


Abstract
This article is a report on a symposium entitled “Physiological Regulation of Drug Metabolism and Transport” sponsored by the American Society for Pharmacology and Experimental Therapeutics and held at the Experimental Biology 2017 meeting in Chicago, IL. The contributions of physiologic and pathophysiological regulation of drug-metabolizing enzymes and transporters to interindividual variability in drug metabolism are increasingly recognized but in many cases are not well understood. The presentations herein discuss the phenomenology, consequences, and mechanism of such regulation. CYP2D6 transgenic mice were used to provide insights into the mechanism of regulation of this enzyme in pregnancy, via hepatocyte nuclear factor 4α, small heterodimer partner, and retinoids. Regulation of intestinal and hepatic drug-processing enzymes by the intestinal microbiota via tryptophan and its metabolites was investigated. The potential impact of parasitic infections on human drug metabolism and clearance was assessed in mice infected with Schistosoma mansoni or Plasmodium chabaudi chabaudi AS, both of which produced widespread and profound effects on murine hepatic drug-metabolizing enzymes. Finally, the induction of Abcc drug efflux transporters by fasting was investigated. This was demonstrated to occur via a cAMP, protein kinase A/nuclear factor-E2-related factor 2/Sirtuin 1 pathway via antioxidant response elements on the Abcc genes.
Introduction
Drug metabolism and transport underlie a large fraction of interindividual variation in drug response. Although genetics accounts for a significant portion, regulation of drug-metabolizing enzymes and transporters by physiologic and pathophysiological factors also contribute to this variation and are relatively poorly understood. The discussions presented here focused on four prevalent physiologic factors (pregnancy, nutritional status, microbiome, and inflammatory disease) that contribute to this variability. By achieving a mechanistic understanding of how each enzyme and transporter is regulated by different physiologic factors, the goal is to be able to better predict drug responses and design rational dosage regimens in special populations and/or individuals.
Mechanisms Underlying Altered Drug Metabolism during Pregnancy (H.J.)
Medication use by pregnant women is prevalent due to pre-existing or newly developing medical conditions. In 2008, nearly 94% of women took at least one medication during pregnancy; 82% used at least one, and a quarter of pregnant women reported taking four or more medications in the first trimester (Mitchell et al., 2011). In estimating the potential negative impacts of these medications on fetal development, pharmacokinetic (PK) information in pregnant women is essential. However, such information is limited for most drugs, in part because pregnant women often have been excluded from clinical studies.
A systematic approach enabling the prediction of PK changes for a drug is critically needed. Considering that pregnancy is a complex physiologic state, the key to accurately predicting PK changes for a given drug in an individual at different gestational time points is to identify the factors that influence drug disposition in pregnancy and understand the mechanisms underlying their effects, followed by clinical verification of the results. This can potentially provide a framework to predict PK changes of any drugs in an individual, based upon which subsequent dosing recommendations can be made.
Pregnancy is accompanied by multiple physiologic changes that can impact drug disposition. Pregnancy leads to ∼30% increases in blood volume and cardiac output (Liu and Arany, 2014). Subsequent increases in renal blood flow trigger enhanced urinary excretion of renally excreted drugs. For example, renal clearance of atenolol, a drug eliminated mainly (90%) by renal excretion, is about 30% higher in second- or third-trimester pregnant women as compared with postpartum controls (Hebert et al., 2005). The effects of pregnancy on hepatic drug metabolism appear to be complex. Based on results obtained from limited clinical PK studies in pregnant women, there are metabolic pathway-dependent changes in the rate and extent of hepatic drug metabolism (Hebert et al., 2005; Tasnif et al., 2016). Drugs metabolized by cytochrome P450 (P450, CYP) enzymes including CYP3A4, 2D6, 2B6, and 2A6 exhibit increased elimination, whereas CYP1A2 metabolism is decreased in pregnant women. For example, unbound oral clearance of midazolam, a commonly used probe substrate for CYP3A4, is about 2-fold higher in the third-trimester pregnant women as compared with women at 6–10 weeks postpartum (Hebert et al., 2008). Factors responsible for these pathway-dependent changes in hepatic drug metabolism remain unclear.
One of most prominent physiologic changes accompanying pregnancy is the rising concentrations of multiple reproductive hormones. For example, the plasma concentrations of female hormones such as estrogen and progesterone increase gradually and peak at term pregnancy to levels >100-fold higher than prepregnancy levels (Liu, 2009). The effects of these hormones on the expression and activities of hepatic drug-metabolizing enzymes (DMEs) have been examined using primary human hepatocytes.
In female hepatocytes, estradiol and progesterone enhance the mRNA expression and enzyme activities of CYP2A6, 2B6, and 3A4 (Fig. 1) (Choi et al., 2013). The directional change for these P450 enzymes is consistent with that reported in clinical studies. Also, significant up-regulation of P450 expression and activity has been observed when the average hormone concentrations were at the levels attained at term pregnancy. Cortisol levels increase approximately 3-fold at term in pregnancy as compared with postpartum women (Soldin et al., 2005), and it also has been shown to increase CYP3A4 expression in primary human hepatocytes (Papageorgiou et al., 2013). Other hormones, including growth hormones, also exhibit increased plasma concentrations during pregnancy; prolactin plasma concentrations increase >10-fold during pregnancy whereas growth hormone variant and placental lactogen are produced only during pregnancy. None of these growth hormones except placental lactogen affected P450 expression/activity (Lee et al., 2014). Interestingly, placental lactogen enhanced CYP2E1 expression in the hepatocytes, although such an increase in CYP2E1-mediated drug metabolism has yet to be reported in pregnant women.

Female hormones up-regulate expression of P450 genes in female human hepatocytes. Primary human hepatocytes (HH) from different donors (n = 3/batch) were treated with (A) estradiol (1 µM), (B) progesterone (10 µM), or vehicle (ethanol) for 72 hours with frequent media changes. The mRNA levels of P450 isoforms were measured by RT-qPCR, and estimated relative to glyceraldehyde 3-phosphate dehydrogenase expression using the ΔΔCt method. Data shown are P450 expression in hormone-treated cells relative to that in vehicle-treated cells (mean ± S.D.). The data are taken from Figs. 2 and 5 of Choi et al. (2013).
Taken together, these results suggest the possibility that multiple hormones are in part responsible for the clinically reported changes in hepatic drug metabolism in pregnancy, especially in enhancing the elimination of CYP2A6, 2B6, or 3A4 substrates. The temporal changes in hepatic metabolism of these drugs predicted based on this information remain to be verified via clinical PK studies in pregnant women.
Multiple clinical studies have reported enhanced CYP2D6-mediated drug metabolism during pregnancy (Hebert et al., 2005; Tasnif et al., 2016). Because CYP2D6 has been considered a noninducible gene, this finding has intrigued many researchers in the field. However, the underlying mechanisms responsible for the changes have remained unknown, which is in part due to a lack of experimental models that could recapitulate the clinical findings. For example, hepatic expression of endogenous CYP2D homologs in rodents (e.g., rat CYP2D2 or mouse Cyp2d22) was found to be decreased (rather than increased) during pregnancy (Dickmann et al., 2008; Koh et al., 2011). Considering the possibility that this is potentially because the promoters that drive expression of CYP2D2 or Cyp2d22 are different from those of CYP2D6, CYP2D6-humanized transgenic (tg-CYP2D6) mice were used as a model. The genome of tg-CYP2D6 mice harbors the 2.5-kb upstream regulatory region of CYP2D6 along with the structural gene. Notably, CYP2D6 expression and activity were 2- to 3-fold higher in mice at term pregnancy as compared with prepregnancy or postpartum mice (Koh et al., 2014). The extent of increase was similar to the clinically reported changes in CYP2D6-mediated drug elimination in pregnant women, suggesting that the transgenic mice may serve as a in vivo model to recapitulate human CYP2D6 induction during pregnancy.
Transcriptional regulation of CYP2D6 had not been extensively studied in part because the gene was considered noninducible. Hepatocyte nuclear factor (HNF) 4α was the most studied transcriptional regulator of CYP2D6; it binds to the proximal promoter region of CYP2D6 and transactivates the promoter. In tg-CYP2D6 mice, pregnancy had minimal effects on the expression (mRNA or protein) of Hnf4α (Koh et al., 2014). Interestingly, however, the results from chromatin immunoprecipitation assays (using mouse liver tissues collected at different gestational time points) showed that Hnf4α recruitment to CYP2D6 promoter increased at term pregnancy, indicating enhanced Hnf4α activity (despite no changes in Hnf4α expression).
Subsequent studies revealed that decreased hepatic expression of the transcriptional repressor, small heterodimer partner (Shp), may underlie the altered Hnf4α activity during pregnancy; reduced Shp expression led to derepression of Hnf4α action on CYP2D6 promoter in mice. Among known regulators of Shp expression, retinoids were found to be potentially responsible for the decrease in Shp expression at term pregnancy. Hepatic levels of bioactive retinoids, including all-trans retinoic acid, were decreased at term pregnancy as compared with prepregnancy levels in tg-CYP2D6 mice (Koh et al., 2014). Taken together, these results suggest that hepatic SHP expression is lower in pregnancy in part due to decreased retinoid levels, and this leads to derepression of the CYP2D6 promoter. Whether such changes in retinoid homeostasis indeed occur in pregnant women and thus cause CYP2D6 induction remains to be examined clinically.
In summary, factors (such as female hormones and retinoids) potentially responsible for altered drug metabolism during pregnancy have been identified through various in vitro and in vivo animal studies. Although these findings remain to be further verified via clinical studies, this information should be useful in performing PK modeling and guiding the design of clinical studies for developing strategies to determine the optimal dosing regimen for pregnant women.
L-Tryptophan and Bacterial Modulation of Intestinal and Hepatic Gene Expression (J.Y.C., J.L.D.)
Gut microbiota are increasingly recognized as being important in regulating the expression of phase I and II DMEs and transporters (together called “drug-processing genes”) in the host liver. Comparison of the expression of liver genes between conventional (CV) mice (mice with a microbiome) and age-matched germ-free (GF) mice (mice lacking a microbiome) revealed that many drug-processing genes are differentially regulated (Bjorkholm et al., 2009; Toda et al., 2009a,b; Selwyn et al., 2015a,b). For example, in adult male mice, Cyp1a2 mRNA increased in livers of GF mice, whereas Cyp3a11 mRNA decreased (Selwyn et al., 2015b). It is well known that Cyp1a2 is a prototypical target gene of the ligand-activated transcription factor aryl hydrocarbon receptor (Ahr) (Denison et al., 1988; Nukaya et al., 2009) whereas Cyp3a11 is a prototypical target gene of the major xenobiotic-sensing nuclear receptor pregnane X receptor (Pxr/Nr1i2) (Kliewer et al., 1998). These findings indicate that certain microbial metabolites may be responsible for the altered expression of drug-processing genes in the liver.
Several tryptophan metabolites produced by intestinal microbiota are Ahr activators, including indole, indole-3-acetic acid, indole-3-aldehyde, tryptamine, and 3-methyl-indole, as well as kynurenic acid which can be derived from both microbiota and host (Kuc et al., 2008; DiNatale et al., 2010; Hubbard et al., 2015a). These metabolites dose-dependently activate human AHR in HepG2 (40/6) cells containing a luciferase reporter under control of the Cyp1a1 enhancer, and in Caco2 cells as evidenced by increased luciferase activity or CYP1A1 mRNA, respectively. Furthermore, activation of AHR by indole in Caco2 cells increased the protein expression of interleukin 6 (IL-6), which regulates cell growth and differentiation as well as immune function. In the presence of IL-1β, this effect was synergistic (Hubbard et al., 2015a).
Indole also has been shown to selectively activate human AHR in primary peritoneal macrophages for immune signaling between microbiota and host intestines (Hubbard et al., 2015a), providing further evidence for the role of AHR in intestinal immune function (Hubbard et al., 2015b). The indole derivatives from intestinal bacteria have a bimolecular (2:1) ligand binding stoichiometry for the activation of human AHR but not mouse Ahr (Hubbard et al., 2015a). This is proposed to be unique for microbiota-host signaling because previously identified non-microbiota–derived AHR activators have unimolecular (1:1) ligand binding stoichiometry (Flaveny et al., 2009; Hubbard et al., 2015a).
Indole-3-propionic acid (IPA), another microbial derivative of tryptophan, did not demonstrate dose-dependent activation of AHR in HepG2 (40/6) cells (Hubbard et al., 2015b). However, IPA increased the mRNA expression of the Pxr-target gene ATP-binding cassette, subfamily B, member 1A (Abcb1a/Mdr1a) in jejunum villi cells in a Pxr-dependent manner (Venkatesh et al., 2014). Because microbial metabolites are absent in GF mice, this led to the hypothesis that exposure to exogenous compounds exclusively metabolized by bacteria could regulate intestinal barrier function (Venkatesh et al., 2014). Although IPA alone is a weak human PXR (hPXR) activator, IPA in the presence of indole dose-dependently activated multidrug-resistance-associated protein 2 (MRP2) luciferase reporter in 293T cells cotransfected with hPXR plasmid (Venkatesh et al., 2014). Moreover, in mouse small intestinal mucosa, IPA increased the mRNA expression of Mdr1a, Cyp3a11, and UDP glucuronosyltransferase family 1 member A1 (Ugt1a1), and this was dependent on both tryptophan and the bacterium Clostridium sporogenes (Wikoff et al., 2009; Venkatesh et al., 2014). Activation of PXR in mouse intestine by IPA was shown to be a potential regulatory route for intestinal barrier function through Toll-like receptor 4, which mediates the innate immune response to the bacterial cell wall component lipopolysaccharide (Venkatesh et al., 2014).
To an extent, several tryptophan metabolites (indole, IPA, etc.) have been shown to activate mouse and/or human AHR and PXR to mediate several intestinal barrier functions. However, in vitro models evaluating AHR and PXR activation in the liver by tryptophan metabolites lack the gut bacteria–liver axis. The tryptophan metabolite IPA activates Pxr in mouse small intestine, but this activity has not been demonstrated in mouse liver or human hepatocytes. In addition, the effect of tryptophan on the intestinal microbiota has not been shown. Therefore, we determined the effect of oral exposure to tryptophan on the intestinal microbiota composition in CV mice as well as hepatic and intestinal Ahr/Pxr-target gene expression in CV mice and in human liver-cancer derived HepaRG cells. To test the hypothesis that the microbial metabolite IPA normalizes the constitutive Pxr signaling in liver, we also determined the effect of oral exposure to IPA in livers of GF mice and in HepaRG cells.
For animal studies, adult CV male mice were orally exposed to tryptophan (10, 20, or 40 mg/kg) for 6 consecutive days, and DNA from the large intestinal content was subjected to 16S ribosomal RNA (rRNA) sequencing targeting the V4 hypervariable region. The data were analyzed using QIIME (Wang et al., 2007; Caporaso et al., 2010a,b; Edgar, 2010; McDonald et al., 2012). The mRNA expression of Ahr- and Pxr-target genes in liver and intestine was quantified by reverse transcription quantitative polymerase chain reaction (RT-qPCR). In a separate study, GF mice were orally exposed to IPA (40 mg/kg) for 4 consecutive days. Tissues for both studies were collected 24 hours after the final treatment, and total RNA was isolated from liver and intestinal tissue sections. For HepaRG cell culture studies, differentiated HepaRG cells were treated with vehicle (0.1% dimethyl sulfoxide), tryptophan (10, 100, or 250 µM), or IPA (100 or 250 µM) for 24 hours followed by total RNA isolation. Additional methodological details are provided in the Supplemental Material.
Oral tryptophan exposure in general had no major effect on the gut microbiota composition, and only the 20 mg/kg dose of tryptophan produced the most prominent effect. For example, two classes of the phylum Firmicutes, namely Clostridia and Erysipelotrichi, were enriched in the 20 mg/kg tryptophan treatment group but were lower in the 40 mg/kg tryptophan treatment group (Fig. 2A). To note, Actinobacteria were particularly enriched in the 20 mg/kg tryptophan treatment group and were lower in all other groups. Conversely, the class Bacteroidia in the phylum Bacteroidetes decreased with 20 mg/kg tryptophan.

(A) Relative abundances of bacteria in large intestinal content of CV mice (n = 3 per group). The 16S ribosomal RNA sequencing data were analyzed by QIIME and are expressed as the percentage of operational taxonomic units (OTUs). Examples of differentially regulated intestinal bacteria with a mean composition greater than 0.01% for at least one treatment group are shown at the class level. Letters (a and b) represent treatment subgroups with significantly differentiated means (analysis of variance followed by Duncan’s post hoc test, P < 0.05). (B) RT-qPCR quantification of mRNAs in various sections of intestine of CV mice treated with corn oil (vehicle) (n = 3) or tryptophan (10, 20, or 40 mg/kg; n = 5 per group) (left panel), as well as GF mice treated with phosphate-buffered saline (PBS, vehicle) or IPA (40 mg/kg; n = 3 per group). Data are expressed as percentage of β-actin. Statistical significance between groups was determined by analysis of variance followed by Duncan’s post hoc test (P < 0.05) for the tryptophan study and Student’s t test (P < 0.05) for the IPA study. Data are presented as mean ± S.E.M. *Statistically significant differences between vehicle and treatment groups.
In intestine of CV mice, the basal expression of the Ahr-target gene Cyp1a2 and the Pxr-target gene Cyp3a11 gradually decreased from duodenum to large intestine. Conversely, the Pxr-target gene Mdr1a gradually increased from duodenum to large intestine (Fig. 2B). Surprisingly, in intestine of CV mice orally exposed to tryptophan, there was no increase in the Ahr-target gene Cyp1a2 mRNA in any sections of intestine; in fact, Cyp1a2 mRNA decreased in jejunum at all three doses of tryptophan and in ileum at the 20 mg/kg tryptophan dose. Cyp3a11 mRNA increased at all doses of tryptophan in duodenum where the highest basal expression was observed and remained unchanged in other sections of intestine. Mdr1a mRNA was up-regulated by the highest dose of tryptophan (40 mg/kg) in all three sections of small intestine and tended to increase in large intestine, although statistical significance was not achieved. However, in the intestine of GF mice IPA had no effect on Cyp3a11 mRNA expression, although similar to the literature report (Venkatesh et al., 2014); the Pxr-target gene Mdr1a was up-regulated by IPA in the small intestine. In addition, IPA had no effect on the Ahr-target gene Cyp1a2 mRNA expression in any sections of the intestine of GF mice.
Contrary to our hypothesis, in the livers of CV mice tryptophan did not increase the mRNA expression of Cyp3a11 (Fig. 3A), and the hepatic IPA concentrations were not significantly different between any groups (Fig. 3B). Hepatic concentrations of other tryptophan metabolites were unchanged (data not shown). This suggests that although IPA has been shown to be a Pxr activator in intestine, its hepatic concentrations in both basal and tryptophan treated groups are not sufficient to activate hepatic Pxr-signaling. Similarly, in livers of GF mice IPA did not alter the hepatic Cyp3a11 or Mdr1a mRNA expression. Surprisingly, in livers of GF mice IPA increased the mRNA expression of the Ahr-target gene Cyp1a2 (Fig. 3A). In HepaRG cells, similar to data obtained from GF mouse livers, IPA at 100 and 250 μM increased CYP1A2 mRNA. Interestingly, IPA increased CYP3A4 mRNA expression, whereas tryptophan alone generally did not affect CYP3A4 mRNA except for a moderate increase at 10 μM tryptophan (Fig. 3C).
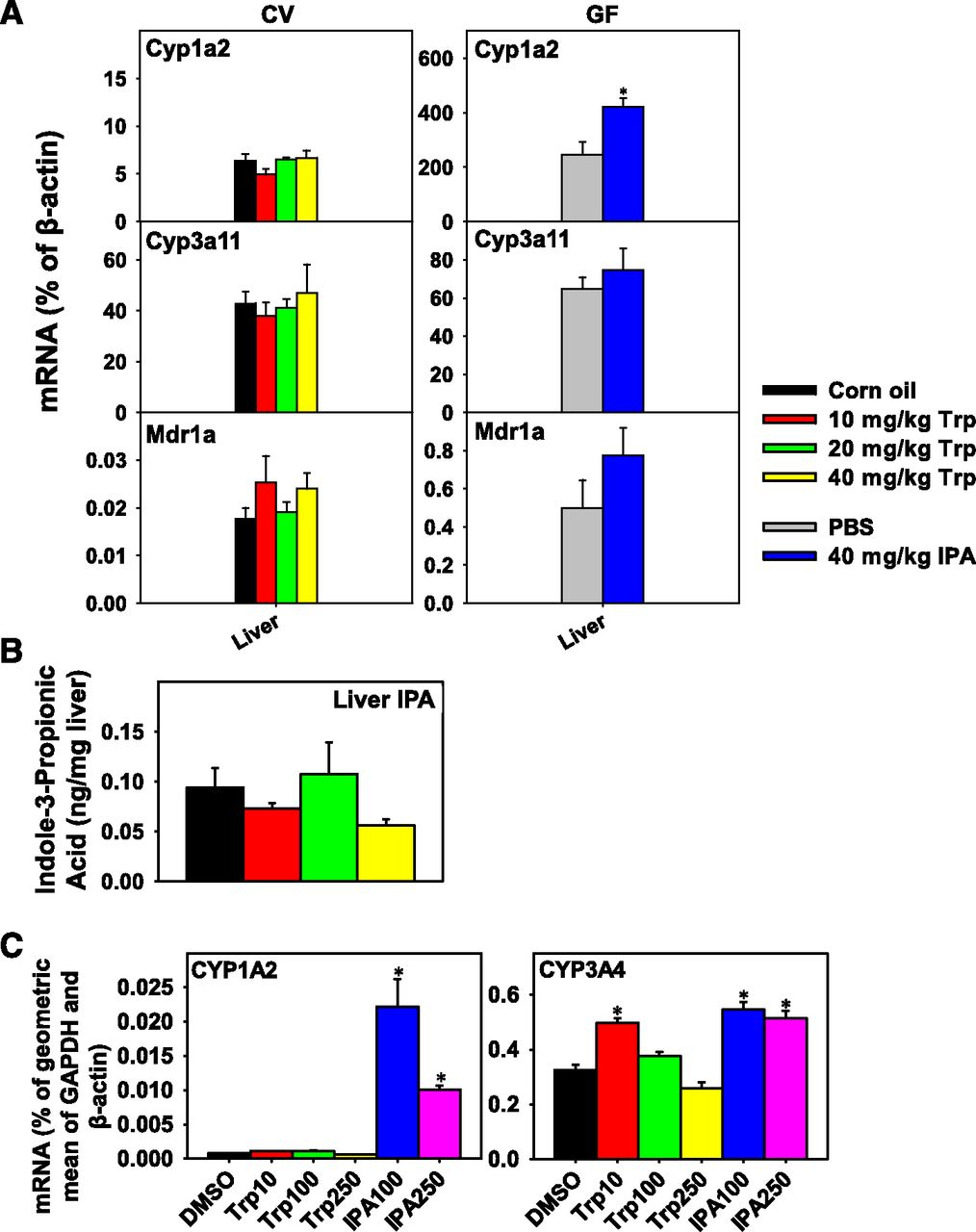
(A) RT-qPCR quantification of mRNAs in the liver of CV mice orally gavaged with corn oil (vehicle) (n = 3) or tryptophan (10, 20, or 40 mg/kg; n = 5 per group), as well as GF mice orally gavaged with PBS (vehicle) or IPA (40 mg/kg; n = 3 per group). Data are expressed as percentage of β-actin. (B) Quantification of IPA by high-performance liquid chromatography coupled with tandem mass spectrometry (HPLC-MS/MS) in liver of CV mice normalized to liver mass. (C) RT-qPCR quantification of mRNAs in HepaRG cells treated with 0.1% dimethyl sulfoxide (DMSO, vehicle), tryptophan (10, 100, or 250 µM), and IPA (100 or 250 µM). Statistical significance between groups was determined by analysis of variance followed by Duncan’s post hoc test (P < 0.05) for the tryptophan study and Student’s t test (P < 0.05) for the IPA study. Data are presented as mean ± S.E.M. *Statistically significant differences between vehicle and treatment groups.
In summary, oral exposure to tryptophan in general had minimal effect on gut microbiota composition at the class level, except for the 20 mg/kg tryptophan dose, which markedly increased Actinobacteria. In addition, at this dose tryptophan also tended to enrich Clostridia and Erysipelotrichi, but tended to decrease Bacteroidia. The class Clostridia contains the bacterium Clostridium sporogenes, which is known to produce IPA; however, enrichment of this class did not affect IPA levels.
Species in both Clostridia and Erysipelotrichi enriched by a high-fat diet can induce obesity independent of diet through greater energy harvesting (Turnbaugh et al., 2006). Profound tissue specificity was observed in tryptophan- and IPA-mediated effects on Ahr- and Pxr-signaling in that oral tryptophan exposure increased the mRNAs of the PXR-target genes Cyp3a11 and Mdr1a in certain sections of intestine but had no effect on the expression of these genes in liver of CV mice. Surprisingly, although various tryptophan host and microbial metabolites were shown to be Ahr-activators (DiNatale et al., 2010; Hubbard et al., 2015a,b), tryptophan at the given doses actually decreased Cyp1a2 mRNA expression, especially in jejunum of CV mice. This suggests that microbial tryptophan metabolites inhibit or are antagonistic to host-specific Ahr activation in intestine.
Venkatesh et al. (2014) concluded that IPA, produced from tryptophan and in the presence of other tryptophan-derived indole compounds and a gut microbiome, can regulate intestinal barrier function through Pxr. In our CV mouse model, tryptophan increased the expression of intestinal Pxr-targeted drug-processing genes; however, in our GF mouse model IPA generally had limited to no effect. Therefore, IPA may be a selective Pxr modulator in that, although it regulates the Pxr-target pathways for immune response, it does not modulate the classic Pxr-target drug metabolism pathways.
In addition, data from the livers of IPA-treated GF mice and HepaRG cells suggest that IPA and/or its host metabolites may be novel Ahr/AHR activators in liver. Previously, IPA-treated HepG2 (40/6) cells assessing human AHR activation had increased luciferase activity at 1 μM, but not at 5 or 10 μM (Hubbard et al., 2015a). This suggests a difference in activator selectivity by human and mouse AHR as well as specific binding differences between cell models.
In addition, the lack of an in vivo effect of IPA on hepatic Pxr signaling may be due to limited bioavailability. Further studies are necessary to evaluate tryptophan metabolism in the small intestines to determine which native bacteria may produce metabolites altering gene expression.
Drug Metabolism in Inflammation and Parasitic Infection (E.T.M., S.M.M., T.J.L.)
In both humans and experimental animals, infections of various types have been associated with changes in drug-metabolizing capacity, accompanied by changes in expression of individual DMEs (Aitken et al., 2006; Morgan, 2017). Stimulation of the innate immune system via Toll-like receptors contributes to some of these changes (Shah et al., 2016), with proinflammatory cytokines such as IL-1β and IL-6, tumor necrosis factor-α, and interferons able to act directly via their respective receptors expressed on hepatocytes to regulate expression of DME genes (Aitken et al., 2006).
Cytokines act by different mechanisms to regulate distinct subsets of DMEs. In addition to cytokines, ligation of integrin receptors may also lead to differential expression of DME genes during viral infections (Jonsson-Schmunk et al., 2016). This study will focus on parasitic infections, which have not been studied as much as bacterial or viral infections with respect to their impact on drug metabolism, perhaps because their main prevalence is in developing countries.
Schistosomiasis.
Schistosomiasis, a disease caused by infection with various species of trematode worms, affects more than 200 million people worldwide, the majority of whom live in endemic areas mostly in Africa, Asia, and South America (WHO, 2018). Humans are exposed to schistosomal cercariae in water contaminated with human feces and urine. The cercariae penetrate the skin and lose their tails, becoming schistosomulae that circulate to the portal blood, where paired male and female worms mature and reproduce in the liver. When the female begins laying eggs, some eggs become deposited in the liver eliciting a TH2-type T-cell response via various egg glycoprotein antigens (Everts et al., 2009; Steinfelder et al., 2009). Granulomas form around the trapped eggs, in turn leading to liver fibrosis, portal hypertension, and hepatomegaly.
As we reviewed recently (Morgan, 2017), 2- to 5-fold increases in half-life or areas under the curve (AUC) of the antischistosomal drug praziquantel have been reported in infected patients with liver disease. Praziquantel is metabolized by CYP2D6 and CYP3A4. The AUC of propranolol, which is metabolized by CYP2D6 and CYP1A2, was reported to be increased by a similar magnitude. Given that, as noted previously, the host response to schistosomal egg-laying is dominated by TH2-type cytokines rather than the classic TH1 proinflammatory cytokines previously shown to modulate DMEs, we determined the impact of infection with Schistosoma mansoni on the expression of DMEs in mouse liver.
Analyses were conducted at 30 days postinfection (dpi), just before the egg-laying stage, and at 45 dpi during the granulomatous stage. A panel of the mRNAs of P450s, Ugts, sulfotransferases (Sult), and nuclear receptors, as well as flavin-containing monooxygenase 3 (Fmo3) were surveyed. At 30 dpi, modest increases (up to 50%) in the expression of Cyp1a2, 2c29, 2e1, 3a11, 2j5, 4f13, and 4f18 were observed, whereas of the phase II enzymes only Sult1a1/2 was induced at this time point (Mimche et al., 2014). These changes were accompanied by 50% increases in expression of the nuclear receptors retinoid X receptor α (Rxra), constitutive androstane receptor (Car), and peroxisome proliferator-activated receptor α (Ppara), but not Pxr. At 45 dpi, the mRNAs of 13 of the 19 Cyp mRNAs studied were dramatically down-regulated, whereas Cyp4f16 and Cyp4f18 were induced. Ugt1a1, 1a9, and 2b5, Sult1b1, 2a1/2 as well as Fmo3 were also down-regulated, as were all of the nuclear receptors measured (by about 60%) (Mimche et al., 2014).
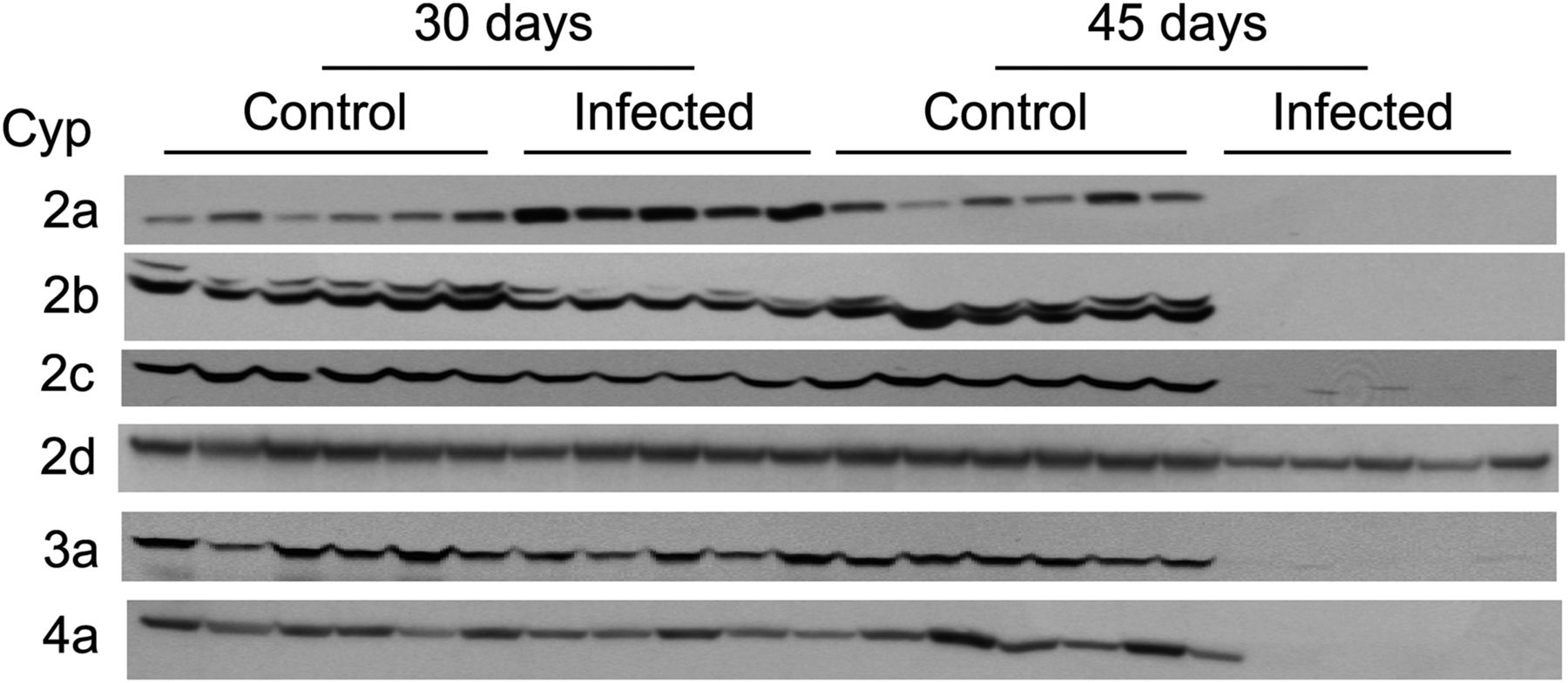
To assess the implications of these results for the expression of DME proteins, we conducted Western blot analysis of P450 protein expression in these livers. As seen in Fig. 4, while Cyp2a proteins were induced about 3-fold at 30 dpi, all P450 proteins except Cyp2d (which was down-regulated by 75%) were virtually absent from the livers of infected animals at 45 dpi (Mimche et al., 2014). This indicates the potential for large, clinically relevant changes in drug metabolism and clearance in schistosomiasis. When we examined the cytokine profiles in the livers of these animals, a TH2-dominated response was confirmed with IL-4, IL-5, and IL-13 all strongly up-regulated. Although this is only correlative, it is suggestive of a role for TH2 cytokines in P450 regulation in schistosomal disease. This hypothesis needs to be investigated, but the possibility that the observed effects are a consequence of liver damage should also be considered.

Effect of S. mansoni infection on protein expression of hepatic P450s in female Swiss Webster mice. Reproduced from Mimche et al. (2014). Mice were infected with S. mansoni and sacrificed at 30 and 45 dpi, and livers were harvested. P450 protein levels were assessed by Western blotting S9 fractions. Each lane corresponds to a different mouse and contains 32 μg S9 protein, except the 45-days-infected group, which has 128 μg. All the primary antibodies used for Western blotting were polyclonal, from rabbit: Cyp2a, anti-human CYP2A13 (Sc-33214) from SantaCruz Biotechnology (Dallas, TX); Cyp 2b and 3a, anti-rat CYP2B1 and CYP3A1 from Dr. James Halpert; Cyp2d9, anti-mouse Cyp2d9 from Dr. Masahiko Negishi; Cyp4a, anti-human CYP4A11 (Ab-140635) from Abcam (Cambridge, MA); and Cyp2c, anti-rat CYP2C11 prepared in our own laboratory. Additional Western blotting details are in Mimche et al. (2014).
Malaria.
Malaria is among the world’s life threatening tropical diseases. In 2015, about 214 million cases and 438,000 deaths were reported (WHO, 2015). The prevalence of malaria around the globe is comparable to that of schistosomiasis, at about 200 million cases per year.
Malaria is spread by infected mosquitos, which, when taking a human blood meal, inject sporozoites in the dermis of the host. These travel to the liver where they infect hepatocytes and mature into schizonts. This initial hepatic stage is clinically silent, and nothing is known about how it may impact drug metabolism. The schizonts rupture, releasing merozoites into the blood that in turn infect red blood cells. Infected red blood cells can sequester by binding to endothelial cells in different organs, including the liver, where eventually they mature to become blood-stage schizonts; these in turn rupture and release merozoites, to perpetuate the asexual erythrocytic cycle.
A summary of the studies that have been performed to date on drug PK in humans with malaria, all of which have been with Plasmodium falciparum infection, are shown in Table 1. Most of these studies were in patients with uncomplicated malaria and concerned measurement of plasma concentrations of the antimalarial drug quinine, which, similarly to the anti-schistosomal drug praziquantel, is metabolized mainly by CYP3A4. Comparison of clearance of quinine in the same patients during infection and in convalescence indicated that uncomplicated malarial infection is associated with increased plasma concentrations of quinine, likely arising from increased AUC, reduced clearance, and slightly reduced free fraction of quinine in the plasma. Some of the changes in PK may be associated with reduced hepatic blood flow, which was measured in one study using indocyanine green. In studies that included malaria with central nervous system involvement, the magnitudes of the effects were greater, which indicates that central nervous system involvement is likely part of a continuum of malaria pathology involving the liver. A few small studies on caffeine clearance suggest that CYP1A2 activity may be reduced as well, indicating that the effects of malaria extend to modulation of several CYP enzymes.
Reported impact of P. falciparum infection on drug pharmacokinetics in humans
Surprisingly few animal studies have been published addressing the effects of Plasmodium infection on DME expression. Of those that have been published, most have used Plasmodium berghei ANKA, a lethal infection in mice of a C57BL/6 background and often employed to model cerebral malaria. In this infection, the mRNAs of Cyp3a11, Cyp1a2, and Cyp2e1 in the liver were down-regulated to less than 20% of control levels accompanied by a prolongation of midazolam sleeping time (Carvalho et al., 2009).
Given that most of the human data available are from people with uncomplicated malaria, we decided to study DME expression and activity in a commonly used nonlethal model of malaria Plasmodium chabaudi chabaudi AS (PccAS). In addition to being nonlethal in many strains of mice, including those on a C57BL/6 background, PccAS infection has several features that make it an attractive model for uncomplicated human malaria, including development of anemia, temperature dysregulation, and hypoglycemia, all of which resolve as the primary infection is controlled (Stephens et al., 2012).
Our preliminary results (data not shown) indicated that PccAS infection produces significant changes in DME expression throughout the course of infection (25 of 27 mRNAs measured), and for most enzymes this correlates with the level of peripheral parasitemia. Of the 25 enzymes affected, 21 were down-regulated, and five were induced. These changes were of similar magnitude to those reported for animals infected with P. berghei ANKA. Furthermore, PK analyses of mice injected with a cocktail of five probe drugs revealed substantial reductions in the clearance of caffeine, bupropion, midazolam, and tolbutamide during PccAS infection. Our results suggest that PccAS may be an attractive model to study the mechanisms by which uncomplicated malaria causes profound effects on drug metabolism and clearance in humans.
The Role of Nuclear Factor E2-Related Factor 2 and Sirtuin 1 in Regulation of Biliary Transporters in Fasting (A.L.S., S.K.)
Fasting increases glucagon and intracellular cAMP and activates protein kinase A (PKA), modulating the transcription of multiple genes in liver that maintain glucose homeostasis, metabolism, and transport (Lavine et al., 1975; Chen et al., 2007). The cAMP/PKA cascade activates downstream expression of gluconeogenic genes, restoring blood glucose levels to normal (Altarejos and Montminy, 2011). Decreased glucose and increased pyruvate concentrations also increase the NAD+/NADH ratio, activating gluconeogenesis. Prolonged fasting activates sirtuin-1 (NAD-dependent deacetylase, SIRT1), which augments the use of free fatty acids as precursors for glucose production via deacetylation and activation of peroxisome proliferator-activated receptor γ coactivator α (PGC-1α) and forkhead box O1 (Foxo1) (Liu et al., 2008; Hayashida et al., 2010).
Along with gluconeogenic genes, fasting induces the transcription of genes involved in biotransformation (e.g., mouse Cyp2b10, and rat CYP2C11, CYP2E1, and CYP2B1/2) and transport (e.g., solute carrier for organic anions Slco1b2) in coordination with nuclear receptors (Chen et al., 2007; Buler et al., 2011). Studies have also indicated increases in gall bladder volume and bile acid concentrations upon fasting. Several studies have established the importance of Abcc2/3/4 in the disposition of bile acids and other endogenous and xenobiotic metabolites via biliary secretion or basolateral efflux. Abcc2–4 belong to the ATP binding cassette superfamily of efflux transporters expressed on the basolateral (Abcc3 and 4) or canalicular (Abcc2) membrane of hepatocytes (Klaassen and Aleksunes, 2010). However, the mechanisms behind the effect of nutritional status on biliary clearance mechanisms or bile flow have yet to be completely established.
Nuclear factor E2-related factor 2 (Nrf2) is an important transcriptional regulator of ABC transporters. Along with the Nrf2-mediated regulation of these transporters, studies have shown that ablation or overexpression of Nrf2 significantly alters bile flow (Maher et al., 2005; Weerachayaphorn et al., 2012). Nrf2 belongs to the basic leucine zipper family of transcription factors, present in the cytoplasm as a complex with Kelch-like ECH-associated protein 1 (Keap1), cullin 3 (Cul3), and ubiquitin-E3 ligase and undergoes continual proteasomal degradation (Kobayashi and Yamamoto, 2005; Taguchi et al., 2011). Upon activation, Nrf2 dissociates from the Keap1–Cul3–ubiquitin-E3 complex, translocates to the nucleus, and activates gene transcription (e.g., NADPH:quinone oxidoreductase [Nqo1] and glutamate-cysteine ligase, catalytic subunit) by binding to an antioxidant response element (ARE) (Kaspar et al., 2009). Our study identified mechanisms underlying fasting and transporter-mediated changes in biliary clearance. We hypothesize that Abcc2/3/4 are inducible via Sirt1-Nrf2 dependent mechanisms that are also driven by signals regulating nutrient homeostasis in the liver.
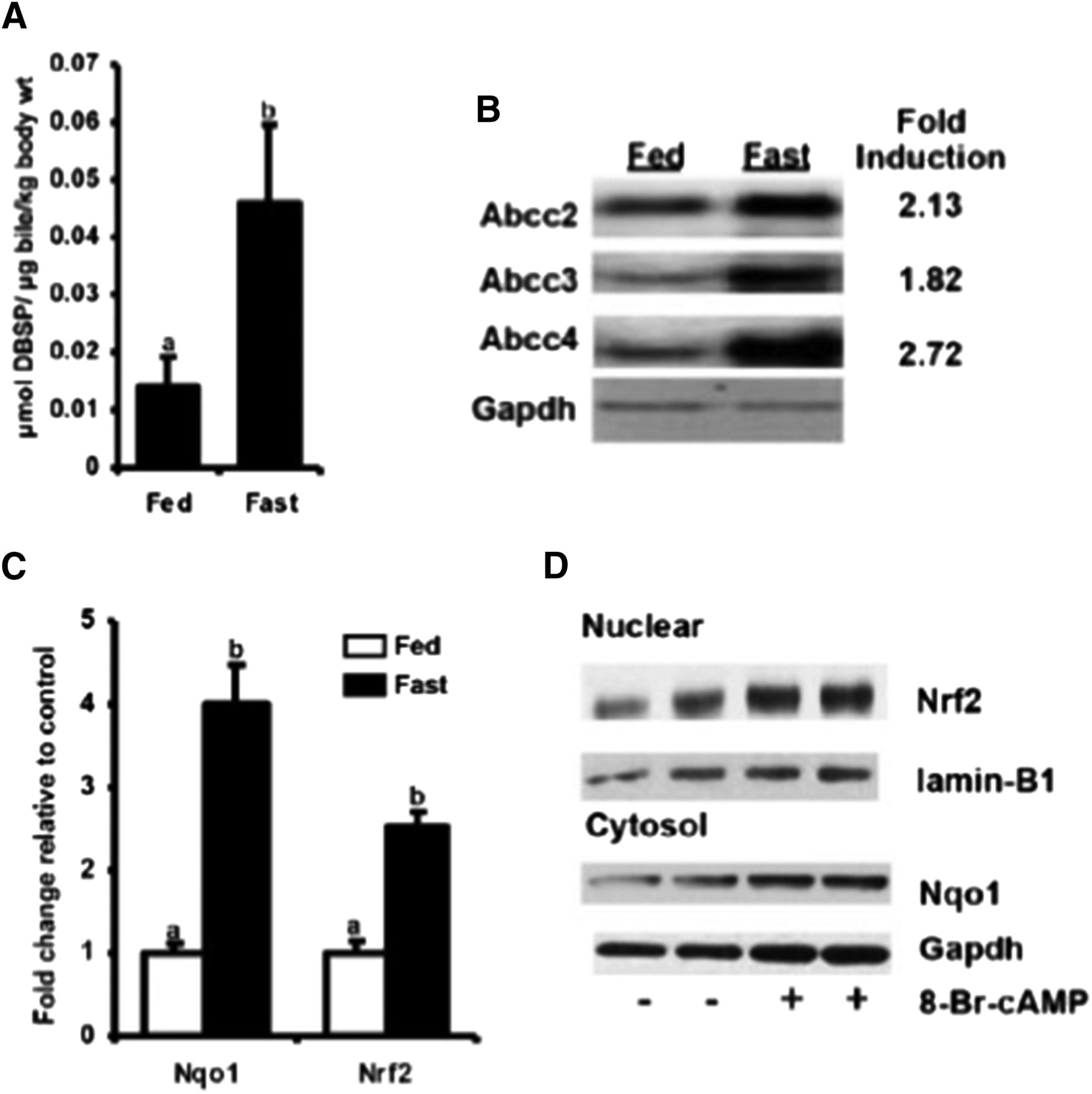
Fasting (24-hour food withdrawal) demonstrated a prototypical induction of hepatic Pgc-1α and Sult2a1 expression. Fasting also demonstrated a 3-fold increase in biliary concentrations of dibromosulfophthalein (DBSP) after intraperitoneal injection (Fig. 5A), a prototypical organic anion that undergoes biliary excretion via Abcc2 (Klaassen and Aleksunes, 2010) as well as increases in hepatic bile acid and cAMP levels (Kulkarni et al., 2014). Increased biliary disposition of organic anions was accompanied by increases in Abcc 2/3/4 protein expression by 1.5- to 2.7-fold (Fig. 5B) along with 4-fold increases in the antioxidant and Nrf2 target gene Nqo1 (Fig. 5C), an observation that was absent in mice deficient in Nrf2.

Fasting-mediated induction of Abcc2-4 is cAMP-PKA and Nrf2 dependent. Adult, male C57BL/6 mice (n = 6–8/group) were fed ad libitum or food withheld (fasted) for 18–24 hours. (A) Biliary concentration of DBSP was measured spectrophometrically 45 minutes after the fed and fasted mice were injected with DBSP (150 μmol/kg per 5 ml i.p.) or saline. (B) Liver protein expression of Abcc2–4. Crude liver membrane preparations were isolated from fed and fasted mice in a sucrose/Tris-based buffer using differential centrifugation followed by Western blot analysis for expression of Abcc2–4. (C) The mRNA expression of Nqo1 and Nrf2. Total RNA was isolated from livers of fed and fasted mice, and mRNA expression of Nqo1 and Nrf2 was quantified using RT-qPCR. (D) Increased nuclear translocation of Nrf2 primary mouse hepatocytes treated with cAMP-PKA activator (8-Br-cAMP). Cytosolic and nuclear fractions were obtained from control and 8-Br-cAMP–treated hepatocytes and immunoblotted for Nqo1 and Nrf2 expression. All data are expressed as average ± S.E.M. (n = 6–8/ group mouse livers, n = 3–4 replicates for in vitro experiments). P ≤ 0.05 was considered statistically significant. Groups without a common letter are statistically significantly different. Figures reprinted with permission from Kulkarni et al. (2014).
These preliminary observations indicate that fasting-mediated induction of Abcc2/3/4 is Nrf2 dependent (Kulkarni et al., 2014). This hypothesis was further supported by 10-fold increases in liver human placental alkaline phosphatase (hPAP) expression of fasted transgenic mice that possess a hPAP reporter downstream of an ARE consensus sequence as well as 2-fold increases in Nrf2 binding to ARE in the livers of fasted mice (Johnson et al., 2002; Kulkarni et al., 2014). This increase in Nrf2 and Abcc2/3/4 activity is potentially to increase the clearance of endogenous metabolites such as bile acid conjugates, bilirubin, cAMP/GMP, and lipid peroxidation products that increase upon fasting and could potentially injure the hepatocytes.
Although downstream activation of Nrf2-target genes has been well described, the upstream pathways, including fasting-mediated activation of Nrf2, are yet unclear. Preliminary studies have indicated a cAMP regulation of Abcc3 and 4 expression. PKA activation mediated by cAMP is a crucial pathway in fasting, as evidenced by the significantly increased hepatic cAMP levels in fasted mice. In accordance with this hypothesis, mouse and human hepatocytes treated with cAMP analogs or forskolin induced Nrf2 along with Nqo1 and Abcc 2/3/4 (Kulkarni et al., 2014). The sample data are shown in Fig. 5D. This induction was diminished upon the introduction of N-[2-[[(E)-3-(4-bromophenyl)prop-2-enyl]amino]ethyl]isoquinoline-5-sulfonamide (H-89), a PKA inhibitor (Fig. 6, A and B), and was nonexistent in Nrf2-deficient hepatocytes. The absence of induction of Nrf2 and Abcc2–4 by AMP kinase activators such as 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside and metformin rules out the possibility of Nrf2 activation by the AMP-activated protein kinase pathway (Canto and Auwerx, 2009; Kulkarni et al., 2014).

The cAMP-PKA pathways activate NRF2 and increase NRF2 and SIRT1 recruitment at ABCC2-ARE: (A) Treatment of hepatic cells with cAMP analog induces dose-dependent activation of antioxidant response element (ARE). Huh7 cells were transiently transfected with an ARE–luciferase pRL–CMV for 24 hours and then treated with 0.1 or 1.0 mM 8-Br-cAMP for 24 hours; luciferase activity was measured using a Dual Luciferase Assay (Promega, Madison, WI). (B) The activation of Nrf2 is diminished when treated with the PKA inhibitor H-89. Primary mouse hepatocytes were obtained from ARE-hPAP reporter mice and treated with 8-Br-cAMP with or without H-89 for 24 hours. Total RNA was isolated, and hPAP expression was quantified using RT-qPCR. (C) NRF2 and SIRT1 enrichment at ABCC2 promoter ARE was determined by chromatin immunoprecipitation assay. Briefly, Huh-7 cells were treated with 8-Br-cAMP (0.1 mM) for 45 minutes. The cells were then crosslinked, sheared, and incubated overnight with ProteinG beads and anti-NRF2 or SIRT1 antibody as well as control preimmune IgG. The chromatins and input DNA were analyzed by PCR for DNA fragments containing NRF2 bound-ARE-like element. All data are expressed as average ± S.E.M. (n = 3–4 replicates for in vitro experiments). P ≤ 0.05 was considered statistically significant. Groups without a common letter are statistically significantly different. Figures reprinted with permission from Kulkarni et al. (2014).
An additional aspect of our study was to identify factors downstream of the cAMP/PKA cascade that mediate activation of the NRF2-ARE pathway. Sirt1 and Pgc-1α act in concert to activate gluconeogenesis pathways in hepatocytes. Pgc-1α is a central transcriptional coactivator for multiple nuclear receptors, and we hypothesized that fasting potentially modulates Nrf2 via Sirt1-related mechanisms. Indeed, liver-specific Sirt1 knockout mice did not demonstrate an induction of Nrf2 and Abcc2/3/4 upon fasting, indicating an involvement of Sirt1 in mediating the cAMP/PKA-Nrf2-ARE-target gene induction in fasting livers (Taguchi et al., 2011). On the other hand, Sirt1-overexpressing mice demonstrated a higher fold induction of hepatic Nrf2 and Abcc4 compared with the wild-type fasted mice (Johnson et al., 2002). Nrf2 expression increases coordinately with Pgc-1α and Sirt1 expression in vitro in hepatocytes treated with cAMP analogs, indicating that Sirt1 potentially increases Nrf2 transcription supported by the higher basal expression of Nrf2 mRNA in Sirt1-overexpressing mouse livers (Buler et al., 2011).
To tie the upstream and the downstream observations together and determine whether fasting induction of cAMP/PKA pathway increases Sirt1-Pgc1α mediated activation of the Nrf2-ARE pathway and the expression of ABCC2/3/4, NRF2 and SIRT1 recruitment to the previously described ABCC2 ARE (Stockel et al., 2000) was determined in vitro (Kulkarni et al., 2014). Huh7 cells treated with cAMP analog demonstrated a higher enrichment of NRF2 as well as SIRT1 at the ABCC2 promoter ARE, indicating that indeed the induction of these transporters upon fasting is NRF2 and SIRT1 mediated (Fig. 6C).
In summary, these observations demonstrate that 1) fasting increases Abcc expression through Nrf2- and Sirt1-dependent mechanisms and 2) cAMP/PKA activators increase ARE activation via Nrf2- and Sirt1-dependent mechanisms in vitro, thus describing Nrf2 as a novel “nutrient-sensitive” responsive transcription factor as well as illustrating that hepatic clearance mechanisms are differentially affected by fasting.
Authorship Contributions
Participated in research design: Morgan, Dempsey, Mimche, Lamb, Kulkarni, Cui, Jeong, Slitt.
Conducted experiments: Dempsey, Mimche, Kulkarni.
Performed data analysis: Morgan, Dempsey, Mimche, Kulkarni, Cui, Jeong, Slitt.
Wrote or contributed to the writing of the manuscript: Morgan, Dempsey, Mimche, Lamb, Kulkarni, Cui, Jeong, Slitt.
Footnotes
- Received December 7, 2017.
- Accepted February 22, 2018.
This work was supported in part by grants from the National Institutes of Health National Institute of General Medical Sciences [Grants R01GM112746, R01GM111381]; Eunice Kennedy Shriver National Institute of Child Health and Human Development [Grant R01HD089455]; and National Institute of Diabetes and Digestive and Kidney Diseases [Grant R01DK072372].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- Abc
- ATP-binding cassette
- Ahr
- aryl hydrocarbon receptor
- ARE
- antioxidant response element
- AUC
- area under the curve
- Cul3
- cullin 3
- CV
- conventional
- Cyp
- cytochrome P450 enzymes
- DBSP
- dibromosulfophthalein
- DME
- drug-metabolizing enzyme
- dpi
- days postinfection
- Fmo3
- flavin-containing monooxygenase 3
- GF
- germ free
- H-89
- N-[2-[[(E)-3-(4-bromophenyl)prop-2-enyl]amino]ethyl]isoquinoline-5-sulfonamide
- Hnf
- hepatocyte nuclear factor
- hPAP
- human alkaline phosphatase
- IL
- interleukin
- IPA
- indole-3-propionic acid
- Keap1
- Kelch-like ECH-associated protein 1
- Nqo1
- NADPH:quinone oxidoreductase 1
- Nrf2
- nuclear factor E2-related factor 2
- P450
- cytochrome P450
- PccAS
- AS clone of Plasmodium chabaudi chabaudi
- Pgc-1α
- peroxisome proliferator-activated receptor γ coactivator α
- PK
- pharmacokinetic
- PKA
- protein kinase A
- Ppar
- peroxisome proliferator-activated receptor
- Pxr
- pregnane X receptor
- RT-qPCR
- reverse transcription quantitative polymerase chain reaction
- Shp
- small heterodimer partner
- Sirt1
- sirtuin1
- Sult
- sulfotransferase
- tg
- transgenic
- Ugt
- UDP glucuronosyltransferase
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}