


Abstract
The intestinal efflux transporter breast cancer resistance protein (BCRP) restricts the absorption of rosuvastatin. Of the transporters important to rosuvastatin disposition, fostamatinib inhibited BCRP (IC50 = 50 nM) and organic anion-transporting polypeptide 1B1 (OATP1B1; IC50 > 10 μM), but not organic anion transporter 3, in vitro, predicting a drug-drug interaction (DDI) in vivo through inhibition of BCRP only. Consequently, a clinical interaction study between fostamatinib and rosuvastatin was performed (and reported elsewhere). This confirmed the critical role BCRP plays in statin absorption, as inhibition by fostamatinib resulted in a significant 1.96-fold and 1.88-fold increase in rosuvastatin area under the plasma concentration–time curve (AUC) and Cmax, respectively. An in vitro BCRP inhibition assay, using polarized Caco-2 cells and rosuvastatin as probe substrate, was subsequently validated with literature inhibitors and used to determine BCRP inhibitory potencies (IC50) of the perpetrator drugs eltrombopag, darunavir, lopinavir, clopidogrel, ezetimibe, fenofibrate, and fluconazole. OATP1B1 inhibition was also determined using human embryonic kidney 293–OATP1B1 cells versus estradiol 17β-glucuronide. Calculated parameters of maximum enterocyte concentration [Igut max], maximum unbound hepatic inlet concentration, transporter fraction excreted value, and determined IC50 value were incorporated into mechanistic static equations to compute theoretical increases in rosuvastatin AUC due to inhibition of BCRP and/or OATP1B1. Calculated theoretical increases in exposure correctly predicted the clinically observed changes in rosuvastatin exposure and suggested intestinal BCRP inhibition (not OATP1B1) to be the mechanism underlying the DDIs with these drugs. In conclusion, solitary inhibition of the intestinal BCRP transporter can result in clinically significant DDIs with rosuvastatin, causing up to a maximum 2-fold increase in exposure, which may warrant statin dose adjustment in clinical practice.
Introduction
Breast cancer resistance protein (BCRP; encoded by ABCG2) is one of the two major clinically relevant human ATP-binding cassette (ABC)–transporter proteins (the other being P-glycoprotein; ABCB1) that use ATP hydrolysis to efflux drugs and xenobiotics out of cells (Giacomini et al., 2010). BCRP is ubiquitously expressed in the intestine (apical brush border membrane of enterocytes), liver (canalicular membrane of hepatocytes), and kidney (apical brush border membrane of renal proximal tubule cells) and can affect the absorption and elimination of drugs that are substrates of this transporter, ultimately defining their exposure (Mao and Unadkat, 2015).
The important role that BCRP plays in drug disposition has been demonstrated clinically in pharmacogenetic studies investigating the ABCG2 single nucleotide polymorphism c.421C>A (Q141K, rs2231142), in which the impaired transport function phenotype (421CA or AA) resulted in increased plasma exposures of several BCRP substrate drugs, including rosuvastatin, atorvastatin, fluvastatin, and diflomotecan (Sparreboom et al., 2004; Keskitalo et al., 2009; Giacomini et al., 2013). Such observations also suggest that interindividual differences in BCRP function due to pharmacogenetics likely contribute to variability in bioavailability (absorption), exposure [area under the plasma concentration–time curve (AUC) and maximum plasma concentration (Cmax)], and efficacy of drugs that are BCRP substrates (Giacomini et al., 2010, 2013). Indeed, it has recently been reported that the observed ethnic difference and variability in exposure of the BCRP substrates rosuvastatin and atorvastatin between Caucasian and Asian populations can be partly explained by the higher frequency of the ABCG2 c.421C>A polymorphism, alongside a hypothesized lower intrinsic BCRP activity, in Asian populations. The resulting overall increase in statin absorption gives rise to the higher plasma exposures observed clinically in Asian compared with Caucasian subjects (Birmingham et al., 2015a).
Aside from genetic polymorphisms, concomitant administration of a drug that is an inhibitor of BCRP can also modulate the pharmacokinetics of a transported substrate, resulting in a drug-drug interaction (DDI), which may lead to toxicity due to increased exposure, or altered efficacy. DDIs attributed to BCRP have been observed clinically between topotecan and elacridar, resulting in increased topotecan absorption (Kruijtzer et al., 2002); between methotrexate and benzimidazoles, resulting in delayed renal elimination of methotrexate (Breedveld et al., 2004); and, in part, between rosuvastatin and cyclosporine, resulting in increased rosuvastatin absorption (Simonson et al., 2004; Tweedie et al., 2013). As highlighted earlier, there is a persuasive body of evidence that supports the clinical importance of the BCRP efflux transporter in drug development.
Rosuvastatin calcium is an 3-Hydroxy-3-methylglutaryl–coenzyme-A reductase inhibitor (statin) that has been developed to treat dyslipidemia by reducing low-density lipoprotein cholesterol (Crestor; AstraZeneca, Cambridge, UK). The critical disposition pathways of rosuvastatin and their individual contributions to overall clearance (fraction excreted values; ƒe), determined by Elsby et al. (2012) and derived from clinical human mass balance, pharmacogenetic, and DDI evidence, are shown in Fig. 1. These critical pathways include the intestinal BCRP efflux transporter as the rate-determining barrier to rosuvastatin absorption (fraction absorbed = 0.5; therefore, BCRP ƒe = 0.5), the hepatic uptake transporter organic anion-transporting polypeptide 1B1 (OATP1B1) responsible for hepatic elimination, and the renal uptake transporter organic anion transporter 3 (OAT3) responsible for the active renal secretion of rosuvastatin (Elsby et al., 2012). Metabolism of rosuvastatin by CYP2C9 constitutes only a minor pathway to overall disposition (≤10%).
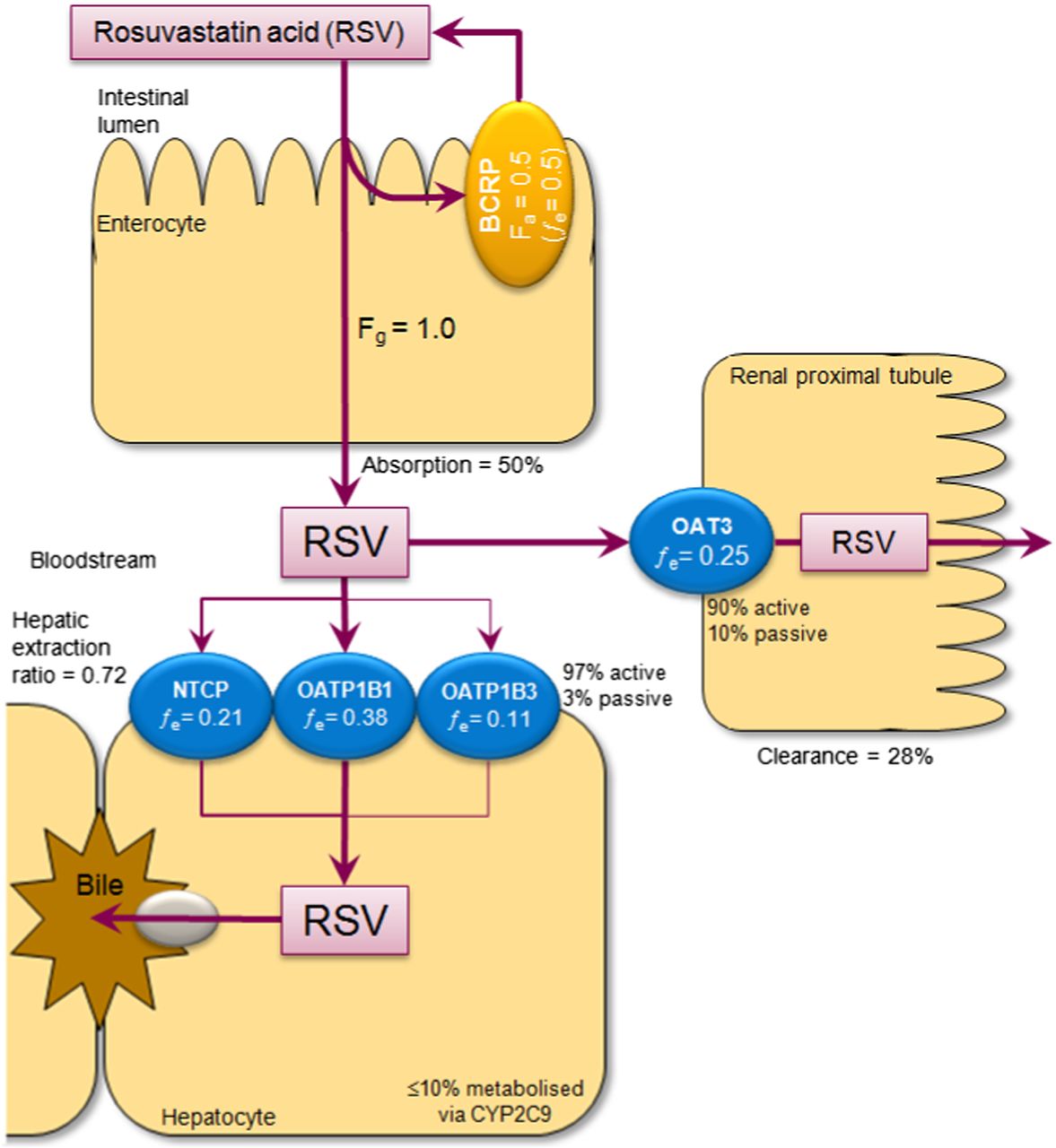
Critical disposition pathways of rosuvastatin as described by Elsby et al. (2012).
Fostamatinib is an oral spleen tyrosine kinase inhibitor that has been investigated as a treatment of rheumatoid arthritis (Weinblatt et al., 2014) and, due to patient comorbidities in this disease population, was likely to be coadministered with statins, such as rosuvastatin and atorvastatin. Fostamatinib is a prodrug (R788; chemical name/structure provided in Sweeny et al., 2010) that is rapidly and essentially completely metabolized by dephosphorylation in the enterocytes of the intestine to the active systemic metabolite R406 (N4-(2,2-dimethyl-3-oxo-4-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethyoxyphenyl)-2,4-pyrimidinediamine) (Sweeny et al., 2010; Baluom et al., 2013). Therefore, the first aim of this study was to investigate fostamatinib and R406 as inhibitors of the principal rosuvastatin transporters BCRP, OATP1B1, and OAT3 in vitro and, on the basis of inhibition predictions, to use fostamatinib as a mechanistic in vivo tool in a rosuvastatin clinical interaction study to confirm the impact that solitary inhibition of BCRP has on rosuvastatin exposure. The second aim was to validate a Caco-2 rosuvastatin BCRP inhibition assay, and then, using determined IC50 data and mechanistic static equations, to evaluate the role BCRP inhibition plays in the clinical DDIs observed between rosuvastatin and a range of perpetrator drugs by comparing predicted theoretical fold increases in exposure to the clinically observed rosuvastatin AUC increases.
Materials and Methods
Materials.
Estrone 3-sulfate, sulfasalazine, estradiol 17β-glucuronide, rifamycin SV, Triton X, novobiocin, Ko143 [(3S,6S,12aS)-1,2,3,4,6,7,12,12a-Octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino-[1′,2′:1,6]pyrido[3,4-b]indole-3-propanoic acid 1,1-dimethylethyl ester], cyclosporin A, pantoprazole, diclofenac, nifedipine, clopidogrel, fluconazole, and fenofibrate were purchased from Sigma-Aldrich (St. Louis, MO; Poole, Dorset, UK). Eltrombopag, darunavir, and lopinavir were purchased from Selleck Chemicals (Houston, TX; Shanghai, China). Fluvastatin was obtained from Sequoia Research Products (Pangbourne, Reading, UK). [3H]estrone 3-sulfate, [3H]estradiol 17β-glucuronide, and Ultima Gold and Optiphase Supermix scintillation cocktails were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA; Buckinghamshire, UK). [3H]methotrexate and methotrexate were purchased from American Radiolabeled Chemicals (St. Louis, MO). Fostamatinib and its active metabolite R406 were supplied by AstraZeneca R&D (Alderley Park, Macclesfield, UK). Metoprolol, rosuvastatin, atorvastatin, and ezetimibe were supplied by AstraZeneca Compound Management Group. [3H]rosuvastatin was custom synthesized by Quotient Bioresearch (Cardiff, UK). Hanks’ balanced salt solution (HBSS; containing CaCl2 and MgCl2) and HEPES were purchased from Life Technologies (Paisley, UK; Grand Island, NY). All other chemicals, solvents, and reagents were purchased from Sigma-Aldrich.
Sodium uptake buffer, ND96 buffer, BD Falcon HTS 96-well flat-bottom tissue culture treated plates, BD Biocoat Poly-d-lysine 24-well multiwell plates, human BCRP (ABCG2) membrane vesicles, human OAT3 (SLC22A8)–expressing Xenopus laevis oocytes, and control (water-injected) oocytes were supplied by BD Biosciences Discovery Labware (Woburn/Bedford, MA; Oxford, UK). UniFilter-96 GF/B filter plates and TopSeal A adhesive sealing film were supplied by PerkinElmer Life and Analytical Sciences. Millicell-96 multiwell cell culture insert plates (with polycarbonate membranes; 0.4 μm pore size, 0.12 cm2 surface area) and Millicell 96-well transport companion plates were purchased from Millipore (Watford, Hertfordshire, UK). Ninety-six–well deep well (2 ml) polypropylene or shallow round-bottomed polypropylene plates were supplied by VWR International Ltd. (Lutterworth, Leicestershire, UK). HTS Transwell-96 permeable supports and associated 96-well plasticware were obtained from Corning Incorporated Life Sciences (Tewksbury, MA). A recombinant cell line expressing OATP1B1 [human embryonic kidney 293 (HEK293)–OATP1B1] was provided by AstraZeneca R&D. Caco-2 cells (HTB37, supplied at passage number 17) were purchased from American Type Culture Collection (Manassas, VA).
In Vitro BCRP Inhibition Assessment of Fostamatinib and R406.
Uptake of the BCRP probe substrate [3H]estrone 3-sulfate (1 μM) was determined (in triplicate wells per condition) according to a previously validated method (Elsby et al., 2011a), using an incubation time of 2 minutes at 32°C and BCRP-expressing membrane vesicles plus/minus ATP (5 mM), in the absence and presence of fostamatinib (0.0009–2 μM), R406 (0.0009–2 μM), and the positive control inhibitor sulfasalazine (0.1–30 μM). Uptake solutions contained ≤2% (v/v) dimethylsulfoxide (DMSO).
In Vitro OATP1B1 Inhibition Assessment of R406.
Uptake of the OATP1B1 probe substrate [3H]estradiol glucuronide (0.02 μM) was determined using 24-well plates according to a previously validated method (Sharma et al., 2010) in HEK293-OATP1B1 cells in the absence and presence of R406 (0.3–10 μM) and the positive control inhibitor rifamycin SV (0.001–100 μM). Incubations (triplicate wells per condition) were conducted in HBSS buffer (containing 10 mM HEPES, pH 7.4) at 37°C for 2 minutes, following a 15-minute preincubation with test inhibitor. Uptake solutions contained ≤1% (v/v) DMSO.
In Vitro OAT3 Inhibition Assessment of R406.
Uptake of the OAT3 probe substrate [3H]methotrexate (10 μM) was determined according to a previously described method (Elsby et al., 2011b) using control and OAT3-expressing oocytes in the absence and presence of R406 (0.11–32 μM) and the positive control inhibitor sulfasalazine (0.3–100 μM). Incubations (pools of 10 oocytes per condition) were conducted at room temperature for 60 minutes. Uptake solutions contained ≤2% (v/v) DMSO.
Data Analysis.
In brief, the amount of radioactivity (disintegrations per minute) taken up by vesicles, HEK293-OATP1B1 cells, or oocytes was determined and used to calculate the amount (picomoles) of probe substrate, which was subsequently converted to uptake rate (pmol/min/mg protein or pmol/h/oocyte) as described previously (Sharma et al., 2010; Elsby et al., 2011b). Background condition uptake rate (uptake in BCRP vesicles minus ATP, uptake in HEK293-OATP1B1 cells in the presence of 100 μM positive control inhibitor where OATP1B1 is 100% inhibited, or uptake in control oocytes) was subtracted from that determined in transporter-expressing vesicles/cells/oocytes to give transporter-specific uptake rate. This was then converted to percentage (vehicle) control transport activity, which was subsequently plotted against nominal inhibitor concentration. Curves were fitted using XLfit 5.1 (ID Business Solutions Ltd., Guildford, Surry, UK) [four-parameter logistic model, Eq. 201, as published for P-glycoprotein inhibition (Elsby et al., 2011c)] to determine the concentration that produces half-maximal inhibition of probe substrate transport (IC50).
Clinical Interaction Study between Fostamatinib and Rosuvastatin.
Recently, a clinical interaction study (NCT01725230, D4300C00039) between fostamatinib and rosuvastatin performed in healthy volunteers (62% Caucasian, 38% black ethnicity; 95% male and 5% female between the ages of 18 and 55 years, with body weight >50 kg and body mass index between 18 and 30 kg/m2) was fully described and reported elsewhere (Martin et al., 2016). In brief, the study was an open-label, fixed-sequence study that assigned eligible subjects to receive 20 mg of rosuvastatin over two treatment periods: alone for period 1 and in combination with fostamatinib at steady state (100 mg twice daily for 5 days) for period 2. Plasma concentrations of rosuvastatin and R406 were quantified by validated bioanalytical liquid chromatography–tandem mass spectrometry (LC-MS/MS) methodologies and used to determine pharmacokinetic parameters (AUC and Cmax) by noncompartmental modeling, as reported in Martin et al. (2016).
Validation of an In Vitro Caco-2 BCRP Inhibition Assay Utilizing Rosuvastatin as BCRP Probe Substrate.
The following experimental properties were investigated in the validation of the Caco-2 BCRP inhibition assay as a method to identify inhibitors of BCRP: 1) the suitability of novobiocin as a positive control inhibitor, 2) interassay variability, and 3) acceptance criteria to quality control future use of the assay. The basolateral-to-apical (B-A) transport of the probe substrate [3H]rosuvastatin was measured across polarized Caco-2 cell monolayers (triplicate wells per condition) in the absence and presence (in both donor and receiver compartments; added at the same time as rosuvastatin) of a range of concentrations (0.03–10, 0.1–30, or 0.3–100 μM) of the known BCRP inhibitors novobiocin, Ko143, cyclosporin A, pantoprazole, sulfasalazine, atorvastatin, diclofenac, fluvastatin, and nifedipine, and of the noninhibitor metoprolol, to determine an apparent IC50 value for the inhibition of BCRP-mediated rosuvastatin transport. Experiments determining the inhibition of BCRP transport by the positive control inhibitor novobiocin were conducted on nine separate occasions spanning five cell passage numbers (24, 25, 26, 27, and 29) to ensure that the data generated were consistent over the passages routinely used for inhibition experiments (20–30). The remaining inhibitors were each assessed on three separate experimental occasions. The methodology used was a slight modification of the previously validated Caco-2 P-glycoprotein inhibition assay (Elsby et al., 2008), which is in agreement with the recommendations of the International P-glycoprotein IC50 Working Group (Bentz et al., 2013). The main modifications were seeding of Caco-2 cells on 96-well multiwell insert plates at a density of 270,000 cells/ml (27,000 cells per well; 245,455 cells/cm2), use of monolayers in transport studies 14–21 days after seeding, and use of a single incubation time point (90 minutes) in the absence of shaking.
In brief, polarized Caco-2 cell monolayers were washed three times and then preincubated with warm transport buffer (HBSS containing 10 mM HEPES, pH 7.4) for 10–15 minutes at 37°C prior to the addition of donor and receiver solutions. Donor solutions of transport buffer containing [3H]rosuvastatin (1 μM; approximately 10-fold lower than Km for BCRP = 10.8 μM; Huang et al., 2006) and the appropriate concentration of test inhibitor or DMSO (vehicle control; at the equivalent volume of solvent vehicle used for test inhibitor) were added to the basolateral compartment of the monolayer (total volume = 300 μl). Receiver solutions of transport buffer containing either the corresponding concentration of test inhibitor or DMSO (vehicle control) and the cell monolayer integrity marker Lucifer yellow (100 μM) were added to the apical compartments (total volume = 100 μl). Donor and receiver solutions contained 0.5% (v/v) DMSO. Following a 90-minute incubation at 37°C, the amount of [3H]rosuvastatin appearing in the receiver compartment was determined by sampling (50 μl), quantified by liquid scintillation counting, and used to calculate apparent permeability (Papp), as described previously (Elsby et al., 2008). The passive permeability of rosuvastatin observed when BCRP is completely inhibited [derived from incubations containing the highest concentration (30 μM) of the positive control inhibitor novobiocin] was subtracted from the determined B-A Papp value in the absence or presence of test inhibitor, to give a corrected BCRP-mediated B-A Papp, which was subsequently converted to percentage (vehicle) control transport activity. The resulting inhibition curves were plotted and fitted as described earlier to determine IC50 values, which are equivalent to Ki values (assuming competitive inhibition). Mass balance (percent recovery) of rosuvastatin was calculated as described previously (Elsby et al., 2011c). The percentage mass (picomoles) transfer of coincubated Lucifer yellow across the cell monolayer was determined, and cell monolayer integrity was deemed acceptable if the transfer was ≤1.5%.
Mechanistic Static Predictions of AUC Changes for Known Clinical DDIs with Rosuvastatin Based upon Determined In Vitro Caco-2 BCRP Inhibitory Data.
Eltrombopag, darunavir, lopinavir, clopidogrel, ezetimibe, fenofibrate, and fluconazole were assessed (over a range of six concentrations) in the validated Caco-2 BCRP inhibition assay (using both radiolabeled and a cross-validated LC-MS/MS endpoint) to determine inhibitory potencies toward BCRP-mediated rosuvastatin transport. The IC50 (equating to Ki) values obtained for the aforementioned compounds and fostamatinib were incorporated into the following adapted Rowland-Matin mechanistic static equation, as described previously by Elsby et al. (2012), to predict the change in rosuvastatin AUC based upon inhibition of a fraction excreted (ƒe) value of 0.5 for intestinal BCRP: where Ki = absolute inhibition constant (equating to IC50 if the probe [S] < <<< Km in the inhibition assay and assuming competitive inhibition, based on the Cheng-Prusoff equation; Cheng and Prusoff, 1973) and [I] = maximum enterocyte concentration (Igut max; as described by Agarwal et al., 2013).
where Ki = absolute inhibition constant (equating to IC50 if the probe [S] < <<< Km in the inhibition assay and assuming competitive inhibition, based on the Cheng-Prusoff equation; Cheng and Prusoff, 1973) and [I] = maximum enterocyte concentration (Igut max; as described by Agarwal et al., 2013).
Additionally, the predicted change in rosuvastatin AUC based upon inhibition of OATP1B1 (ƒe = 0.38) was also determined using Ki values derived from either the aforementioned in vitro OATP1B1 inhibition assay (at 0.3, 1, 3, 10, 30, and 100 μM; using LC-MS/MS as the analytical endpoint) or the literature, in context with the unbound maximum hepatic inlet concentration as [I], to understand the contribution (if any) of OATP1B1 inhibition toward the overall magnitude of exposure change observed clinically through DDI.
Results
Assessment of Fostamatinib and R406 as Inhibitors of BCRP In Vitro.
Inhibition of estrone 3-sulfate uptake by the positive control inhibitor sulfasalazine (IC50 = 0.61 μM) passed the acceptance criteria set during the assay validation (Elsby et al., 2011a) and therefore indicated that the vesicle test system was capable of detecting inhibitors of BCRP. Both fostamatinib and R406 inhibited BCRP-mediated transport of estrone 3-sulfate with IC50 values of 0.050 and 0.031 μM, respectively (Fig. 2).

Inhibition of BCRP-, OATP1B1-, and OAT3-mediated probe substrate transport activity by fostamatinib, R406, and positive control inhibitors. Data are expressed as the mean (n = 3 wells or n ≥ 5 oocytes).
Assessment of R406 as an Inhibitor of OATP1B1 and OAT3 In Vitro.
Inhibition of probe substrate uptake by the positive control inhibitors rifamycin SV (IC50 = 0.02 μM) or sulfasalazine (IC50 = 4.3 μM) demonstrated that the transfected cell or oocyte test systems were able to detect inhibitors of OATP1B1 or OAT3, respectively. Although R406 did not inhibit OAT3-mediated transport, it did inhibit OATP1B1-mediated transport of estradiol 17β-glucuronide with an IC50 > 10 μM (27% inhibition at 10 μM; Fig. 2).
Calculation of [I]/Ki Ratios and R-Value to Predict the DDI Potential of Fostamatinib.
The calculated [I]/Ki ratios and R-value for the expected therapeutic dose of fostamatinib (100 mg) are shown in Table 1. The mean total Cmax value of R406 (at steady state), used toward the calculation of hepatic inlet concentration, is derived from the plasma concentration data determined from multiple clinical studies. The [I2]/Ki ratio predicted that there was a potential for a DDI with BCRP substrate drugs in vivo through inhibition of intestinal BCRP. In contrast, R406 was unlikely to cause a DDI through inhibition of hepatic OATP1B1 in vivo (as R-value < 1.25), nor through renal OAT3, as R406 was not an inhibitor of OAT3.
Summary of fostamatinib (100 mg) dose-related concentration and inhibitory kinetic parameters for calculation of [I]/Ki ratios and R-value
R-value is equal to (1 + [Iinlet max u]/Ki).
Effect of Fostamatinib on the Pharmacokinetic Parameters of the BCRP Substrate Rosuvastatin in Healthy Volunteers.
The mean plasma concentration–time profiles (0–96 hours) of a single oral dose of rosuvastatin (20 mg) when administered alone or concomitantly with 100 mg of fostamatinib at steady state (dosed twice daily for 5 days) are shown in Fig. 3 (reprinted from Martin et al., 2016). Fostamatinib increased the geometric least-squares mean AUC and Cmax of rosuvastatin by 96% (90% confidence interval, 78–115) and 88% (90% confidence interval, 69–110), respectively, indicating a clinical pharmacokinetic interaction (Martin et al., 2016).
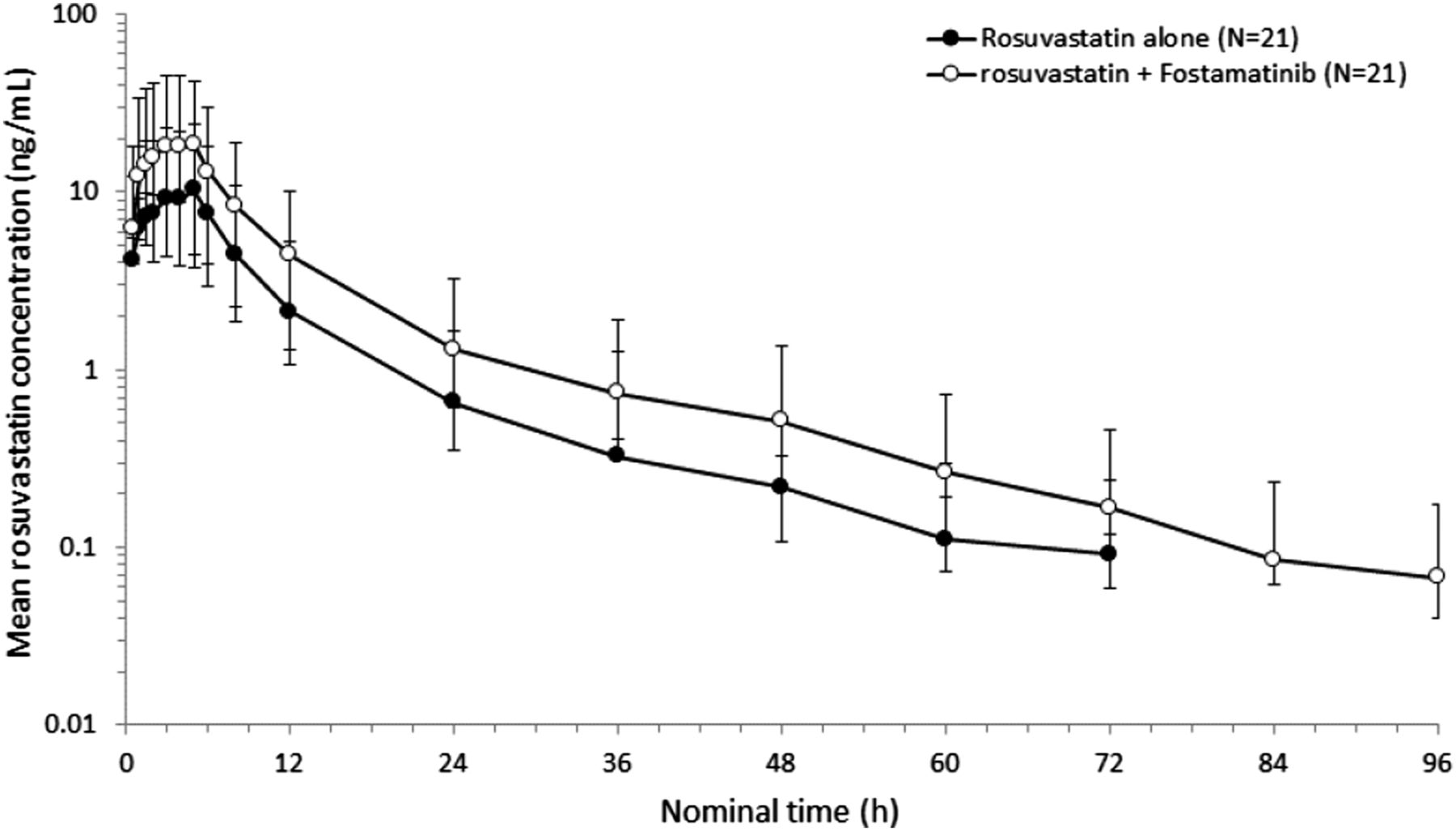
Semilogarithmic plot of the geometric mean (± S.D.) rosuvastatin plasma concentration versus time following administration of a single oral dose of rosuvastatin (20 mg) in the presence and absence of 100 mg of fostamatinib. The figure is reprinted with permission from Martin et al. (2016).
Validation of an In Vitro Caco-2 BCRP Inhibition Assay Utilizing Rosuvastatin as BCRP Probe Substrate.
Initial comparison of bidirectional apparent permeabilities confirmed that rosuvastatin (1 μM) exhibited suitable BCRP-mediated efflux in the Caco-2 cell test system: mean ± S.D. (n = 3 wells) A-B Papp = 0.39 ± 0.05 and B-A Papp = 15.6 ± 1.75 × 10−6 cm/s, giving an efflux ratio of 40. BCRP is solely responsible for rosuvastatin efflux in Caco-2 cells, as the efflux ratio returned to unity in the presence of the BCRP inhibitor novobiocin (30 μM; data not shown), which did not inhibit P-glycoprotein (up to 300 μM) in MDCK-MDR1 cells (R. Elsby, unpublished observation). The rosuvastatin B-A Papp values observed at cell passage numbers 24, 25, 26, 27, and 29 were 16.4 ± 1.80, 14.2 ± 1.50, 16.1 ± 3.10, 20.6 ± 1.9, and 12.5 ± 0.90 × 10−6 cm/s, respectively, indicating consistent functional expression of BCRP over the range of cell passages used for inhibition experiments.
The determined interassay mean IC50 values for the inhibition of BCRP-mediated rosuvastatin transport by the positive control inhibitor novobiocin from nine separate occasions over five different cell passage numbers, and by Ko143, cyclosporin A, pantoprazole, sulfasalazine, atorvastatin, diclofenac, fluvastatin, and nifedipine are shown in Table 2, proving that the Caco-2 cell test system is able to correctly identify known literature inhibitors of BCRP. Mean IC50 curves are shown in Fig. 4, which also shows that metoprolol did not inhibit BCRP-mediated transport of rosuvastatin. The mean IC50 calculated for the positive control inhibitor novobiocin was 1.4 ± 0.5 μM (precision = 36%), giving an acceptance range of ≤2.9 μM for future quality control of the assay (derived from the mean ± 3 S.D.). For all BCRP inhibitors tested, the IC50 values determined from individual experimental occasions (Table 2) gave good interassay reproducibility, demonstrating that future assessment of compounds as inhibitors of BCRP in this test system can be acceptably performed just once, and need not be repeated multiple times. The validated inhibition assay was determined to be robust, reproducible, and fit for the routine evaluation of compounds as inhibitors of BCRP.
Determined IC50 values for inhibition of rosuvastatin basolateral-to-apical flux across Caco-2 cell monolayers by known inhibitors of BCRP

Mean concentration-dependent inhibition of the BCRP-mediated transport of [3H]rosuvastatin (1 μM) by a range of known literature BCRP inhibitors, and the noninhibitor metoprolol, using polarized Caco-2 cell monolayers. Data are expressed as the mean (± S.D.) for n ≥ 3 experimental occasions per inhibitor.
Predicted versus Observed AUC Changes for Known Clinical DDIs with Rosuvastatin Based upon Determined In Vitro Caco-2 BCRP Inhibitory Data.
Eltrombopag, darunavir, lopinavir, clopidogrel, ezetimibe, and fenofibrate inhibited BCRP-mediated rosuvastatin transport with IC50 values of 2.1, 75, 8.7, 63, 2.9, and 170 μM, respectively (Table 3). In HEK293-OATP1B1 cells, darunavir, lopinavir, clopidogrel, and ezetimibe inhibited OATP1B1-mediated estradiol 17β-glucuronide uptake with IC50 values of 4.3, 0.43, 1.8, and 2.2 μM, respectively (Table 3). In contrast, fluconazole did not inhibit BCRP or OATP1B1 over the concentration ranges tested (data not shown).
Inhibitory properties and pharmacokinetic parameters of drugs coadministered with rosuvastatin in a clinical interaction study
Using the previously determined drug inhibitory affinities and their pharmacokinetic parameters given in Table 3, calculations were performed with mechanistic static equations to predict the theoretical fold increase in rosuvastatin AUC that would occur as a consequence of these drugs’ inhibition of intestinal BCRP and/or hepatic OATP1B1. The calculated theoretical fold increases in exposure due to inhibition of each transporter alone, and in combination, are shown in Table 4. For each individual drug highlighted, the theoretical fold increase in AUC due to inhibition of OATP1B1 alone was lower (ranging from 1.01- to 1.25-fold) than the corresponding increase due to sole inhibition of BCRP, which ranged from 1.15- to 2-fold (Table 4).
Predicted versus observed AUC changes of rosuvastatin with various coadministered drugs based upon inhibition of intestinal BCRP (fe = 0.5) or hepatic OATP1B1 (fe = 0.38) transport
Discussion
The BCRP transporter plays an important role in clinically observed DDIs, the majority of which are attributable to inhibition of intestinal BCRP, resulting in increased absorption of substrates such as topotecan and rosuvastatin (Kruijtzer et al., 2002; Elsby et al., 2012). Studying the potential for a drug to perpetrate a BCRP-mediated DDI in the clinic involves initial in vitro evaluation of the drug as an inhibitor of BCRP and, where warranted, a follow-up interaction study with a clinical BCRP probe substrate, such as rosuvastatin, to confirm interaction potential in vivo (Giacomini et al., 2010; Food and Drug Administration draft DDI guidance 2012, (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf). However, rosuvastatin would not be a reliable probe for such a study, as BCRP is not the only pathway that is critical to the disposition of rosuvastatin in vivo, since both OATP1B1 and OAT3 pathways are also important (Elsby et al., 2012). Consequently, any clinical study using rosuvastatin would simply be a broad drug interaction study unless the “interacting” drug is proven not to inhibit OATP1B1 and OAT3 in vitro or, if an in vitro inhibitor, not predicted to inhibit in vivo, and only then would it become a mechanistic study for assessing BCRP inhibition.
On the basis of fulfilling these criteria, following assessment of IC50 values toward BCRP (R788; [I2]/Ki = 13,820; DDI likely), OATP1B1 (R406; R-value = 1.03; no DDI potential), and OAT3 (R406; no inhibition), it was recognized that fostamatinib could be used as an in vivo tool for a mechanistic clinical BCRP interaction study with rosuvastatin. Such a study would confirm the critical role BCRP plays in rosuvastatin absorption by evaluating, through DDI, the impact solitary inhibition of BCRP has on statin exposure. Although rosuvastatin was not used as the in vitro probe to assess OATP1B1 and BCRP inhibition potential, estradiol 17β-glucuronide is a surrogate for OATP1B1-mediated rosuvastatin transport (Izumi et al., 2015), and for BCRP, even if a fostamatinib IC50 value using rosuvastatin was 100-fold higher than that obtained versus estrone 3-sulfate, it would not change DDI potential or predictions. The data reproduced from the clinical interaction study by Martin et al. (2016) demonstrated that sole inhibition of BCRP by fostamatinib resulted in a clinically significant approximately 2-fold increase in rosuvastatin exposure (AUC and Cmax). Examination of rosuvastatin concentration–time profiles (Fig. 3) with and without fostamatinib suggested that there was no change in rosuvastatin elimination rate (0.044 or 0.049 hour−1, respectively), as elimination phases were parallel. Coupled with the finding that there was no change in rosuvastatin half-life (15.7 vs. 14.2 hours, respectively), these data indicated that the DDI with fostamatinib was attributable to increased rosuvastatin absorption due to inhibition of intestinal BCRP (Martin et al., 2016). Furthermore, the magnitude of the observed exposure change is consistent with a doubling in rosuvastatin absorption, which not only suggests complete inhibition of intestinal BCRP (ƒe = 0.5) by fostamatinib (conforming to Ki being >1000 times lower than anticipated intestinal concentration), but also confirms that BCRP efflux restricts absorption of rosuvastatin to 50% in humans.
Eltrombopag, darunavir, lopinavir, clopidogrel, ezetimibe, fenofibrate, and fluconazole are drugs that are listed on the Crestor FDA label as perpetrating pharmacokinetic DDIs upon coadministration with rosuvastatin. Of the rosuvastatin disposition pathways, OAT3 inhibition is unlikely to contribute to these DDIs because darunavir and fluconazole do not inhibit OAT3 in vitro, and eltrombopag (Ki = 7.8 μM), lopinavir (Ki > 10 μM), and fenofibric acid (Ki = 2.2 μM) are not predicted to inhibit OAT3 in vivo based on Cmax u/Ki ratios being <0.1 (Chu et al., 2007; Yoshida et al., 2012). Additionally, inhibition of OATP1B3 is unlikely to play a role since, although eltrombopag (Ki = 25.6 μM), darunavir (Ki = 4.51 μM), and ezetimibe (Ki > 4 μM) are inhibitors, R-value extrapolations indicate their potential for DDI as unlikely (<1.25), and lopinavir is not an inhibitor (De Bruyn et al., 2011, 2013; Takeuchi et al., 2011; Vildhede et al., 2014). Finally, inhibition of sodium/taurocholate cotransporting peptide (NTCP) can be ruled out as a contributory factor since 1) despite being an NTCP inhibitor in vitro (Ki = 25 μM; Dong et al., 2013), ezetimibe is not expected to cause a DDI via this mechanism in vivo (R = 1.00); 2) lopinavir does not inhibit NTCP (Vildhede et al., 2014); and 3) although it is unknown whether the remaining drugs are NTCP inhibitors, even if they were to have similar potencies to those described earlier for OATP1B3, then R-values would still indicate that DDI potential by this mechanism is unlikely. Consequently, this reasoning indicates inhibition of BCRP and/or OATP1B1 as being the predominant transporter pathway(s) to focus on to decipher the underlying mechanisms behind observed DDIs. The IC50 values of these drugs toward BCRP were determined in the Caco-2 BCRP inhibition assay to obtain clinically relevant parameters that could be used, alongside OATP1B1 potencies, in calculations of maximum theoretical increases in rosuvastatin exposure to aid understanding of the mechanism(s) behind each DDI. Apart from fluconazole, which did not inhibit BCRP, all of the drugs were inhibitors of BCRP-mediated rosuvastatin transport and, other than fenofibrate, were predicted to cause a DDI through intestinal BCRP inhibition in vivo ([I2]/Ki ratios >10; Table 3). Interestingly, none of these inhibitors were anticipated to inhibit biliary BCRP in vivo (ratios of [Iinlet max u]/Ki were all ≤0.1; Table 3), supporting the notion that inhibition of intestinal BCRP is clinically important for BCRP-mediated DDIs. However, although all the drugs (except fluconazole) were OATP1B1 inhibitors, only darunavir, lopinavir, and clopidogrel (300 mg) gave R-values >1.25 (= 1.29, 2.07, and 1.69, respectively), indicating potential for an interaction in vivo via OATP1B1. Collectively, these data suggested that inhibition of intestinal BCRP may be the principal cause of the clinically observed DDIs perpetrated by eltrombopag, darunavir, lopinavir, clopidogrel, ezetimibe, and fenofibrate (Table 4).
Predictions of AUC change were performed using determined IC50 values and the adapted Rowland-Matin equation, as described by Elsby et al. (2012). Previously, a maximum 2-fold increase in exposure due to complete inhibition of intestinal BCRP efflux was predicted when [I2]/Ki > 10. However, such an approach likely overpredicts the impact due to BCRP inhibition (particularly for those DDIs where the increase in observed AUC is <2-fold) because of high [I2] values, which may not reflect true concentrations once intestinal solubility is accounted for. An alternative parameter, maximum inhibitor concentration in the enterocyte [Igut max], may be a more accurate representation of intestinal concentration as it is independent of solubility (Agarwal et al., 2013) (see Table 3 for equation). Consequently, in this study, we explored the use of [Igut max] in the adapted Rowland-Matin equation, alongside a BCRP fe = 0.5, to establish whether taking into account varying degrees of intestinal BCRP inhibition (rather than assuming 100% inhibition) resulted in more accurate predictions of clinical exposure.
Comparison of predicted increases in rosuvastatin AUC due to inhibition of composite transporter pathways BCRP and OATP1B1 confirmed intestinal BCRP inhibition to be the primary mechanism underlying each DDI; as for each individual drug, the AUC increase due to BCRP was higher than that attributed to OATP1B1 inhibition alone (Table 4). Additionally, all of the predicted AUC increases due to only OATP1B1 inhibition were ≤1.25. Overall, calculated fold increases in AUC (the product of the major AUC change from BCRP with the minor from OATP1B1) accurately predicted the clinically observed dose-dependent fold increases in rosuvastatin exposure due to clopidogrel and those increases due to eltrombopag, darunavir, lopinavir, ezetimibe, and fenofibrate, as all predictions were within 10% of the clinical value (Table 4). Furthermore, the accuracy of the predictions not only support our earlier assumption that BCRP and (to a lesser extent) OATP1B1 are the dominant pathways involved in the majority of rosuvastatin clinical DDIs, but also that [Igut max] appears to be the correct concentration parameter for use in predictions with rosuvastatin.
The clinical role of intestinal BCRP inhibition in causing rosuvastatin DDIs implies that the magnitude of such DDIs may be reduced in individuals who are polymorphic for ABCG2 c.421C>A, due to these individuals having less functional BCRP activity to inhibit. This may translate to an ethnic difference in DDI susceptibility if the higher frequency of ABCG2 polymorphism in a particular group results (in part) in lower functional BCRP activity at the population level, as has been demonstrated in Asian compared with Caucasian populations (Sakiyama et al., 2014; Birmingham et al., 2015a,b). Interestingly, such an ethnic difference has been observed clinically with eltrombopag, where there was a larger 1.88-fold increase in rosuvastatin AUC in non-Asian subjects compared with only a 1.3-fold increase in Asian subjects following coadministration of eltrombopag (Allred et al., 2011). Thus, the much smaller magnitude of AUC change for the Asian group is consistent with this group having less functional BCRP activity to be inhibited by eltrombopag. It was for this reason that the non-Asian group AUC change was used as the clinically observed AUC change for comparison with predictions in this study, as it more accurately reflects the impact of inhibiting fully functioning BCRP.
Finally, taking into account the analyses from both this study and that of Elsby et al. (2012) toward understanding the likely mechanisms behind clinically observed DDIs with rosuvastatin, it is interesting to note that out of a total of 12 reported clinical DDIs (the first 10 listed on the Crestor label, plus clopidogrel and fostamatinib) for which increases in AUC were ≥1.2-fold, six (50%; Table 4) can be attributed to BCRP inhibition as the primary mechanism, two (17%; cyclosporine and atazanavir) can be attributed to complete inhibition of BCRP combined with inhibition of OATP1B1, and one (8%; gemfibrozil) can be attributed to the inhibition of both OATP1B and OAT3. The remaining three DDIs (tipranavir, dronedarone, and itraconazole) have not been studied, and their mechanisms wait to be determined. These findings confirm the importance of intestinal BCRP inhibition toward manifesting clinical DDIs with rosuvastatin.
In conclusion, solitary inhibition of the intestinal BCRP efflux transporter can result in a clinically significant DDI with rosuvastatin causing a maximum 2-fold increase in exposure, which may warrant statin dose adjustment or dose capping in clinical practice.
Acknowledgments
The authors acknowledge the contract research services provided by BD Gentest (now Corning Gentest; Woburn, MA) and Pharmaron Beijing Co. (Beijing, China) in performing some of the in vitro transporter experiments.
Authorship Contributions
Participated in research design: Elsby, Surry, Martin.
Conducted experiments: Elsby, Sharma, Surry.
Performed data analysis: Elsby, Sharma, Surry, Martin, Fenner.
Wrote or contributed to the writing of the manuscript: Elsby, Martin, Surry, Sharma, Fenner.
Footnotes
- Received August 13, 2015.
- Accepted December 18, 2015.
↵1 Current affiliation: Drug Transporter Services, Cyprotex Discovery Ltd., Biohub at Alderley Park, Macclesfield, Cheshire, United Kingdom.
↵2 Current affiliation (DS): Skill Supply, Sandbach, Cheshire, United Kingdom.
This research was funded by AstraZeneca. P.M., P.S., and K.F. are employees of AstraZeneca, and R.E. and D.S. are former employees of AstraZeneca.
Abbreviations
- ABC
- ATP-binding cassette
- AUC
- area under the plasma concentration–time curve
- B-A
- basolateral to apical
- BCRP
- breast cancer resistance protein
- DDI
- drug-drug interaction
- DMSO
- dimethylsulfoxide
- HBSS
- Hanks’ balanced salt solution
- HEK293
- human embryonic kidney 293
- Ko143
- (3S,6S,12aS)-1,2,3,4,6,7,12,12a-Octahydro-9-methoxy-6-(2-methylpropyl)-1,4-dioxopyrazino-[1′,2′:1,6]pyrido[3,4-b]indole-3-propanoic acid 1,1-dimethylethyl ester
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- NTCP
- sodium/taurocholate cotransporting peptide
- OAT3
- organic anion transporter 3
- OATP1B1
- organic anion-transporting polypeptide 1B1
- R406
- N4-(2,2-dimethyl-3-oxo-4-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethyoxyphenyl)-2,4-pyrimidinediamine
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}