ultracentrifugation.
Expression Plasmids. Preparation of expression plasmids containing cDNAs for human NTCP, OATP1A2 (OATP-A), OATP1B1, rat Oatp1a1 (oatp1), Oatp1a4 (oatp2) and oct1 has been described previously (Tirona et al., 2003). Human OATP1A2 and rat Oatp1a5 (oatp3) cDNAs were kindly provided by Drs. Peter Meier (University Hospital, Zurich, Switzerland) and Paul Dawson (Wake Forest, NC). All other expression plasmids were obtained by reverse transcription-polymerase chain reaction followed by insertion of cDNAs into pEF6/V5-His vector (Invitrogen, Carlsbad, CA). All plasmids were sequence-verified and, when expressed in cells, were shown to be transport-competent toward prototypical substrates.
Transient Transfection and Uptake Transport Assays. Transient transfection assays were performed using the recombinant vaccinia virus (VTF-7) expression method detailed previously (Cvetkovic et al., 1999). Briefly, human cervical carcinoma cells (HeLa) (American Type Culture Collection, Manassas, VA) were seeded into 12-well plates, infected with vaccinia virus, and then transfected with expression plasmids or vector control using Lipofectin reagent (Invitrogen). Sixteen hours thereafter, cells were washed with transport media (Optimem; Invitrogen) and treated with the radiolabeled drug in the presence or absence of transport inhibitors. At various time intervals, cells were washed three times with ice-cold medium and then lysed with 1% SDS. Retained cellular radioactivity was quantified by liquid scintillation spectrometry.
Stable and Inducible Expression of Human OATP1B1 and OATP1B3 (OATP8) in Tet-on HeLa Cells and Uptake Transport Assays. The Tet-on gene expression system allows the high-level expression of target genes in a regulated fashion upon the addition of doxycycline. The Tet-on HeLa cell line (stably transfected with the tetracycline-controlled reverse transactivator) was purchased from BD Biosciences Clontech (Palo Alto, CA). Pilot studies were carried out to determine the optimal plating density and to titrate the selection agent, hygromycin (Hyg). Tet-on HeLa cells were then transfected with the response plasmid constructs containing human OATP1B1 or OATP1B3 using Lipofectin reagent (Invitrogen). The transfected cells were plated at the optimal density of 2 × 105 cells in 10-cm culture dishes, and stable cell lines were selected in the presence of Hyg (200 μg/ml). After 2 to 3 weeks, large and healthy colonies resistant to Hyg were isolated and tested for functional expression of target genes upon the addition of doxycycline (1 μg/ml, 48 h). Tet-on cell lines expressing OATP1B1 or OATP1B3 were then used for the uptake transport assays as described in the previous section.
In Vitro Inhibition of Caspofungin on Cellular Accumulation of [3H]VBL in KB-V1 and KB-3-1 Cells. The drug-sensitive human epidermoid carcinoma cell line KB-3-1 and its vinblastine (VBL)-selected multidrug-resistant variant KB-V1 were provided by Dr. Michael M. Gottesman (National Cancer Institute, Bethesda, MD) and used under license agreement. The inhibition of caspofungin on cellular accumulation of [3H]VBL in KB-V1 and KB-3-1 cells was carried out as described previously (Yamazaki et al., 2001). Briefly, to prepare cell monolayers, 5 ml of cell suspension containing 106 cells in Dulbecco's modified Eagle's medium (low glucose without phenol red; Invitrogen) with 10% fetal calf serum was added to a six-well plate (NUNC A/S, Roskilde,, Denmark). After an overnight incubation at 37°C, cell monolayers were washed once with a transport medium consisting of serum-free Hanks' balanced salt solution with 10 mM Hepes (pH 7.4). An aliquot of 2 ml of transport buffer containing [3H]VBL (1 μM) in the presence or absence of caspofungin (10 or 100 μM) was then added to each well. For comparison, cellular accumulation of [3H]VBL in the presence of cyclosporin A (10 μM), a typical P-glycoprotein (P-gp) inhibitor, was examined simultaneously. After incubation at 37°C for 4 h, the medium was removed and the cell monolayers were rinsed twice with 5 ml of cold phosphate-buffered saline. Cells were lysed in 1 ml of 1 N NaOH and transferred to vials containing 0.5 ml of 2 N HCl and 15 ml of scintillation cocktail. The total radioactivity was measured by liquid scintillation spectrometry.
Data Fitting and Statistical Analysis. Analysis was performed using Student's t test or ANOVA followed by the Newman-Keuls test. A value of p < 0.05 was considered statistically significant.
Results
Tissue Distribution of [3H]Caspofungin in Rats. The distribution of [3H]caspofungin was studied extensively in rats. The results from the whole-body exposure of [3H]caspofungin in rats are presented in a separate paper by the authors (Stone et al., 2004b). Briefly, total radioactivity (i.e., attributed to both drug and metabolites) was widely distributed in the body of rats following a 2 mg/kg i.v. dose of [3H]caspofungin. At 30 min, the tissues with the highest levels of radioactivity were liver, lung, kidney, and spleen. In general, the concentration of radioactivity peaked within 2 h of dosing and declined thereafter, except in the liver. A detailed follow-up study was designed to investigate the time course of radioactivity in the liver of rats. Data from this study following a 2 mg/kg i.v. administration of [3H]caspofungin to rats are presented in Table 1. The initial hepatic uptake of radioactivity was slow, with approximately 8% of the dose in the liver at 30 min, and the radioactivity levels peaked at 24 to 48 h (approximately 35–39%). Elimination of radioactivity from the liver also occurred slowly with approximately 14.2 and 2.8% of the dose still in the liver at 120 h (day 5) and 288 h (day 12), respectively. These results suggest that hepatic uptake and elimination of caspofungin are very slow processes and that the equilibration between blood and liver tissue is not established rapidly. The liver thus appears to play an important role in the overall disposition of caspofungin.
Time course of radioactivity in the liver of rats following an intravenous dose of 2 mg/kg [3H]caspofungin
The values are presented as mean percentage (n = 2–4) of dose in the liver.
Hepatic Uptake of [3H]Caspofungin in Rats. To elucidate the underlying mechanism responsible for the slow liver uptake and elimination, the hepatic uptake of [3H]caspofungin was studied using an in situ rat liver perfusion preparation. When [3H]caspofungin (10 μg/ml) was recirculated through the liver in a 0.5% albumin solution, radioactivity in the perfusate declined in a biphasic fashion; a rapid decrease in the initial 5 min was followed by a very slow decline thereafter (Fig. 2). The radioactivity retained in the liver at the end of a 60-min perfusion accounted for approximately 20% of the dose. The biliary excretion of radiolabeled drug was negligible within 1 h of the perfusion. This result suggests that the hepatic uptake of caspofungin is a two-step process with a rapid initial phase, possibly due to the adsorption of the drug to the cell surface, followed by a slow transport process of the drug across the cell membrane (Xu et al., 1996). This biphasic trend is consistent with earlier observations from the tissue distribution study (Stone et al., 2004b) as well as with data presented in Table 1 indicating that the time course of hepatic uptake and elimination of [3H]caspofungin is a slow process. Additional studies showed that the amount of radioactivity retained in the perfused rat liver was highly dependent on the unbound fraction in the perfusate; i.e., the higher the unbound fraction of caspofungin in the perfusate, the greater the amount of drug retained in the liver (Table 2). These findings support the cell surface hypothesis and also indicate that binding of caspofungin to albumin affects the drug available for hepatic uptake in that the initial adsorption step in this uptake depends upon the free fraction of caspofungin present in the perfusate. Furthermore, the majority of the radioactivity in the liver (approximately 81%) following the initial rapid uptake process was removed by reperfusing the liver with a drug-free, 4% albumin solution (Table 3), presumably via competitive binding.

[3H]Caspofungin radioactivity in the perfusate from the in situ rat liver perfusion study. Rat livers were recirculated with 100 ml of perfusate containing 0.5% albumin and 10 μg/ml [3H]caspofungin.
Effect of protein concentration on the hepatic uptake of [3H]caspofungin in the perfused rat liver
Radioactivity in the liver values are the mean ± S.D. (n = 3). The total concentration of [3H]caspofungin added to the perfusate was 10 μg/ml.
Percentage of dose of [3H]caspofungin in rat livers in situ and in vivo pre- and postwash with perfusate containing albumin
For the in situ group, rat livers were perfused with [3H]caspofungin (10 μg/ml) in a recirculated mode for 60 min. For the in vivo group, rats received an intravenous bolus of [3H]caspofungin (2 mg/kg) and livers were sampled 0.5 and 24 h postdosing. For the washout, livers were perfused in a single-pass mode for 60 min with the perfusate containing a high concentration of albumin (4%). Data shown are the mean ± S.D. (n = 3).
In additional experiments, the extent of radioactivity removed from the liver of rats dosed in vivo and subsequently perfused in situ with a drug-free, 4% albumin solution was dependent on the length of time elapsed between dosing and in situ perfusion. At 30 min following i.v. administration of [3H]caspofungin (2 mg/kg) to rats, the majority (66%) of the hepatic radioactivity was removed by subsequent perfusion of the liver with a 4% albumin solution. A considerably smaller fraction (19%) of radioactivity, however, was removed from the liver at 24 h postdose (Table 3). Collectively, both in situ and in vivo results are consistent with the hypothesis that the hepatic uptake of caspofungin is a two-step process involving an initial rapid binding of the drug to the cell surface that is followed by a slow mechanism of transport into the cell.
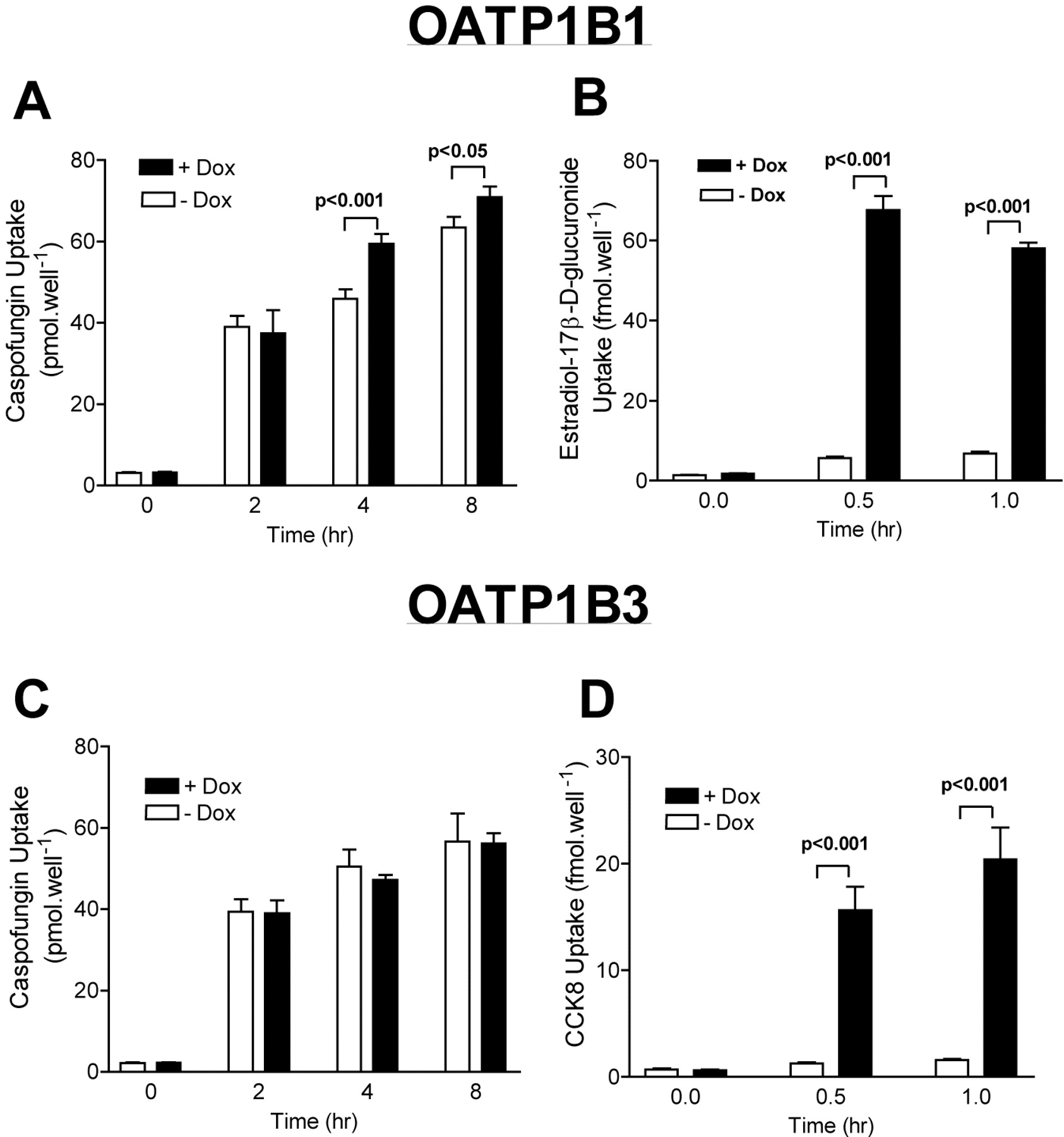
Caspofungin Uptake Is Mediated by OATP1B1. To identify the proteins responsible for the hepatic uptake of caspofungin, transport experiments were performed in HeLa cells transiently transfected with a variety of cDNAs coding for members of human (OATP1A2, OATP2B1, OATP1B1, OATP4A1, and OATP1B3) and rat Oatp (Oatp1a1, 1a4, and 1a5), OAT (OAT1 and OAT3), OCT (OCT1 and 2), and NTCP families. When tested using a recombinant vaccinia system, a statistically significant difference in caspofungin uptake (up to 60 min) was not observed for any of the tested transporters, although a trend for greater cellular caspofungin accumulation was observed in cells expressing OATP1B1. Because data from the perfused liver experiments suggested that the absolute amount of caspofungin taken up by the liver is relatively modest within 1 h, we created and utilized Tet-on HeLa cells, a cell line expressing a tetracycline-responsive nuclear receptor (Tet-on), to express OATP1B1 and OATP1B3. Unlike the recombinant vaccinia system, in which the system is optimized for assessing function during a defined time period after target gene transfection, the Tet-on HeLa cells can be used to assess transport function over long periods of time. Using this system, caspofungin uptake appeared to be enhanced in the Tet-on HeLa-OATP1B1 cell line, but not in Tet-on HeLa-OATP1B3 cells (Fig. 3). The absolute amount transported, however, was modest for caspofungin, even with the Tet-on HeLa cells. Thus, the affinity (Km) for caspofungin uptake by OATP1B1 could not be accurately determined due to the low apparent transport efficiency.

[3H]Caspofungin uptake by Tet-on HeLa cells expressing OATP1B1 and OATP1B3. Stable Tet-on HeLa cells expressing OATP1B1 or OATP1B3 in an inducible manner by the addition of doxycycline were tested for caspofungin uptake (A and C). Cellular uptake of prototypical substrates for OATP1B1 (estradiol-17β-d-glucuronide; B) and for OATP1B3 (cholecystokinin 8; D) is also shown. Data are shown as the mean ± S.E. (n = 4) from a representative experiment performed on two separate occasions.
Evaluation of Caspofungin as a Potential Inhibitor of Hepatic Transporters. To further evaluate the interactions between caspofungin and other hepatic transporters, transport inhibition studies were performed. OATP1B1-specific transport of the prototypical substrate estradiol-17β-d-glucuronide was inhibited by caspofungin, although a significant effect was seen only at a high concentration of 100 μM. A similar type of effect was observed for NTCP-mediated uptake of [3H]taurocholate, hOCT1-mediated uptake of [14C]tetraethylammonium, and hOAT1-mediated uptake of [3H]para-aminohippurate (Fig. 4). Lovastatin, ursodeoxycholic acid, quinidine, and indomethacin were used as prototypical inhibitors of OATP1B1, NTCP, OCT1, and OAT1, respectively. These findings suggest that caspofungin may be a relatively weak inhibitor of multiple systems.

Inhibitory effect of caspofungin on uptake transporters. OATP1B1, NTCP, OCT1, and OAT1 were expressed using a recombinant vaccinia system. Inhibition of prototypical substrate uptake by a known inhibitor of each transporter or caspofungin was assessed. Data are shown as the mean ± S.E. of experiments performed on two separate occasions (n = 2). *, p < 0.05, **, p < 0.01, *** p < 0.001 compared with control (no added inhibitor).
Inhibition of Caspofungin on P-gp Activity. The inhibitory effect of caspofungin on P-glycoprotein activity, as assessed using [3H]vinblastine efflux, appeared to be minimal. [3H]VBL accumulation in KB-V1 cells was 7% of that in the control KB-3-1 cells. Cyclosporin A markedly increased (ca. 17-fold) the cellular accumulation of [3H]VBL in KB-V1 cells to the levels comparable to those in control KB-3-1 cells. In contrast, caspofungin (10 and 100 μM) displayed minimal effects on the cellular accumulation of [3H]VBL in KB-V1 cells (Fig. 5).

Inhibitory effect of caspofungin on P-glycoprotein. Effects of caspofungin on the cellular accumulation of [3H]vinblastine (1 μM), a positive marker substrate of P-gp, in KB cell lines at 4 h. The concentrations used were 10 μM for cyclosporin A (a typical inhibitor of P-gp-mediated transport), and 10 μM and 100 μM for caspofungin. Data are shown as mean ± S.E. (n = 4). **, p < 0.01, ***, p < 0.001 compared with KV-V1 control.
Discussion
It is now increasingly recognized that structural complexities which add additional specificity or potency to a drug must be made with the recognition that such changes can lead to unexpected or unfavorable pharmacokinetic profiles of the drug. Remarkable advances in our understanding of drug-metabolizing enzymes, especially members of the cytochrome P450 superfamily, and their role in drug metabolism have resulted in the application of such knowledge to the design of drugs that are less likely to be dependent on certain cytochrome P450 enzymes or that, in some cases, bypass such metabolic steps. However, it now appears that other processes, particularly drug transporters expressed in organs such as the liver, intestine, and kidney, can become a major pathway of drug disposition. In this study, the hepatic processes governing the uptake of caspofungin were characterized in vivo and in situ as well as by assessing the role of various transporters. Caspofungin is a semisynthetic lipopolypeptide produced from a fermentation product of the fungus Glarea lozoyensis (Letscher-Bru and Herbrecht, 2003). The drug molecule is large and structurally very complex (Fig. 1). In addition, this drug is known to be water-soluble and to have poor oral bioavailability attributed to poor intestinal absorption, which might be due to a combination of the large molecular weight and low lipophilicity (log P of –1.2) of the molecule. Preliminary pharmacokinetic studies in several animal species indicated that caspofungin possesses slow plasma clearance (Hajdu et al., 1997). The plasma clearance in humans was also observed to be low following single-dose administration of the compound (Stone et al., 2002). Disposition studies in human subjects indicated that the metabolism and excretion of caspofungin were very slow processes with little excretion or biotransformation taking place in the first few days following drug administration (Stone et al., 2004b). Although very little metabolism or excretion occurred early on, plasma concentrations of caspofungin dropped by more than an order of magnitude in the first 2 days postdose (Stone et al., 2002), suggesting that plasma pharmacokinetics are affected by nonelimination processes, such as tissue distribution.
Metabolism of caspofungin is thought to occur, partly, via nonenzymatic hydrolysis. The known metabolic pathways of caspofungin further support the involvement of these nonenzymatic processes (Balani et al., 2000; Sandhu et al., 2004). The presence in the liver of a large percentage of the intravenous dose administered to rats (Table 1; Stone et al., 2004b) indicates that the liver plays an integral role in the metabolism of caspofungin. However, since most of the metabolism of caspofungin is attributed to nonenzymatic hydrolytic processes, metabolism can likely occur in other tissues as well.
In this study, we assessed the hepatic uptake of caspofungin in rats in vivo as well as by utilizing an in situ rat liver perfusion model. The results indicated that the hepatic uptake and elimination of caspofungin were slow processes (Table 1; Fig. 2) and supported the pharmacokinetic and disposition profiles observed in vivo. This finding was also consistent with the observed long elimination half-life and time to reach steady state for this drug in humans (Stone et al., 2002). Accordingly, we hypothesized that liver-specific transporter(s) might be involved in the hepatic uptake of this drug. To identify such a mechanism, cell lines with heterologous expression of various transporters were utilized. Initially, we expressed an array of transporters known to be expressed in the liver such as OATP2B1 (OATP-B), OATP1B1, OATP1B3, OCT1, and the bile acid transporter NTCP, using a recombinant vaccinia expression system. Using this system, we observed no discernable difference in the uptake of caspofungin by any of the transporters tested, relative to the control vector transfected cells. It should be noted that the vaccinia-based expression model is a transient expression system, optimized for peak expression within a few hours of starting the experiments. If the rate of uptake is slow, it is possible that a statistically significant uptake may not be observed, since the efficiency of transport/cellular viability is known to decline over time (Blakely et al., 1991).
To study relevant transport processes over prolonged periods of time, we then developed novel stable cell lines in which the gene of interest can be expressed in a regulated fashion. Specifically, a HeLa cell line expressing a tetracycline-responsive nuclear receptor (Teton) was utilized to transduce the liver-specific OATP transporters, OATP1B1 and OATP1B3. These transporters were selected for use in the creation of a Tet-on-based stable cell line since these transporters are liver-specific and have been shown to possess broad substrate specificities including many drugs in clinical use. A number of laboratories, including ours, have shown that rifampin is transported by OATP1B1 and OATP1B3 (Cui et al., 2001; Vavricka et al., 2002; Tirona et al., 2003). Furthermore, rifampin appears to be a potent inhibitor of OATP1B1 (Tirona et al., 2003). Moreover, these data are consistent with the clinical studies, which have shown that coadministration of rifampin to patients can acutely elevate caspofungin plasma levels (Stone et al., 2004a). We have shown that the Tet-on-based OATP1B1 and OATP1B3 cells are functional in that both are able to avidly transport prototypical substrates such as estradiol-17β-d-glucuronide and cholecystokinin 8. Interestingly, when uptake of caspofungin was studied for prolonged time periods, we observed a modest but statistically significant uptake of the drug by cells expressing OATP1B1, but not OATP1B3 (Fig. 3). However, due to the remarkably slow uptake process, we were not able to define relevant transport kinetics in terms of Km and Vmax. We then assessed a potential inhibitory effect of caspofungin on hepatic transporters such as OATP1B1, NTCP, OCT1, and renal organic anion transporter OAT1 and the well known drug efflux pump, P-glycoprotein. Caspofungin appeared to be a modest inhibitor of the uptake transporters tested since, at the higher concentration of 100 μM, significant inhibition of OATP1B1, NTCP, OCT1, and OAT1 was observed (Fig. 4). Similarly, caspofungin appeared to possess only a minimal inhibitory effect on P-gp (Fig. 5). Since the plasma concentrations of caspofungin at clinically effective doses are 1 to 10 μM, a clinically relevant inhibition of the above-mentioned transporters appears unlikely, assuming such in vitro data can be extrapolated to the in vivo situation. It is also possible that other processes such as endocytosis/pinocytosis may have a role in the hepatic uptake of this compound. However, observed clinical data showing a nearly 2-fold increase in the plasma level of caspofungin after rifampin (a known OATP1B1 substrate and inhibitor) coadministration suggest that OATP1B1 may in fact be relevant to the in vivo disposition of this compound (Stone et al., 2004a). Moreover, we are not aware of any studies that suggest that rifampin can acutely inhibit a hepatic endocytotic pathway(s). We have noted that in human liver, OATP1B1 is highly expressed, and when assessed using Western analysis, the proportion of the highly glycosylated/larger molecular weight form of the transporter is far greater than that observed using in vitro expression systems (unpublished data). Therefore, another possibility to account for the moderate and slow rate of uptake of caspofungin observed in this study could be the fact that currently available in vitro systems, although useful, underestimate the relative expression or function of OATP1B1 in vivo. However, if OATP1B1 is a major determinant of caspofungin hepatic uptake in vivo, a potential source of intersubject variability could result from coadministration of compounds that inhibit and/or induce OATP1B1 such as rifampin, thereby affecting the pharmacokinetics of caspofungin and leading to possible drug-drug interactions. Another source of intersubject variability in the elimination of this compound may relate to single nucleotide polymorphisms (SNPs) in this transporter. We have reported recently that OATP1B1 has a number of SNPs (Tirona et al., 2001). Furthermore, a number of functionally relevant SNPs in OATP1B1 were found to be relatively common among a population of European and African-American subjects (Tirona et al., 2001).
In conclusion, we demonstrate that the hepatic uptake of the novel antifungal agent caspofungin is a remarkably slow process, likely mediated, at least in part, by the uptake transporter OATP1B1. Accordingly, targeted in vitro and in vivo studies of drug transporters are likely to further enhance our understanding of the mechanisms of drug disposition and some of the observed drug-drug interactions.
Acknowledgments
We thank Y. Tang and F. DeLuna of Merck Research Laboratories for synthesis of radiolabeled caspofungin and for assistance with animal studies, respectively.
Footnotes
-
This work was supported in part by United States Public Health Service Grants GM54724, GM31304, and a Grant-in-Aid from Merck Research Laboratories.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.104.003244.
-
ABBREVIATIONS: NTCP, sodium-dependent taurocholate-cotransporting polypeptide; OAT, organic anion transporter; OATP, organic anion-transporting polypeptide; OCT, organic cation transporter; Hyg, hygromycin; P-gp, P-glycoprotein; VBL, vinblastine; SNP, single nucleotide polymorphism.
-
↵1 Current address: Department of Drug Safety and Metabolism, Wyeth Pharmaceuticals, 1 Burtt Rd., Andover, MA.
-
↵2 Current address: Department of Pharmacokinetics and Drug Metabolism, Amgen, Thousand Oaks, CA.
- Received December 10, 2004.
- Accepted February 14, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}