


Visual Overview

Abstract
Galeterone and abiraterone acetate are antiandrogens developed for the treatment of metastatic castration-resistant prostate cancer. In the present study, we investigated the effect of these drugs on dehydroepiandrosterone (DHEA) sulfonation catalyzed by human liver and intestinal cytosols and human recombinant sulfotransferase enzymes (SULT2A1, SULT2B1b, and SULT2E1) and compared their effects to those of other antiandrogens (cyproterone acetate, spironolactone, and danazol). Each of these chemicals (10 μM) inhibited DHEA sulfonation catalyzed by human liver and intestinal cytosols. Enzyme kinetic analysis showed that galeterone and abiraterone acetate inhibited human liver cytosolic DHEA sulfonation with apparent Ki values at submicromolar concentrations, whereas cyproterone acetate, spironolactone, and danazol inhibited it with apparent Ki values at low micromolar concentrations. The temporal pattern of abiraterone formation and abiraterone acetate depletion suggested that the metabolite abiraterone, not the parent drug abiraterone acetate, was responsible for the inhibition of DHEA sulfonation in incubations containing human liver cytosol and abiraterone acetate. Consistent with this proposal, similar apparent Ki values were obtained, regardless of whether abiraterone or abiraterone acetate was added to the enzymatic incubation. Abiraterone was more effective than abiraterone acetate in inhibiting DHEA sulfonation when catalyzed by human recombinant SULT2A1 or SULT2B1b. In conclusion, galeterone and abiraterone are novel inhibitors of DHEA sulfonation, as determined in enzymatic incubations containing human tissue cytosol (liver or intestinal) or human recombinant SULT enzyme (SULT2A1, SULT2B1b, or SULT1E1). Our findings on galeterone and abiraterone may have implications in drug-drug interactions and biosynthesis of steroid hormones.
Introduction
Cytosolic sulfotransferases (SULTs) are a family of Phase II drug-metabolizing enzymes that sulfonate various endogenous and xenobiotic substrates via the transfer of a sulfuryl group from the cosubstrate 3′-phosphoadenosine-5′-phosphosulfate (PAPS) (James and Ambadapadi, 2013; Wang et al., 2014). The human SULT2 hydroxysteroid sulfotransferase family consists of three isoforms, SULT2A1, SULT2B1a, and SULT2B1b (Blanchard et al., 2004). SULT2A1 is expressed predominantly in the liver and adrenal glands, but also in the small intestines and lungs (Riches et al., 2009). In contrast, SULT2B1a and SULT2B1b mRNA have been shown to be preferentially expressed in extrahepatic tissues, including prostate, uterus, ovary, and placenta (Her et al., 1998; Geese and Raftogianis, 2001; Javitt et al., 2001; Falany and Rohn-Glowacki, 2013). Although SULT2B1b protein is expressed in various tissues, SULT2B1a protein has not been detected in tissues (Falany and Rohn-Glowacki, 2013). Endogenous substrates of SULT2A1 include dehydroepiandosterone (DHEA), pregnenolone, androsterone, and bile acids (Falany et al., 1989; Radominska et al., 1990). Although DHEA is sulfonated to dehydroepiandosterone sulfate (DHEA-S) by SULT2A1 in human liver, it is also sulfonated by SULT2B1a, SULT2B1b (Geese and Raftogianis, 2001; Meloche and Falany, 2001), and SULT1E1 (Falany et al., 1995) in extrahepatic tissues such as prostate (Her et al., 1998; Geese and Raftogianis, 2001; Nakamura et al., 2006; Takase et al., 2007). Sulfotransferases are involved not only in detoxification of drugs and other chemicals (e.g., environment toxicants) (Liu et al., 2006; James and Ambadapadi, 2013), but also bioactivation of certain drugs to give toxic reactive intermediates (Shibutani et al., 1998; Glatt, 2000). Inhibition of sulfotransferases by xenobiotics may lead to drug-drug interactions and perturbation in homeostasis of endogenous substances, such as sex steroids (Wang and James, 2006).
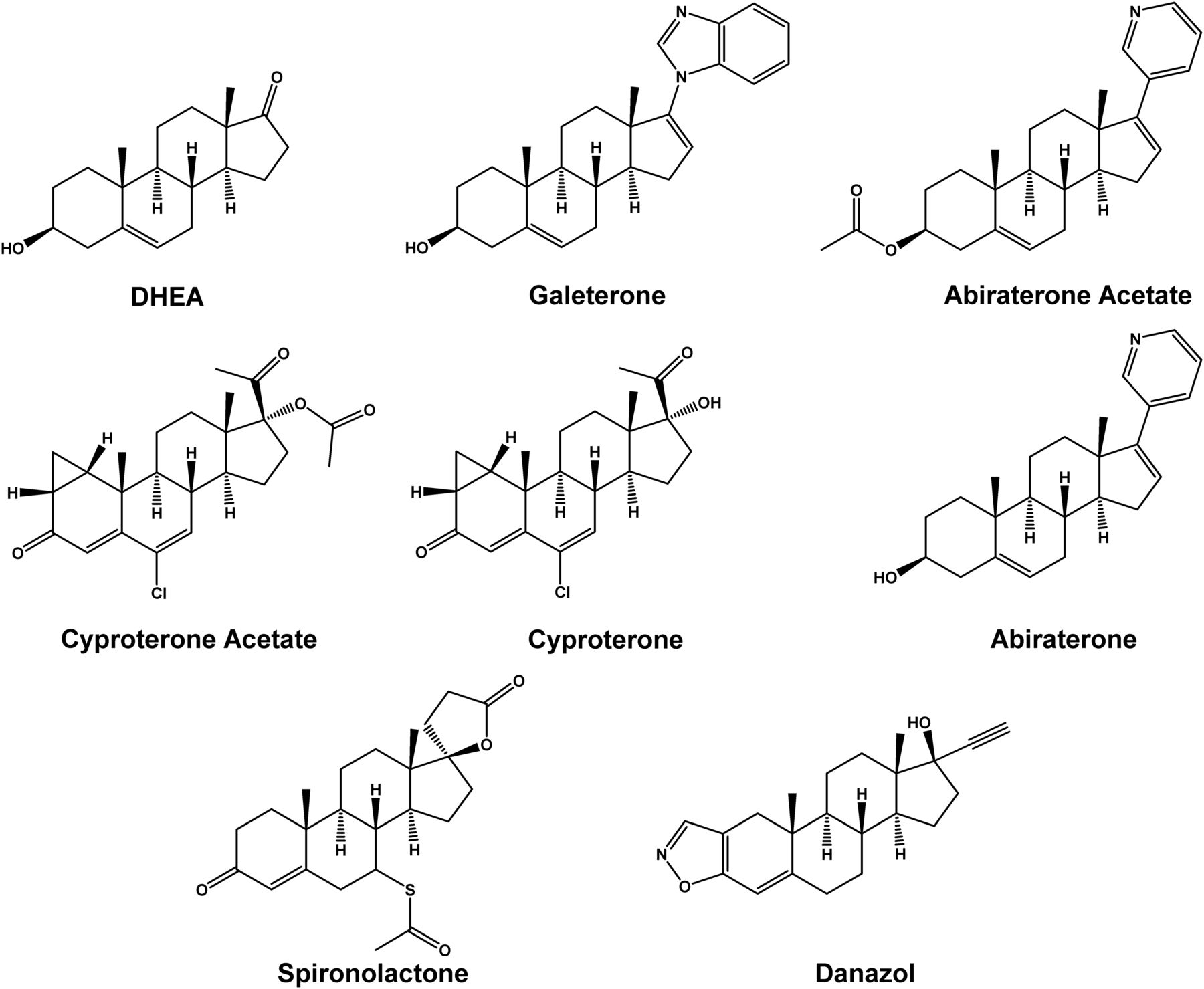
For advanced or metastatic prostate cancer, one of the treatment options is androgen deprivation therapy by chemical castration using antiandrogen drugs (Thompson et al., 2007), which may be categorized as steroidal and nonsteroidal compounds (Chen et al., 2009). Among the steroidal members, the older drugs include cyproterone acetate, spironolactone, and danazol, whereas the newer drugs include abiraterone acetate and galeterone (Fig. 1). Abiraterone acetate, which was approved by US Food and Drug Administration in 2012 for the treatment of metastatic castration-resistant prostate cancer, is the first approved inhibitor of CYP17A1 that also antagonizes androgen receptor and inhibits 3β-hydroxysteroid dehydrogenase (Beckett et al., 2012). Structurally similar to abiraterone (Fig. 1), galeterone was developed as a multitargeting agent that targets multiple points of the androgen biosynthesis and androgen receptor signaling pathway, including CYP17A1 inhibition, androgen receptor antagonism, and degradation of androgen receptor protein, for the treatment of metastatic castration-resistant prostate cancer (Montgomery et al., 2016). Currently, it is not known whether galeterone and abiraterone regulate androgen homeostasis by modulating sulfotransferases that deactivate DHEA, which is an endogenous multifunctional hormone with many biological functions, including neuroprotection (Maninger et al., 2009), antiaging (Yen, 2001), and as a precursor in the biosynthesis of steroidal androgens, namely androstenedione, testosterone, and dihydrotestosterone (Prough et al., 2016).

Chemical structures of DHEA, galeterone, abiraterone acetate, and the other chemicals investigated in the present study.
In the present study, we investigated the effect of galeterone and abiraterone acetate on DHEA sulfonation catalyzed by human tissue cytosol and human recombinant SULT enzymes (namely SULT2A1, SULT2B1b, and SULT1E1) and compared the effect with those of other antiandrogens (cyproterone acetate, spironolactone, and danazol) (Fig. 1). To provide insight into the structural elements involved in the inhibition of DHEA sulfonation by antiandrogens, we compared the effect of abiraterone acetate (prodrug) versus abiraterone (pharmacologically active metabolite) and the effect of cyproterone acetate (pharmacologically active) versus cyproterone (metabolite) on DHEA sulfonation. Our findings indicate that galeterone and abiraterone are novel inhibitors of DHEA sulfonation, as assessed in enzymatic assays with human liver and intestinal cytosol and human recombinant SULT2A1, SULT2B1b, and SULT1E1.
Materials and Methods
Chemicals and Reagents.
Galeterone, spironolactone, danazol, amoxicillin, quercetin, cortisol (hydrocortisone), 3′-phosphoadenosine 5′-phosphosulfate lithium salt hydrate (PAPS; purity of ≥60%), magnesium chloride hexahydrate (MgCl2), and dimethyl sulfoxide (DMSO) were bought from Sigma-Aldrich Corporation (St. Louis, MO), and 3′-phosphoadenosine 5′-phosphosulfate solution [PAPS; supplied in 25 mM Tris, 23% ethanol, pH 7.5, at a stock concentration of 8.5 or 9.9 mM (lot dependent); purity of >90%] was from R&D Systems, Inc. (Minneapolis, MN). Abiraterone acetate and cyproterone acetate were purchased from Cayman Chemical Company (Ann Arbor, MI). Abiraterone was purchased from Toronto Research Chemicals, Inc. (Toronto, ON, Canada). Cyproterone was bought from Santa Cruz Biotechnology, Inc. (Dallas, TX). DHEA and DHEA-S were purchased from Steraloids, Inc. (Newport, RI). Methanol (HPLC grade) and acetonitrile (HPLC grade) were purchased from Tedia Company, Inc. (Fairfield, OH), and formic acid was from VWR International, LLC (Radnor, PA). All other commercially available chemicals were of analytical grade.
Cytosols and Recombinant Enzymes.
Human liver cytosol (mixed sex; pool of 150 donors; catalog #452115, lot #38290, Gentest brand; age of 18–79 years; 75 women and 75 men) was purchased from Corning, Inc. (Corning, NY). Human male liver cytosol (pool of 10 donors; catalog #H1000.C, lot #0710493; age of 21–58 years), human female liver cytosol (pool of 10 donors; catalog #H1500.C; lot #0710245; age of 31–78 years), human intestinal cytosol (mixed sex; pool of 13 donors; catalog #H0610.IC, lot #1210439; age of 18–55 years), human lung cytosol (mixed sex; pool of 4 donors; catalog #H0610.PC(NS), lot #1310100; age of 12–65 years) were bought from Sekisui XenoTech, LLC (Kansas City, KS). Human recombinant SULT2A1 (catalog #CYP104, lot #INT044E2B) and SULT1E1 (catalog #CYP103, lot #INT044E1B) enzymes and control cytosol (isolated from Escherichia coli host cells) were purchased from Cypex Ltd. (Dundee, Scotland, UK). Previous drug metabolism studies have used these SULT2A1 and SULT1E1 enzymes (Gong et al., 2012; Diao et al., 2014). Human recombinant SULT2B1b enzyme (catalog #6174-ST-020, lot #DADE0616011), which contained Met-1 to Glu-311 amino acids of SULT2B1b expressed in Escherichia coli host cells and contained a C-terminal 6-histidine tag, was purchased from R&D Systems, Inc. This recombinant SULT2B1b enzyme has been shown to be functional and has a high enzyme activity [>10 (nmol/min)/mg protein] (R&D Systems, Inc.).
DHEA Sulfonation Assay.
The DHEA sulfonation assay was optimized previously (Bansal and Lau, 2016). The total volume of each standard incubation was either 100 (human intestinal cytosol and human lung cytosol) or 200 μl (human liver cytosol, human male liver cytosol, human female liver cytosol, human recombinant SULT2A1 enzyme, human recombinant SULT2B1b enzyme, and human recombinant SULT1E1 enzyme). Each standard incubation mixture contained potassium phosphate buffer (100 mM, pH 7.4), magnesium chloride (2.5 mM), DHEA (0.5 μM), and various amounts of cytosols, as specified in each figure legend. The final concentration of methanol was at a concentration (0.5% v/v) not known to affect the DHEA sulfonation assay (Ma et al., 2003). Each incubation mixture was prewarmed for 3 minutes at 37°C in a shaking water bath. The enzymatic reaction was initiated by adding a cosubstrate known as PAPS (James, 2014), and the mixture was incubated for a specific duration, as specified in each figure legend. In the present study, a saturating concentration of PAPS was used (Bansal and Lau, 2016), either 20 μM PAPS lithium salt or 1 μM PAPS solution, which yielded a similar extent of DHEA sulfonation, as assessed in a preliminary experiment involving human liver cytosol. The reaction was terminated by adding an equal volume of ice-cold acetonitrile containing cortisol (0.05 μM final concentration; internal standard). Each sample was mixed immediately, placed on ice, and subsequently centrifuged at 16,000 g for 15 minutes at 4°C. The supernatant was then transferred into a 96-well microplate for ultra-high performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) analysis.
Quantification of DHEA-S by UPLC-MS/MS.
The amount of DHEA-S and cortisol (internal standard) was quantified using a validated UPLC-MS/MS method shown to be specific, easier, safer, and faster than radiometric-based sulfotransferase enzyme assays (Bansal and Lau, 2016). Calibration standards were prepared by adding freshly prepared DHEA-S stock solutions (1–1000 μM in DMSO) to a standard incubation to give final concentrations of 1–1000 nM DHEA-S (equivalent to 0.2–200 pmol in 200 μl).
Enzyme Kinetic Analysis of DHEA Sulfonation.
DHEA sulfonation by human tissue cytosols was determined at DHEA concentrations ranging from 0.0025 to 5 μM. The values of Vmax and apparent Km were obtained by nonlinear least-squares regression analysis (SigmaPlot 12.5; Systat Software Inc., San Jose, CA) of the velocity of product formation (V) and substrate concentration (S) data using the equations for Michaelis-Menten, Hill, substrate inhibition, and substrate activation models. The goodness of fit for each model was evaluated by considering the Akaike information criterion, R2, and visual inspection of the data in the Michaelis-Menten plot. Based on the above evaluation criteria, DHEA sulfonation by cytosols, recombinant SULT2A1 enzyme, and recombinant SULT2B1b enzyme was determined using the substrate inhibition model: Based on the above evaluation criteria, DHEA sulfonation by recombinant SULT1E1 enzyme was determined using the Hill model:
Based on the above evaluation criteria, DHEA sulfonation by recombinant SULT1E1 enzyme was determined using the Hill model: where S represents the substrate concentration, Vmax represents the apparent maximum reaction velocity, Km and S50 represent the substrate concentration at which the reaction rate is half of Vmax (James, 2014), Ki represents the inhibitory constant of the substrate, and n represents the Hill coefficient.
where S represents the substrate concentration, Vmax represents the apparent maximum reaction velocity, Km and S50 represent the substrate concentration at which the reaction rate is half of Vmax (James, 2014), Ki represents the inhibitory constant of the substrate, and n represents the Hill coefficient.
Enzyme Inhibition Experiments.
Enzyme inhibition was determined by conducting the DHEA sulfonation assay in the presence of a test chemical, cytosol, and DHEA at an amount or concentration stated in each figure legend. Concentration-response experiments for each of the test chemicals were conducted in the presence of varying concentrations of a test chemical, DHEA (0.5 μM), human liver cytosol (mixed sex; 60 μg), and PAPS (20 μM). Concentration-response curves were plotted, and half-maximal inhibitory concentration (IC50) was determined by nonlinear regression analysis (SigmaPlot 12.5) using the sigmoidal dose-response (variable slope) model: where min is the minimum inhibitory effect, max is the maximum inhibitory effect, x is the inhibitor concentration, and Hill slope is the Hill coefficient.
where min is the minimum inhibitory effect, max is the maximum inhibitory effect, x is the inhibitor concentration, and Hill slope is the Hill coefficient.
To characterize the kinetics of the inhibition, the DHEA sulfonation assay was conducted in the presence of varying concentrations (0.2, 0.4, 0.6, or 0.8 μM) of DHEA and varying concentrations of a test chemical, as indicated in the figure legend. The apparent inhibitory constant (Ki) and the mode of inhibition for each test chemical was determined by nonlinear least-squares regression analysis of the rate of DHEA-S formation at varying concentrations of DHEA and test chemical, using equations for full and partial competitive, noncompetitive, uncompetitive, and mixed-mode inhibition (SigmaPlot 12.5). The goodness of fit for each model was evaluated by considering the Akaike information criterion, R2, and visual inspection of the data in the Lineweaver-Burk plot. Based on the above evaluation criteria, the Ki values of galeterone and abiraterone acetate were determined using the competitive inhibition model: Based on the above evaluation criteria, the Ki value of cyproterone acetate was determined using the partial competitive inhibition model:
Based on the above evaluation criteria, the Ki value of cyproterone acetate was determined using the partial competitive inhibition model: Based on the above evaluation criteria, the Ki values of spironolactone and danazol were determined using the partial mixed inhibition model:
Based on the above evaluation criteria, the Ki values of spironolactone and danazol were determined using the partial mixed inhibition model: where S represents the substrate concentration, I represents the inhibitor concentration, Vmax represents the apparent maximum reaction velocity, Km represents the substrate concentration at which the reaction rate is half of Vmax, and Ki represents the apparent inhibitory constant.
where S represents the substrate concentration, I represents the inhibitor concentration, Vmax represents the apparent maximum reaction velocity, Km represents the substrate concentration at which the reaction rate is half of Vmax, and Ki represents the apparent inhibitory constant.
Formation of Abiraterone and Depletion of Abiraterone Acetate in Human Liver Cytosol.
Each 200 μl standard incubation mixture contained potassium phosphate buffer (100 mM, pH 7.4), magnesium chloride (2.5 mM), abiraterone acetate (0.1 μM), and 60 μg of human liver cytosol. The final concentration of methanol was at a concentration (0.5% v/v). Each incubation mixture was prewarmed for 3 minutes at 37°C in a shaking water bath. The enzymatic reaction was initiated by adding human liver cytosol, and the mixture was incubated for various durations, as specified in the figure legend. At the end of the incubation period, an 100 μl aliquot of the incubation mixture was removed and added to 400 μl of ice-cold acetonitrile containing cortisol (0.25 μM final concentration; internal standard) for the analysis of abiraterone (i.e., dilution of 5×), whereas another 20 μl aliquot of the incubation mixture was added to 980 μl of ice-cold acetonitrile containing cortisol (0.25 μM final concentration; internal standard) for the analysis of abiraterone acetate (i.e., dilution of 50×). Each sample was immediately mixed, placed on ice, and subsequently centrifuged at 16,000 g for 15 minutes at 4°C. The supernatant was then transferred into a 96-well microplate for UPLC-MS/MS analysis. Control incubations containing only human liver cytosol, only vehicle (0.5% v/v methanol), or only abiraterone acetate (substrate) were also prepared and analyzed.
Quantification of Abiraterone and Abiraterone Acetate by UPLC-MS/MS.
The amount of abiraterone and abiraterone acetate was quantified using a newly developed UPLC-MS/MS method. The instrument consisted of an Agilent 1290 Infinity LC System (Agilent Technologies, Santa Clara, CA) coupled to a AB Sciex 3500 triple quadrupole mass spectrometer (Applied Biosystems, Foster City, CA). Chromatographic separation was carried out on an ACQUITY UPLC BEH C18 column (2.1 × 50 mm, 1.7 µm), which was protected by an ACQUITY UPLC BEH C18 VanGuard Pre-column (2.1 × 5 mm, 1.7 µm). The column temperature was maintained at 45°C. The mobile phases were (A) water containing 0.1% formic acid and (B) acetonitrile containing 0.1% formic acid. The mobile phase flow rate was 0.5 ml/min and the gradient elution was optimized as follows: 5% B at 0.0–1.0 minute, linear increase from 5% to 95% B at 1.0–2.0 minutes, 95% B at 2.0–3.5 minutes, linear increase from 95% to 100% B at 3.5–3.6 minutes, 100% B at 3.6–5.0 minutes, linear decrease from 95% to 5% B at 5.0–5.1 minutes, and 5% B at 5.1–6.0 minutes. The autosampler compartment was kept at 4°C and the injection volume was 5 µl. The chromatographic effluent was introduced directly into the mass spectrometer from 1.5 to 3.5 minutes. The mass spectrometer was operated in the positive electrospray ionization mode. Abiraterone, abiraterone acetate, and cortisol (internal standard) were quantified in the multiple reaction monitoring mode using the mass-to-charge transition of m/z 350.3 → 156.1, 392.2 → 332.3, and 363.2 → 121.0, respectively. The optimized compound-dependent MS parameters for abiraterone were as follows: declustering potential, 145.26 V; entrance potential, 10 V; collision energy, 68.20 V; collision cell exit potential, 11.46 V; and dwell time, 200 ms. The optimized compound-dependent MS parameters for abiraterone acetate were as follows: declustering potential, 107.45 V; entrance potential, 10 V; collision energy, 46.82 V; collision cell exit potential, 10.33 V; and dwell time, 200 ms. The optimized compound-dependent MS parameters for cortisol were as follows: declustering potential, 120 V; entrance potential, 4.6 V; collision energy, 30 V; collision cell exit potential, 8 V; and dwell time, 150 ms. Nitrogen gas was used as the ion source and collision gas. The ion spray voltage was 3500 V, ion source temperature was 650°C, curtain gas was 40 psi, collision activated dissociation gas setting was 8 U, and ion source gas 1 and gas 2 were 40 psi. Data acquisition and processing were performed using Analyst software version 1.6.2 (Applied Biosystems).
Calibration standards were prepared by adding freshly prepared abiraterone (1–3000 μM in DMSO) or abiraterone acetate (1–1000 μM in DMSO) stock solutions into the incubation mixture to give final concentrations of 1–3000 nM abiraterone (equivalent to 0.1–300 pmol in 100 µl) and 1–1000 nM abiraterone acetate (equivalent to 0.02–20 pmol in 20 µl). To determine the lower limit of quantification (LLOQ), abiraterone and abiraterone acetate stock solutions were diluted and added to the 100 or 20 µl incubation mixture, respectively. Six replicate samples were prepared for each concentration of each chemical. The LLOQ was established based on a signal-to-noise ratio of more than 5:1, a precision of ±20%, an accuracy of 80%–120%, and at least four out of the six replicates should fulfill the above criteria (Food and Drug Administration, 2013).
Statistical Analysis.
Data were analyzed by one-way analysis of variance and, where appropriate, was followed by the Student-Newman-Keuls post hoc test (SigmaPlot 12.5). The level of statistical significance was set a priori at P < 0.05.
Results
Optimization of the Assay Conditions for DHEA Sulfonation Catalyzed by Human Liver, Intestinal, and Lung Cytosols.
Initial experiments were performed to establish the appropriate conditions for the DHEA sulfonation assay. First, the linear range of the DHEA sulfonation assay with respect to amount of cytosol and incubation time was determined for each type of cytosol. The rate of DHEA-S formation was linear up to 100 µg of human liver cytosol (mixed sex), 200 µg of human liver cytosol (male), 200 µg of human liver cytosol (female), 100 µg of human intestinal cytosol (mixed sex), and 200 µg of human lung cytosol (mixed sex) (Supplemental Fig. S1). The extent of DHEA-S formation was linear up to 25 minutes for human liver cytosol (mixed sex), 45 minutes for human liver cytosol (male), 45 minutes for human liver cytosol (female), 45 minutes for human intestinal cytosol (mixed sex), and 45 minutes for human lung cytosol (mixed sex) (Supplemental Fig. S2). The differences in the linearity range with respect to enzyme amount and incubation time among the mixed-sex human liver cytosols (150 donors) versus the male or female liver cytosols (10 donors) may reflect differences in the number of donors in each of these cytosol preparations. Based on those data, subsequent experiments were performed under linear condition with respect to the amount of cytosolic protein and incubation time. Control experiments showed that DHEA-S was not formed in the absence of either substrate (DHEA) or cosubstrate (PAPS). DHEA-S and cortisol were below the lower limit of quantification or not detected in blank incubation containing cytosol and potassium phosphate buffer.
To optimize the substrate concentration for use in the inhibition assays, we determined the enzyme kinetics of DHEA sulfonation catalyzed by various cytosolic preparations. As shown in Fig. 2A and Table 1, DHEA sulfonation by pooled mixed-sex human liver cytosol yielded an apparent Km (mean ± S.D.) of 0.45 ± 0.14 μM, Vmax of 143 ± 40 (pmol/min)/mg protein, Vmax/Km of 323 ± 15 (μl/min)/mg protein, and the kinetics followed substrate inhibition model with an apparent Ki of 2.5 ± 1.6 μM. Cytosols from male liver (Fig. 2B) and female liver (Fig. 2C) gave comparable kinetic values (apparent Km, Vmax, Vmax/Km, and substrate inhibition Ki) (Table 1). Compared with mixed-sex human liver cytosol, mixed-sex human intestinal cytosol sulfonated DHEA with comparable apparent Km and Ki, but smaller Vmax and Vmax/Km values (Fig. 2D; Table 1). In contrast, mixed-sex human lung cytosol sulfonated DHEA to a very little extent and kinetic values could not be determined (Fig. 2E). Our findings are consistent with those reported previously that sulfotransferases are susceptible to inhibition at high substrate concentrations (Gamage et al., 2006; James, 2014).
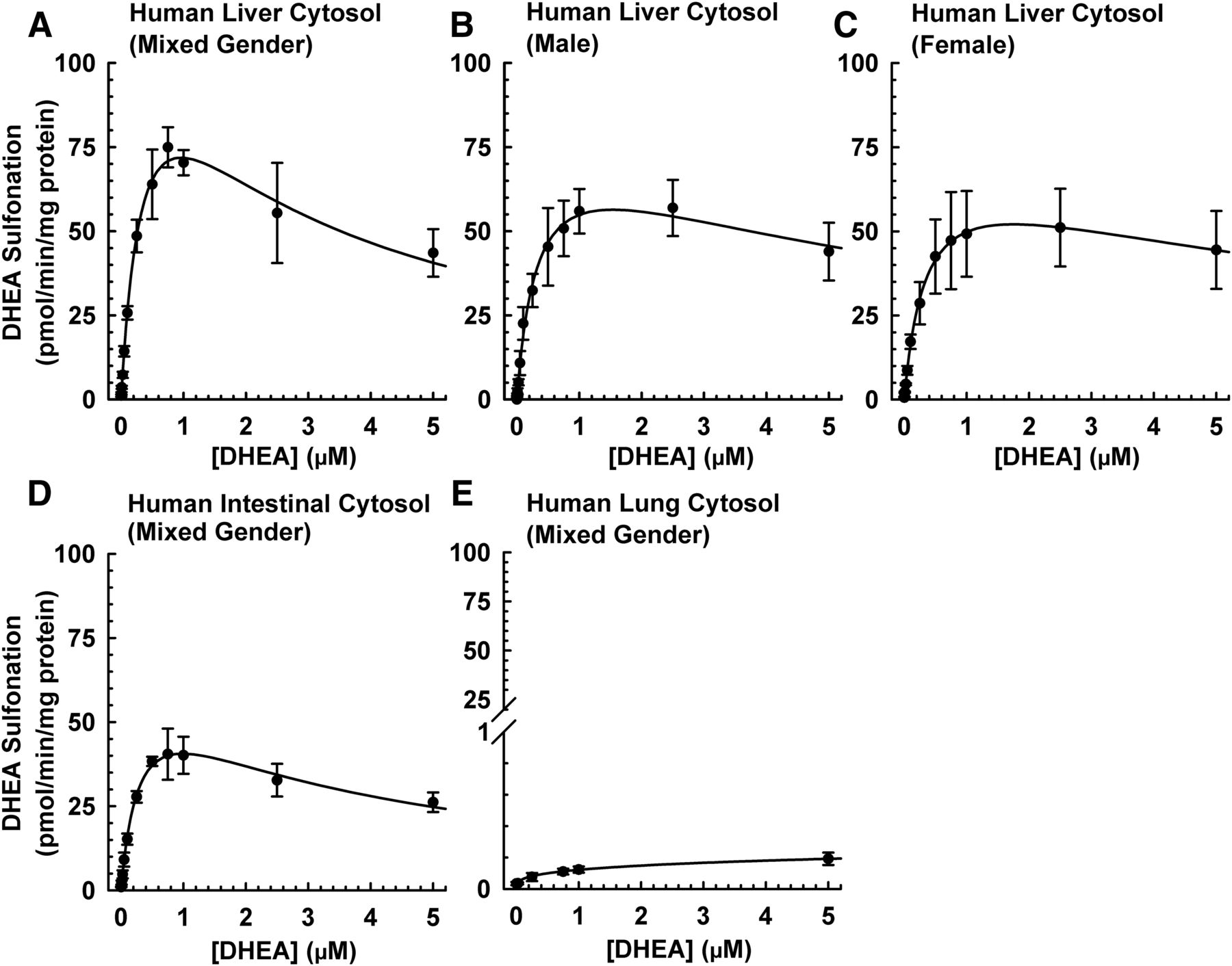
DHEA sulfonation catalyzed by cytosol isolated from human liver, intestine, and lung at various concentrations of DHEA. Human liver cytosol (mixed sex; 60 μg protein) (A), human liver cytosol (male; 60 μg protein) (B), human liver cytosol (female; 60 μg protein) (C), human intestinal cytosol (mixed sex; 60 μg protein) (D), or human lung cytosol (mixed sex; 80 μg protein) (E) was incubated at 37°C for 15 minutes (A–D) or 45 minutes (E) in an incubation mixture as indicated in Materials and Methods. The concentrations of DHEA were 0.0025, 0.005, 0.01, 0.025, 0.05, 0.1, 0.25, 0.5, 0.75, 1, 2.5, and 5 μM. The amount DHEA-S was quantified by UPLC-MS/MS. Velocity of product formation (V) and substrate concentration (S) data were analyzed by nonlinear least-squares regression and fitted to the substrate inhibition model. Data are expressed as mean ± S.D. of three independent experiments conducted on separate occasions.
Enzyme kinetic analysis of DHEA sulfonation catalyzed by human liver and intestinal cytosol
Velocity of product formation (V) and substrate concentration (S) data were analyzed by non-linear least-squares regression and fitted to the substrate inhibition model. Data are expressed as mean ± S.D. based on the data shown in Fig. 2.
Comparative Effect of Galeterone and Abiraterone Acetate on DHEA Sulfonation Catalyzed by Human Liver and Intestinal Cytosols.
To determine whether galeterone and abiraterone acetate differentially inhibited DHEA sulfonation, human liver or intestinal cytosol was incubated with DHEA (0.5 μM) and an equimolar concentration (10 μM) of galeterone, abiraterone acetate, or 0.5% v/v vehicle. The concentration of DHEA was selected based on the apparent Km values and the linear range of the velocity-substrate curves obtained for each type of cytosol (Fig. 2; Table 1). As shown in Fig. 3A, galeterone and abiraterone acetate inhibited DHEA sulfonation catalyzed by human liver cytosol (mixed sex) to a similar extent (>95%). Control experiments showed that cyproterone acetate, spironolactone, and danazol inhibited the activity by 69%, 54%, and 59%, respectively. The inhibition pattern by the five chemicals was similar for human male liver cytosol (Fig. 3B), human female liver cytosol (Fig. 3C), and human intestinal cytosol (mixed sex) (Fig. 3D). Amoxicillin, which did not inhibit human liver cytosolic DHEA sulfonation in a previous study (Bamforth et al., 1992), was used as a negative control. As expected, amoxicillin did not inhibit DHEA sulfonation by various cytosols (Fig. 3). Blank samples containing a known amount of DHEA-S and each of the five inhibitors were analyzed by UPLC-MS/MS. None of the inhibitors interfered with the quantification of DHEA-S by the analytical method (data not shown).
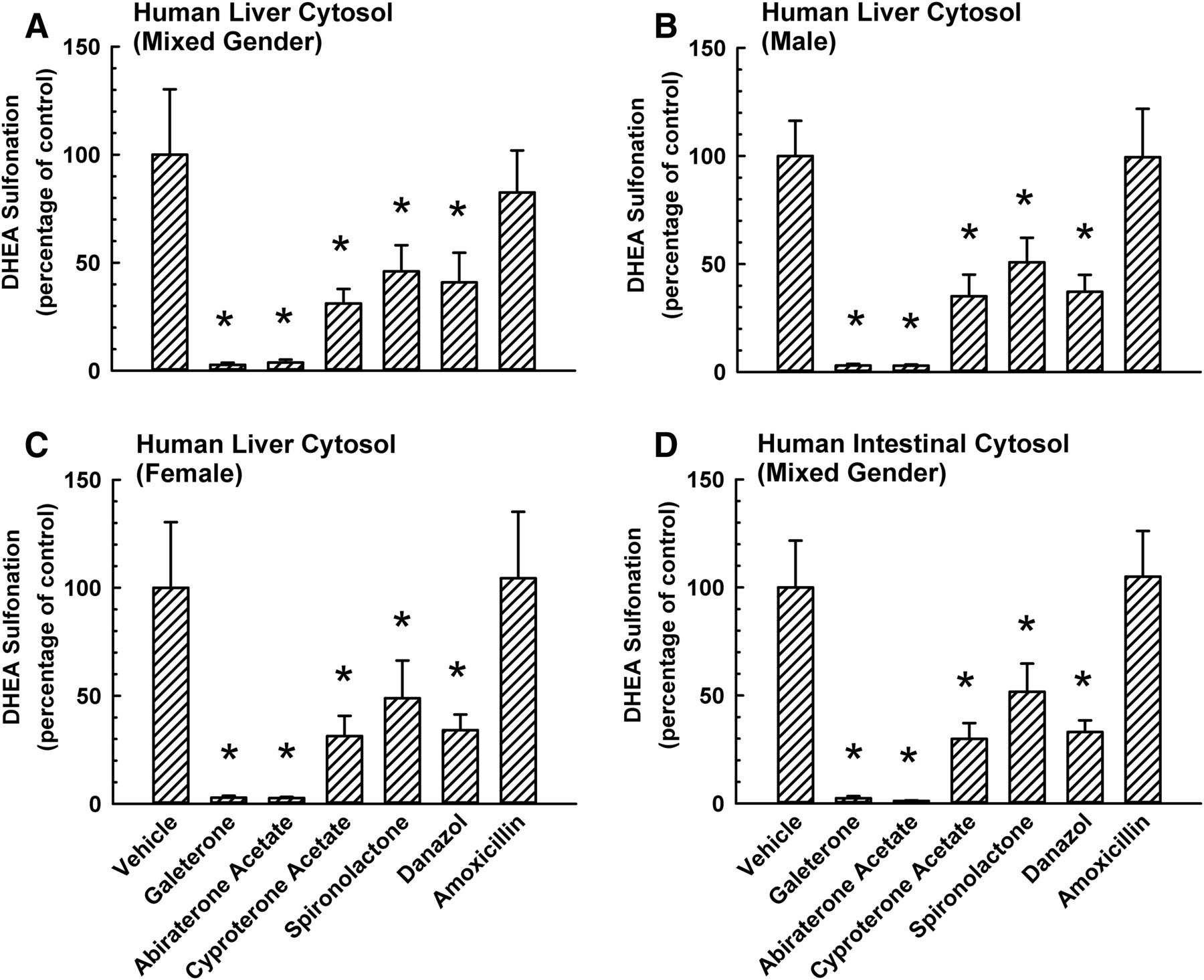
Comparative inhibitory effect of galeterone, abiraterone acetate, and other steroidal antiandrogens on DHEA sulfonation catalyzed by cytosol isolated from human liver and intestine. Human liver cytosol (mixed sex) (A), human liver cytosol (male) (B), human liver cytosol (female) (C), or human intestinal cytosol (mixed sex) (D) was incubated at 37°C for 15 minutes with DHEA (0.5 μM; 0.25% v/v methanol) and in the presence of 10 μM galeterone, abiraterone acetate, cyproterone acetate, spironolactone, danazol, amoxicillin (negative control), or vehicle (0.25% v/v methanol or acetonitrile) in an incubation mixture as described in Materials and Methods. The amount of DHEA-S was quantified by UPLC-MS/MS. Data were normalized to the amount of DHEA-S formation in the respective vehicle-treated control group and expressed as mean ± S.D. for three independent experiments conducted on separate occasions. *Significantly different from the vehicle-treated control group (P < 0.05). DHEA sulfonation in the vehicle-treated control group was 119 (pmol/min)/mg protein for human liver cytosol (mixed sex) (A), 87 (pmol/min)/mg protein for human liver cytosol (male) (B), 80 (pmol/min)/mg protein for human liver cytosol (female) (C), and 63 (pmol/min)/mg protein for human intestinal cytosol (mixed sex) (D).
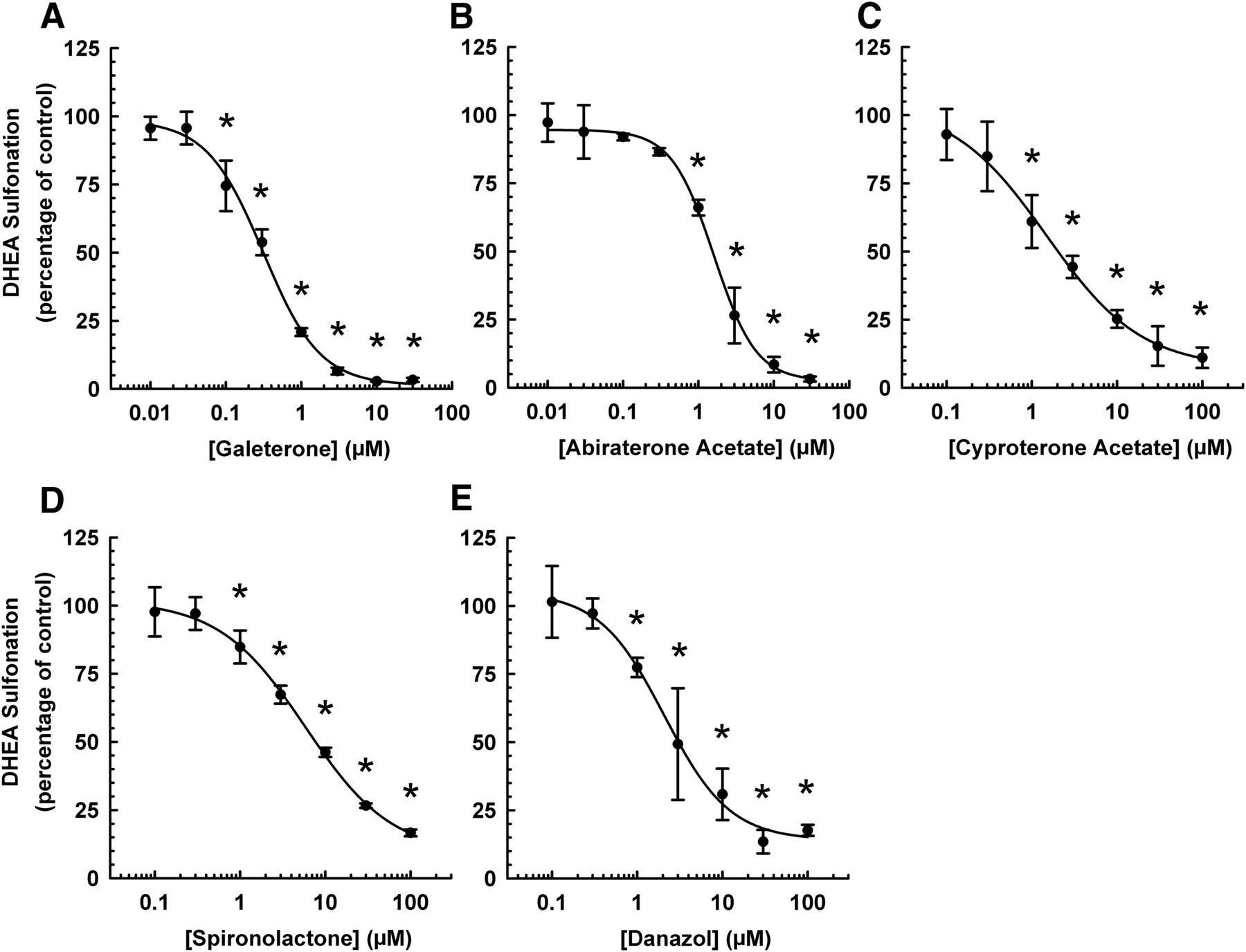
Concentration-Response Relationship in the Inhibition of Human Liver Cytosolic DHEA Sulfonation by Galeterone and Abiraterone Acetate.
Given that galeterone and abiraterone acetate inhibited DHEA sulfonation by various cytosols obtained from different sex and tissues in a similar pattern, subsequent experiments were performed with human mixed-sex liver cytosol. The next aim was to determine the minimum inhibitory concentration and inhibitory potency (IC50 values) of each chemical in the inhibition of human liver cytosolic DHEA sulfonation. As shown in Fig. 4, the minimum inhibitory concentration was 0.1 μM for galeterone and 1 μM for abiraterone acetate, whereas it was 1 μM for the positive controls (cyproterone acetate, spironolactone, and danazol). The IC50 values (mean ± S.D.) for the inhibition of human liver cytosolic DHEA sulfonation by galeterone, abiraterone acetate, cyproterone acetate, spironolactone, and danazol were 0.30 ± 0.06, 1.68 ± 0.39, 1.55 ± 0.67, 5.90 ± 1.44, and 2.16 ± 0.49 μM, respectively.

Concentration-response relationship in the inhibitory effect of galeterone, abiraterone acetate, and other steroidal antiandrogens on human liver cytosolic DHEA sulfonation. Human liver cytosol (mixed sex; 60 μg) was incubated at 37°C for 15 minutes with DHEA (0.5 μM; 0.25% v/v methanol), and with varying concentrations of galeterone (A), abiraterone acetate (B), cyproterone acetate (C), spironolactone (D), or danazol (E) in an incubation mixture as described in Materials and Methods. The concentrations were 0.1, 0.3, 1, 3, 10, 30, and 100 μM for cyproterone acetate, spironolactone, and danazol, and 0.01, 0.03, 0.1, 0.3, 1, 3, 10, and 30 μM for galeterone and abiraterone acetate. Inhibitors were replaced with the respective solvents (0.25% v/v methanol for galeterone, cyproterone acetate, and danazol; 0.25% v/v acetonitrile for spironolactone and abiraterone acetate) in the vehicle-treated control groups. The amount of DHEA-S was quantified by UPLC-MS/MS. Data were normalized to the amount of DHEA-S formation in the respective vehicle-treated control groups and expressed as mean ± S.D. of three independent experiments conducted on separate occasions. *Significantly different from the vehicle-treated control group (P < 0.05).
Mode of Inhibition of Human Liver Cytosolic DHEA Sulfonation by Galeterone and Abiraterone Acetate.
To determine the mode of inhibition and apparent inhibitory constant (Ki) for each chemical, enzyme kinetics experiments were performed with four concentrations of each inhibitor (selected based on the IC50 values, as determined from the concentration-response experiments) and four concentrations of the substrate (DHEA). Nonlinear least-squares regression analysis indicated that galeterone (Fig. 5A) and abiraterone acetate (Fig. 5B) inhibited human liver cytosolic DHEA sulfonation by competitive mode with apparent Ki values of 0.19 ± 0.07 and 0.47 ± 0.20 μM, respectively (Table 2). As shown in Fig. 5C, cyproterone acetate exhibited partial competitive inhibition with an apparent Ki value of 1.24 ± 0.41 μM (Table 2). Spironolactone (Fig. 5D) and danazol (Fig. 5E) showed partial mixed mode inhibition, with apparent Ki values of 4.63 ± 1.79 and 1.90 ± 0.82 μM, respectively (Table 2).

Lineweaver-Burk plots for the inhibition of human liver cytosolic DHEA sulfonation by galeterone, abiraterone acetate, and other steroidal antiandrogens. Human liver cytosol (mixed sex; 60 μg) was incubated at 37°C for 15 minutes with varying concentrations of galeterone (0.5, 1, or 2 μM) (A), abiraterone acetate (0.5, 1, or 2 μM) (B), cyproterone acetate (3, 10, or 30 μM) (C), spironolactone (10, 30, or 100 μM) (D), or danazol (2, 5, or 10 μM) (E) in an incubation mixture as described in Materials and Methods. The concentrations of DHEA were 0.2, 0.4, 0.6, and 0.8 μM. Inhibitors were replaced with the respective solvents (0.25% v/v methanol for galeterone, cyproterone acetate, and danazol; 0.25% v/v acetonitrile for spironolactone and abiraterone acetate) in the vehicle-treated control groups. The amount of DHEA-S was quantified by UPLC-MS/MS. Data were fitted to various inhibition models using nonlinear regression and expressed as mean ± S.D. for three independent experiments conducted on separate occasions.
Potential in vivo relevance of the in vitro inhibitory effect of galeterone, abiraterone, and other antiandrogens on DHEA sulfonation catalyzed by human liver cytosol
Shown are mode of inhibition, apparent Ki values, literature values of plasma drug concentrations [I], and calculated R values. Apparent Ki data are expressed as mean ± S.D. based on the data shown in Fig. 5.
Differential Effect of Galeterone and Abiraterone Acetate on DHEA Sulfonation Catalyzed by Human Recombinant SULT2A1, SULT2B1b, and SULT1E1.
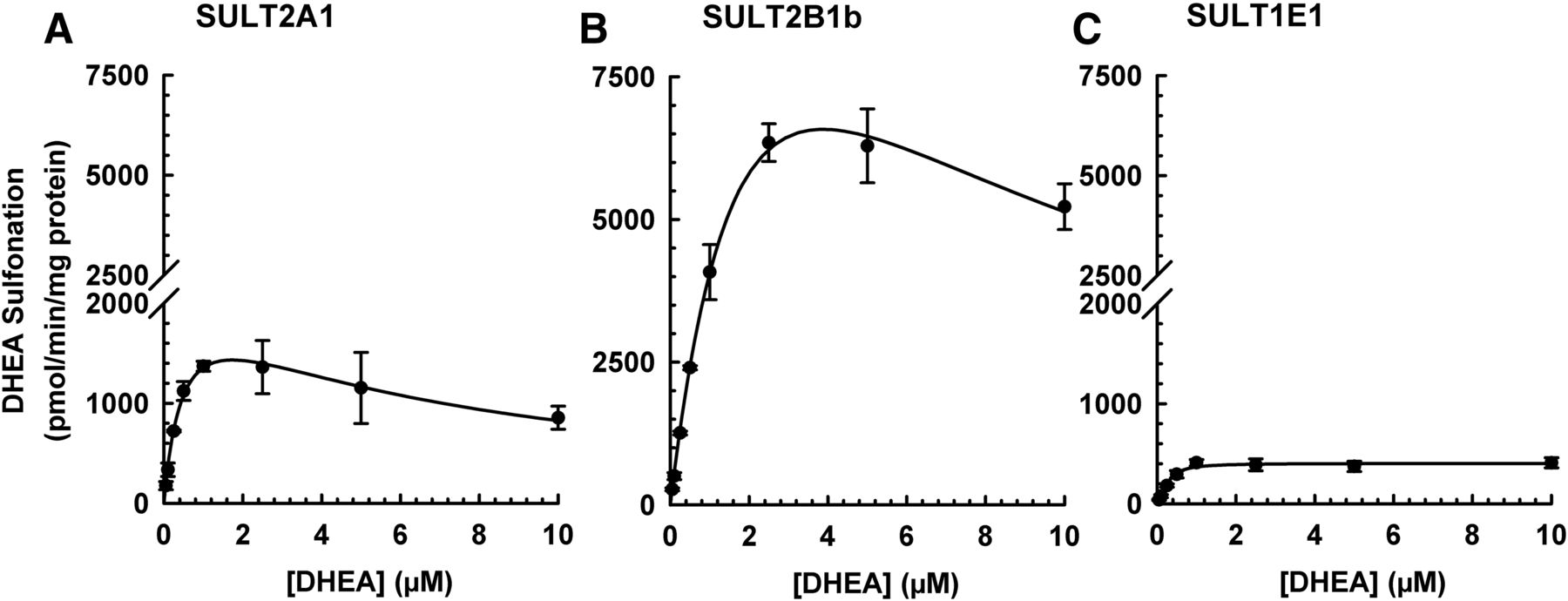
DHEA, which is a prototypical substrate of SULT2A1, has also been reported to be metabolized by SULT2B1 (Geese and Raftogianis, 2001; Meloche and Falany, 2001) and SULT1E1 (Falany et al., 1995). Therefore, an initial experiment was performed to compare the enzyme kinetics of DHEA sulfonation by human recombinant SULT2A1, SULT2B1b, and SULT1E1 to determine the optimal substrate concentration to use for inhibition experiments. The DHEA sulfonation assay was linear with respect to amount of enzyme (up to 5 μg of SULT2A1, 0.3 μg of SULT2B1b, and 5 μg of SULT1E1) and incubation time (up to 45 minutes for SULT2A1, 60 minutes for SULT2B1b and SULT1E1). Enzyme kinetic analysis showed that the kinetics of DHEA sulfonation catalyzed by SULT2A1 and SULT2B1b followed the substrate inhibition model, whereas DHEA sulfonation catalyzed by SULT1E1 followed the Hill model (Fig. 6). The apparent Vmax was 2220 ± 388 (pmol/min)/mg protein and the apparent Km was of 0.50 ± 0.15 μM for SULT2A1-catalyzed DHEA sulfonation. By comparison, the apparent Vmax was 15,934 ± 2450 (pmol/min)/mg protein and the apparent Km was 2.73 ± 0.39 μM for SULT2B1b-catalyzed DHEA sulfonation. In the case of SULT1E1-catalyzed DHEA sulfonation, the apparent Vmax was 406 ± 56 (pmol/min)/mg protein and the apparent S50 was 0.27 ± 0.07 μM. Analysis with control cytosol (corresponding to the one used for expressing the human recombinant enzymes) did not yield any metabolite (data not shown). Based on the results obtained, the substrate concentration of 0.5 μM DHEA was selected for subsequent inhibition experiments.

DHEA sulfonation catalyzed by human SULT2A1, SULT2B1b, and SULT1E1 at various concentrations of DHEA. Recombinant SULT2A1 (2.5 μg protein) (A), SULT2B1b (0.2 μg protein) (B), or SULT1E1 (5 μg protein) (C) enzyme was incubated at 37°C for either 30 (A) or 45 minutes (B and C) in an incubation mixture as described in Materials and Methods. The concentrations of DHEA were 0.0025, 0.005, 0.01, 0.025, 0.05, 0.1, 0.25, 0.5, 0.75, 1, 2.5, and 5 μM. The amount of DHEA-S was quantified by UPLC-MS/MS. Data were analyzed by nonlinear least-squares regression and fitted to the substrate inhibition (A and B) or Hill model (C). Data are expressed as mean ± S.D. of three or four independent experiments conducted on separate occasions.
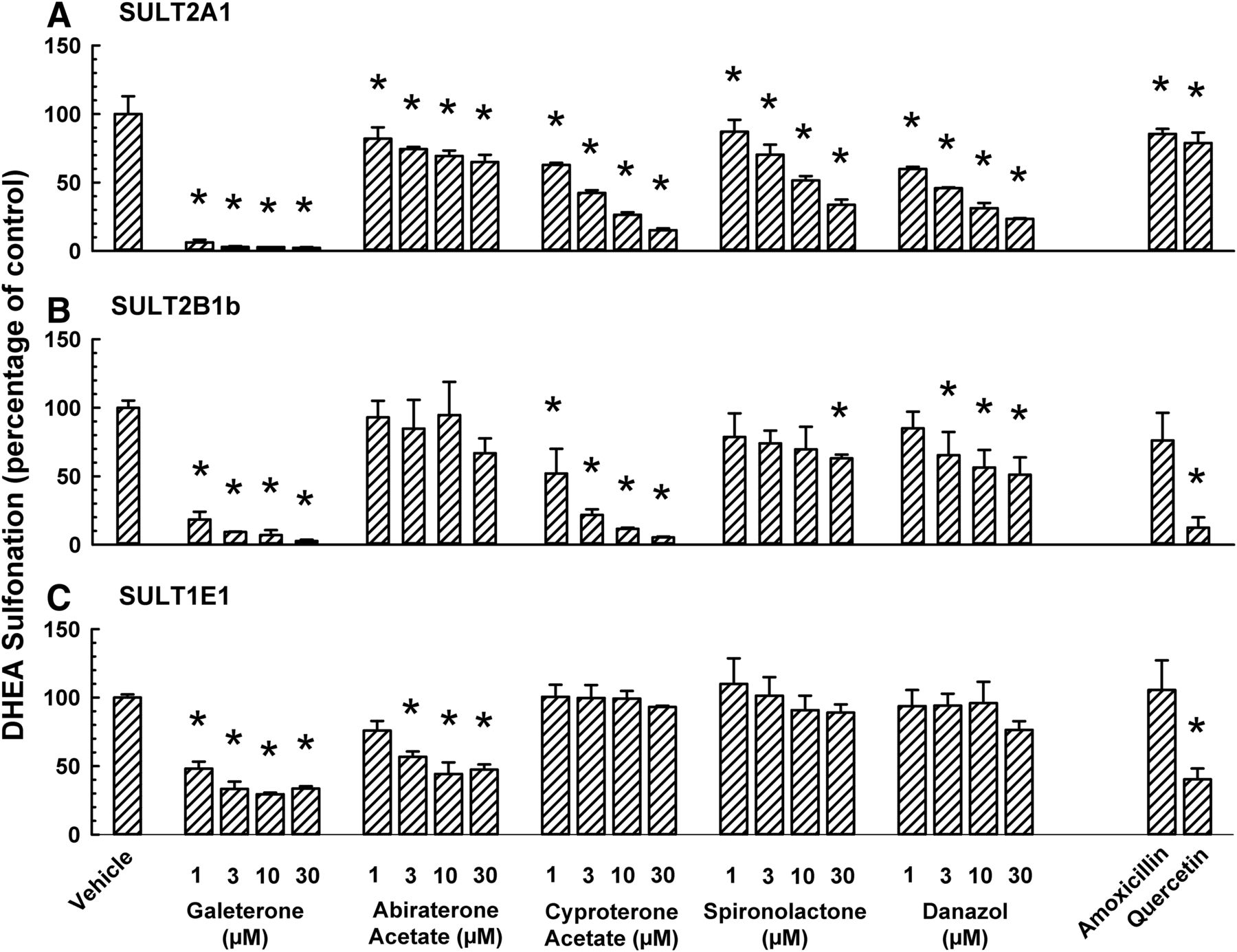
It is not known whether galeterone and abiraterone acetate differentially inhibits DHEA sulfonation catalyzed by SULT2A1, SULT2B1b, and SULT1E1. Therefore, we compared the effect of galeterone and abiraterone acetate on DHEA sulfonation catalyzed by these enzymes and compared with that of cyproterone acetate, spironolactone, and danazol. Galeterone, abiraterone acetate, cyproterone acetate, spironolactone, and danazol inhibited SULT2A1-catalyzed DHEA sulfonation (Fig. 7A). With galeterone as an inhibitor, it appears that the extent of SULT2A1 (Fig. 7A) and SULT2B1b (Fig. 7B) inhibition was greater than the extent of SULT1E1 inhibition (Fig. 7C). By comparison, abiraterone acetate inhibited SULT1E1 to a greater extent than SULT2A1 and SULT2B1b. Compared with galeterone, abiraterone acetate was a much less efficacious inhibitor of SULT2A1 (Fig. 7A) and SULT2B1b (Fig. 7B). This was in direct contrast to the data from human liver cytosol where both galeterone and abiraterone acetate inhibited the activity to a similar extent (Fig. 3A). Amoxicillin, which did not inhibit human liver cytosolic DHEA sulfonation in a previous study(Bamforth et al., 1992), was used as a negative control. It had little or no effect on SULT2A1, SULT2B1b, or SULT1E1. Quercetin was reported to be an inhibitor of ethinyl estradiol 3-O-sulfonation and inhibited SULT1E1 but not SULT2A1 (Schrag et al., 2004). Therefore, it was used as a positive control for SULT1E1 and negative control for SULT2A1. In our experiments, quercetin inhibited SULT2B1b and SULT1E1 to a far greater extent than SULT2A1 (Fig. 7).

Differential inhibition of human SULT2A1-, SULT2B1b-, and SULT1E1-catalyzed DHEA sulfonation by galeterone, abiraterone acetate, and other steroidal antiandrogens. Human recombinant SULT2A1 (2.5 μg protein) (A), SULT2B1b (0.2 μg protein) (B), or SULT1E1 (5 μg) (C) enzyme was incubated at 37°C for either 30(A) or 45 minutes (B and C) with DHEA (0.5 μM; 0.25% v/v methanol), and an antiandrogen (galeterone, abiraterone acetate, cyproterone acetate, spironolactone, or danazol; each at 1, 3, 10, or 30 μM), amoxicillin (10 μM, negative control), quercetin (1 μM, positive control), or vehicle (0.25% v/v methanol or acetonitrile) in an incubation mixture as described in Materials and Methods. The amount of DHEA-S was quantified by UPLC-MS/MS. Data are normalized to the amount of DHEA-S formation in the respective vehicle-treated control group and expressed as mean ± S.D. for three independent experiments conducted on separate occasions. *Significantly different from the vehicle-treated control group (P < 0.05). DHEA sulfonation in the vehicle-treated control group was 1893 (pmol/min)/mg protein (A), 5000 (pmol/min)/mg protein (B), and 689 (pmol/min)/mg protein (C).
Comparative Effect of Abiraterone Acetate and an Active Metabolite Abiraterone on Human Liver Cytosolic-, SULT2A1-, SULT2B1b-, and SULT1E1-Catalyzed DHEA Sulfonation.
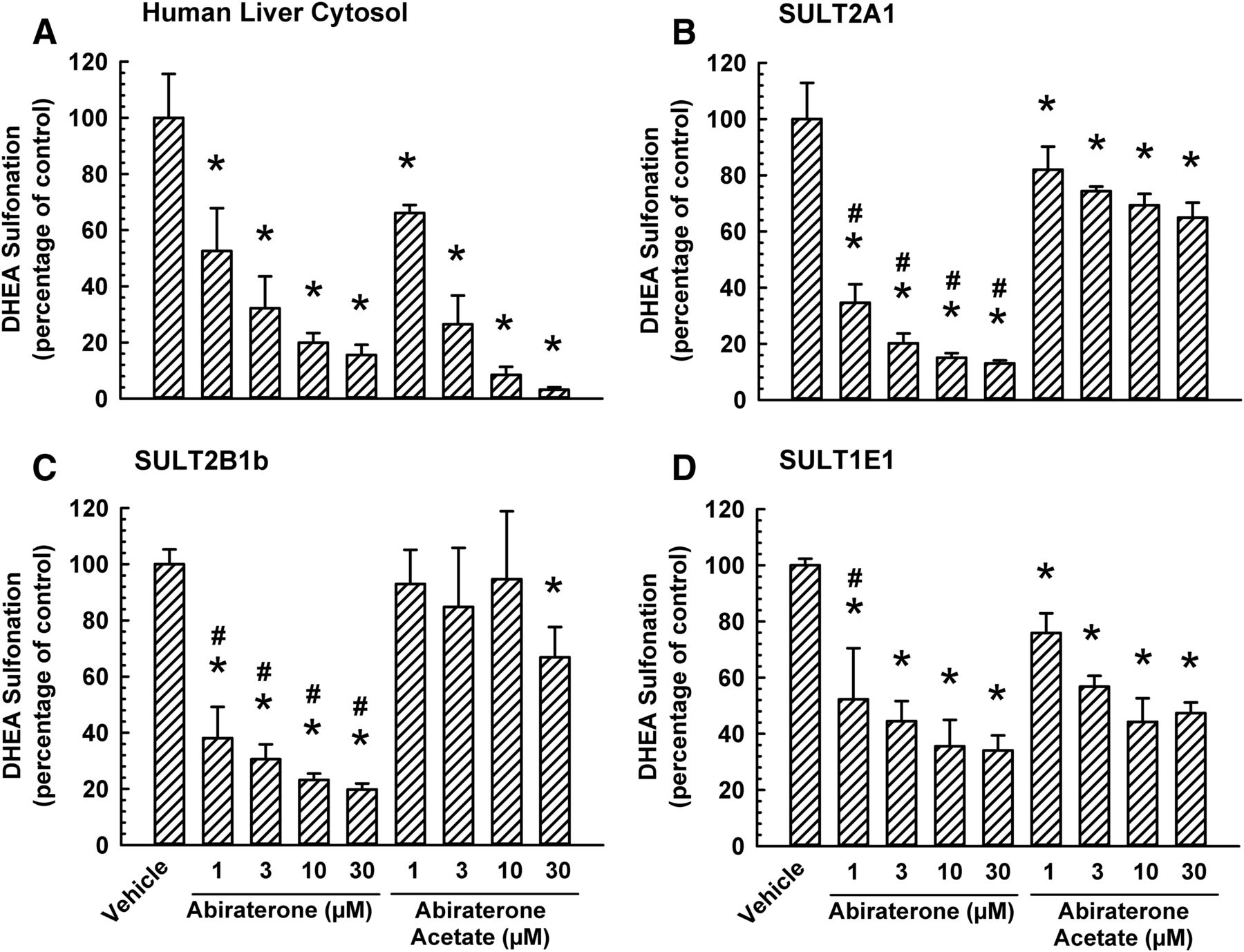
To compare the apparent inhibitory constant (Ki) for the effect of abiraterone acetate and abiraterone on DHEA sulfonation catalyzed by human liver cytosol, an enzyme inhibition experiment was conducted. Nonlinear least-squares regression analysis of enzyme kinetic data obtained from the same experiment yielded comparable apparent Ki values in incubations containing abiraterone acetate (0.51 ± 0.20 μM) or abiraterone (0.60 ± 0.09 μM). However, abiraterone acetate inhibited human liver cytosolic DHEA sulfonation to a far greater extent than its inhibition of SULT2A1- and SULT2B1-catalyzed DHEA sulfonation (Figs. 3 and 7), whereas this difference between cytosol and SULT2A1/SULT2B1 was not apparent for other inhibitors (galeterone, cyproterone acetate, spironolactone, or danazol) (Figs. 3 and 7). Therefore, we hypothesized that the prodrug abiraterone acetate was metabolized to abiraterone (an active metabolite) in incubations containing human liver cytosol, but not in incubations containing human recombinant sulfotransferase enzyme, and that the inhibition of human liver cytosolic DHEA sulfonation was due to abiraterone rather than abiraterone acetate. To test our hypothesis, we compared the effect of abiraterone acetate with abiraterone on DHEA sulfonation catalyzed by human liver cytosol and human recombinant SULT2A1, SULT2B1b, and SULT1E1. As shown in Fig. 8A, abiraterone inhibited human liver cytosolic DHEA sulfonation to a similar extent as that by abiraterone acetate. In contrast, abiraterone, at 1, 3, 10, and 30 μM, inhibited SULT2A1 and SULT2B1b to a greater extent than the corresponding concentration of abiraterone acetate (Fig. 8, B and C). As shown in Fig. 8D, 1 μM abiraterone was more effective than 1 μM abiraterone acetate in the inhibition of SULT1E1-catalyzed DHEA sulfonation, whereas at 3, 10, and 30 μM, the extent of inhibition by both chemicals was similar (Fig. 8D).

Comparative inhibitory effect of abiraterone acetate and its active metabolite abiraterone on DHEA sulfonation catalyzed by human liver cytosol, SULT2A1, SULT2B1b, and SULT1E1. Human liver cytosol (60 μg protein) (A), SULT2A1 (2.5 μg protein) (B), SULT2B1b (0.2 μg protein) (C), or SULT1E1 (5 μg) (D) enzyme was incubated at 37°C for 15 (A), 30 (B), or 45 minutes (C and D) with DHEA (0.5 μM; 0.25% v/v methanol) and with various concentrations of abiraterone acetate (1, 3, 10, or 30 μM), abiraterone (1, 3, 10, or 30 μM), or vehicle (0.25% v/v methanol or acetonitrile) in an incubation mixture as described in Materials and Methods. The amount of DHEA-S was quantified by UPLC-MS/MS. Data are normalized to the amount of DHEA-S formation in the respective vehicle-treated control group and expressed as mean ± S.D. for three independent experiments conducted on separate occasions. *Significantly different from the vehicle-treated control group (P < 0.05). #Significantly different from the abiraterone acetate-treated group (P < 0.05).
Formation of Abiraterone and Depletion of Abiraterone Acetate in Enzymatic Incubations Containing Human Liver Cytosol.
To gain additional insight as to whether DHEA sulfonation in incubations containing human liver cytosol and abiraterone acetate was due to abiraterone rather than abiraterone acetate, the formation of abiraterone (metabolite) and depletion of abiraterone acetate (substrate) in human liver cytosol was quantified. A new UPLC-MS/MS method was developed to accurately quantify abiraterone and abiraterone acetate. The bioanalytical method was sensitive, yielding a LLOQ of 0.2 nM (0.1 pmol in 100 µl incubation mixture) for abiraterone and 0.02 nM (0.02 pmol in 20 µl incubation mixture) for abiraterone acetate. As shown in Fig. 9A, the formation of abiraterone increased with incubation time and reached a plateau after 10–15 minutes. Correspondingly, in the same incubation mixture, the amount of abiraterone acetate decreased to a negligible level by 10 minutes (Fig. 9B). Control incubations containing only human liver cytosol, only vehicle (0.5% v/v methanol), or only abiraterone acetate (substrate) did not yield any abiraterone or abiraterone acetate peak. Taken together, these findings indicated that abiraterone acetate was converted almost completely to abiraterone in human liver cytosol after 15 minutes incubation time, which was the incubation time used in other experiments (Figs. 3–5 and 8).
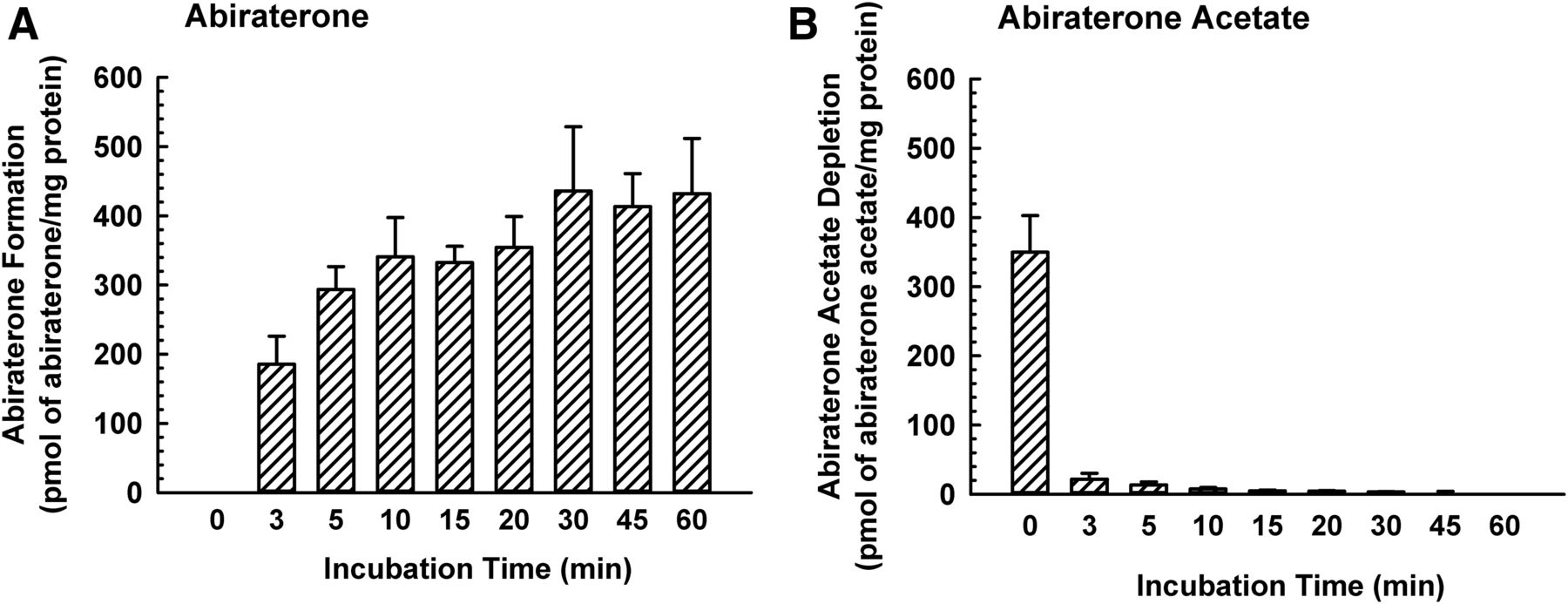
Formation of abiraterone and depletion of abiraterone acetate in human liver cytosol. Human liver cytosol (60 μg protein) was incubated with abiraterone acetate (0.1 μM) or vehicle (0.5% v/v methanol) in an incubation mixture (as described in Materials and Methods) at 37°C for 0, 3, 5, 10, 15, 20, 30, 45, or 60 minutes. The amount of abiraterone (A) and abiraterone acetate (B) was quantified by UPLC-MS/MS. Data are expressed as mean ± S.D. for three independent experiments conducted on separate occasions.
Comparative Inhibition of Human Liver Cytosolic-, SULT2A1-, SULT2B1b-, and SULT1E1-Catalyzed DHEA Sulfonation by Cyproterone Acetate and Cyproterone.
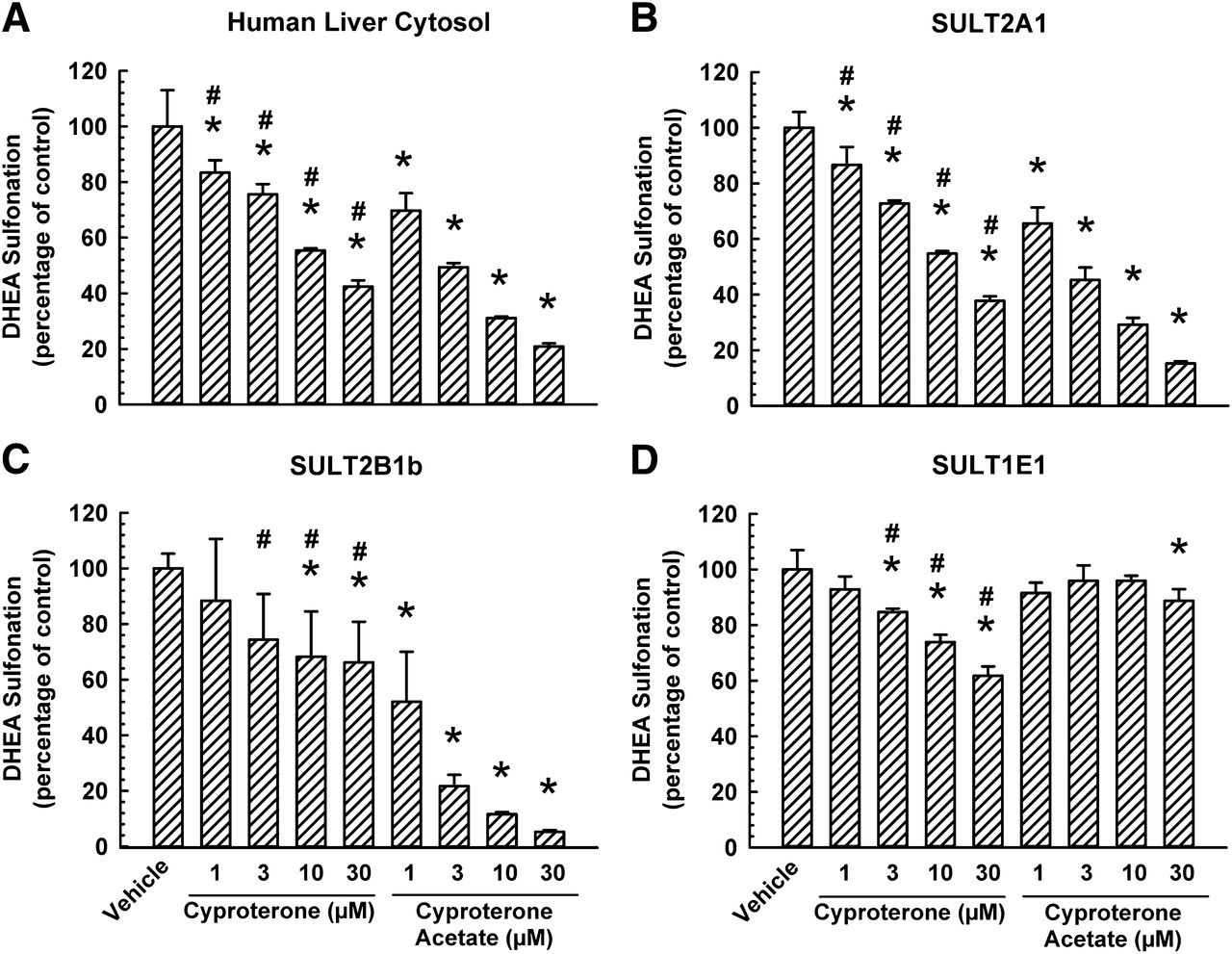
Given that abiraterone and abiraterone acetate differ by a hydroxyl group at the C3 position of the steroidal ring and that abiraterone was a more potent inhibitor of SULT2A1 than abiraterone acetate (Fig. 8B), the next question was whether the substituent (hydroxyl vs. acetate group) at the C17 position of cyproterone acetate also plays a role in the inhibition of DHEA sulfonation. Therefore, we compared the effect of cyproterone on human liver cytosol-, SULT2A1-, SULT2B1b-, and SULT1E1-catalyzed DHEA sulfonation with that of cyproterone acetate. As shown in Fig. 10A and 10B, 1, 3, 10, and 30 μM of cyproterone decreased human liver cytosol- and SULT2A1-catalyzed DHEA sulfonation to a lesser extent than that by cyproterone acetate. Similarly, 3, 10, and 30 μM of cyproterone decreased SULT2B1b-catalyzed DHEA sulfonation to a far lesser extent than that by cyproterone acetate (Fig. 10C). In contrast, SULT1E1-catalyzed DHEA sulfonation was inhibited to a greater extent by 3, 10, and 30 μM of cyproterone compared with the corresponding concentrations of cyproterone acetate (Fig. 10D).

Comparative inhibitory effect of cyproterone acetate and its metabolite cyproterone on DHEA sulfonation catalyzed by human liver cytosol, SULT2A1, SULT2B1b, and SULT1E1. Human liver cytosol (60 μg protein) (A), SULT2A1 (2.5 μg protein) (B), SULT2B1b (0.2 μg protein) (C), or SULT1E1 (5 μg) (D) enzyme was incubated at 37°C for 15 (A), 30 (B), or 45 minutes (C and D) with DHEA (0.5 μM; 0.25% v/v methanol) and with various concentrations of cyproterone acetate (1, 3, 10, or 30 μM), cyproterone (1, 3, 10, or 30 μM), or vehicle (0.25% v/v methanol or acetonitrile) in an incubation mixture as described in Materials and Methods. The amount of DHEA-S was quantified by UPLC-MS/MS. Data are normalized to the amount of DHEA-S formation in the respective vehicle-treated control group and expressed as mean ± S.D. for three independent experiments conducted on separate occasions. *Significantly different from the vehicle-treated control group (P < 0.05). #Significantly different from the cyproterone acetate-treated group (P < 0.05).
Discussion
A novel finding from our study is that the steroidal antiandrogens galeterone and abiraterone inhibited DHEA sulfonation catalyzed by human tissue cytosol, including liver cytosol (male, female, and mixed sex) and intestine cytosol (mixed sex). These drugs are relatively potent inhibitors of human liver cytosolic DHEA sulfonation, with apparent Ki values at submicromolar concentrations (Table 2). Given that abiraterone is a substrate of SULT2A1 (https://www.zytiga.com/shared/product/zytiga/zytiga-prescribing-information.pdf), this supports our finding that abiraterone inhibited DHEA sulfonation by competitive mode. The structural similarities between abiraterone and galeterone suggest that galeterone may also be a substrate of SULT2A1. As shown in the present study, galeterone inhibited DHEA sulfonation by a competitive mode. Overall, the hydroxyl group at the C3 position of the steroidal backbone, as evident in the prototypical substrate DHEA, galeterone, and abiraterone, may be essential for the interaction with amino acid moieties in the active site of SULT2A1 and to facilitate sulfonation.
Human SULT2A1 (Falany et al., 1989), SULT2B1a (Geese and Raftogianis, 2001; Meloche and Falany, 2001), SULT2B1b (Geese and Raftogianis, 2001; Meloche and Falany, 2001), and SULT1E1 (Falany et al., 1995) have been reported to catalyze DHEA sulfonation. As shown by the catalytic efficiency (ratio of Vmax/Km) calculated in the present study, SULT2B1b and SULT2A1 were most efficient in catalyzing DHEA sulfonation, whereas SULT1E1 was the least efficient. This is the first systematic enzyme kinetic analysis of DHEA sulfonation by these three enzymes in a single study. Prior to the present study, no chemical inhibitors have been reported for SULT2B1b, and only a few chemical inhibitors have been identified for human SULT2A1 and SULT1E1 enzymes. Based on studies conducted with human SULT2A1 and SULT1E1 recombinant enzymes, hydroxylated polychlorinated biphenyls (Liu et al., 2006; Ekuase et al., 2011), celecoxib (Ambadapadi et al., 2015), and tamoxifen metabolites (endoxifen, N-desmethyltamoxifen, 4-hydroxytamoxifen, tamoxifen N-oxide) (Squirewell et al., 2014) are the known chemical inhibitors of SULT2A1, whereas hydroxylated metabolites of polyhalogenated aromatic hydrocarbons (Kester et al., 2002), quercetin (Schrag et al., 2004), triclosan (Wang et al., 2004), nonsteroidal anti-inflammatory agents (e.g., meclofenamic acid, sulindac) (King et al., 2006), and trans-resveratrol and a metabolite (piceatannol) (Furimsky et al., 2008) are inhibitors of SULT1E1. In the present study, we identified galeterone, abiraterone, and cyproterone acetate as the first chemical inhibitors of SULT2B1b. Furthermore, in the case of galeterone, the extent of inhibition was greater when DHEA sulfonation was catalyzed by SULT2A1 or SULT2B1b than when it was catalyzed by SULT1E1. By comparison, cyproterone acetate, spironolactone, and danazol inhibited SULT2A1 and SULT2B1b, but not SULT1E1. This preferential inhibition may be related to the greater amino acid sequence similarity between SULT2A1 and SULT2B1b (Tibbs et al., 2015). Interestingly, among the various inhibitors, only abiraterone acetate elicited a slightly greater inhibitory effect on SULT1E1 than SULT2A1 and SULT2B1b.
The extent of the inhibition of DHEA sulfonation in incubations containing abiraterone acetate and either SULT2A1 or SULT2B1b was less than that in incubations containing abiraterone acetate and human liver cytosol. Abiraterone acetate is a prodrug formulated to improve bioavailability (Acharya et al., 2012). In vivo, upon administration, abiraterone acetate is rapidly deacetylated to its active form, abiraterone, by intestinal esterases (Stappaerts et al., 2015). In contrast to incubations containing recombinant SULT enzyme, incubations containing liver or intestinal cytosol would contain esterases (Jewell et al., 2007; Laizure et al., 2013), which are capable of metabolizing abiraterone acetate to abiraterone, resulting in inhibition of DHEA sulfonation. Time course experiment indicated that abiraterone acetate was almost completely metabolized to abiraterone by human liver cytosol within 15 minutes of incubation time, suggesting that abiraterone was the chemical responsible for the inhibition of SULT enzymes in incubations in which abiraterone acetate was added. Therefore, the presence of the larger acetyl group at the C3 position appears to hinder binding of abiraterone acetate to the active site, and the lack of a C3 hydroxyl group for sulfonation renders the acetate form of abiraterone less effective as an inhibitor of sulfotransferases. This explanation is consistent with the following findings: 1) comparable inhibitory effect of the active metabolite, abiraterone, on DHEA sulfonation catalyzed by human liver cytosol, SULT2A1, and SULT2B1b; and 2) galeterone, which is not acetylated at the C3 position, yielded similar inhibitory efficacy on DHEA sulfonation catalyzed by human liver cytosol, SULT2A1, and SULT2B1b.
As mentioned above, the hydroxyl group at the C3 position of the steroidal ring in the abiraterone molecule was required for the inhibitory effect of abiraterone on sulfotransferase-catalyzed DHEA sulfonation. By comparison, the hydroxyl group at the C17 position of cyproterone rendered this chemical to be less effective in inhibiting DHEA sulfonation when catalyzed by human liver cytosol, SULT2A1, and SULT2B1b, but somewhat more effective in inhibiting DHEA sulfonation when catalyzed by SULT1E1. This suggests differences in the structural requirement among SULT2A1, SULT2B1b, and SULT1E1 in the interaction between cyproterone and DHEA.
The potential in vivo relevance of the in vitro inhibition of sulfotransferase-catalyzed DHEA sulfonation by galeterone, abiraterone acetate, abiraterone, and other antiandrogens such as cyproterone acetate, danazol, and spironolactone depends on the in vivo plasma drug concentrations achieved in humans. By using the in vivo human plasma drug concentrations (obtained from the literature and summarized in Table 2) and the apparent Ki values obtained in the present study, the R values [1+ [I]/Ki)] were calculated (Food and Drug Administration, 2012). Based on the calculated R values (Table 2) and according to a US Food and Drug Administration guidance for drug interaction studies (Food and Drug Administration, 2012), galeterone and abiraterone are candidate inhibitors of DHEA sulfonation in vivo and future investigations in humans are warranted. Our results show that galeterone and abiraterone inhibit not only hepatic SULT2A1, but also intestinal sulfotransferases, such as SULT2B1b and SULT1E1. In the gastrointestinal tract, the intraluminal concentration of abiraterone acetate was reported to reach a peak of approximately 200 μM in healthy human volunteers who were administered with a single 250-mg dose of abiraterone acetate tablet (Zytiga, Janssen Biotech, Inc., Horsham, PA) (Stappaerts et al., 2015). This concentration is approximately 426 times greater than the apparent Ki value of approximately 0.5 μM (obtained with hepatic cytosol in the present study), suggesting a greater probability for in vivo interactions with intestinal sulfotransferases, thereby providing a potential extrahepatic mechanism for an abiraterone-based drug interaction.
Due to high expression of SULT2A1 in the liver and adrenal gland, these tissues predominantly conjugate DHEA to DHEA-S and serve as a major source of systemic DHEA-S (Mueller et al., 2015). Therefore, the present study was conducted predominantly in human liver cytosol to determine the impact of antiandrogen drugs on DHEA-S levels in the liver. As unconjugated DHEA is less metabolically stable than DHEA-S (Hornsby, 1997; Rutkowski et al., 2014), the major circulating form in the plasma is DHEA-S. Our findings suggest that galeterone and abiraterone inhibit the formation of DHEA-S, which is the form of DHEA that is circulated and transported to peripheral tissues, such as prostate, thereby decreasing the transport of DHEA-S to peripheral tissues and leading to a decrease in the in situ formation of DHEA and androgens in peripheral tissues (Mueller et al., 2015). Therefore, inhibition of hepatic sulfonation of DHEA to DHEA-S by galeterone and abiraterone may confer a beneficial effect in prostate cancer because local formation of androgens in the prostate is one of the reasons for drug resistance and progression of prostate cancer (Chang et al., 2014). Future in vivo studies will be needed to determine the overall impact of antiandrogen drugs on DHEA-S transport from liver to prostate and whether a decrease in hepatic DHEA-S, which is the main source of DHEA reaching peripheral tissues, leads to a subsequent decrease in androgen levels in the prostate.
In conclusion, galeterone and abiraterone competitively inhibited human liver cytosolic DHEA sulfonation. As no known chemical inhibitors of SULT2B1b have been reported to date, our study identified the first class of chemicals that inhibit the catalytic activity of SULT2B1b. In view of the likelihood of in vivo inhibition of DHEA sulfonation by galeterone and abiraterone (Table 2), there may be potential interactions with other drugs or chemicals sulfonated predominantly by SULT2A1 or SULT2B1b.
Authorship Contributions
Participated in research design: Yip, Bansal, Wong, Lau.
Conducted experiments: Yip, Bansal, Wong.
Performed data analysis: Yip, Bansal, Wong, Lau.
Wrote or contributed to the writing of the manuscript: Yip, Lau.
Footnotes
- Received October 16, 2017.
- Accepted February 2, 2018.
This research was supported by the Singapore Ministry of Education Academic Research Fund Tier 1 [Grant R-148-000-218-112 to A.J.L.], the National University of Singapore [Start-Up Grant R-148-000-185-133 to A.J.L.], and the Singapore Ministry of Health’s National Medical Research Council under its Cooperative Basic Research Grant scheme [Grant R-148-000-225-511 to A.J.L.]. S.B. was supported by a National University of Singapore Research Scholarship.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- DHEA
- dehydroepiandrosterone
- DHEA-S
- dehydroepiandrosterone sulfate
- DMSO
- dimethyl sulfoxide
- IC50
- half-maximal inhibitory concentration
- Ki
- inhibition constant or equilibrium dissociation constant for the enzyme-inhibitor complex
- Km
- Michaelis-Menten constant
- LLOQ
- lower limit of quantification
- MS
- mass spectrometry
- PAPS
- 3′-phosphoadenosine 5′-phosphosulfate
- S50
- substrate concentration at half-maximal activity
- SULT
- sulfotransferase
- UPLC
- ultra-high performance liquid chromatography
- Vmax
- maximum velocity
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}