


Abstract
The induction of cytochrome P450 (P450) enzymes is one of the risk factors for drug-drug interactions (DDIs). To date, the human pregnane X receptor (PXR)-mediated CYP3A4 induction has been well studied. In addition to CYP3A4, the expression of CYP2C subfamily is also regulated by PXR, and the DDIs caused by the induction of CYP2C enzymes have been reported to have a major clinical impact. The purpose of the present study was to investigate whether chimeric mice with a humanized liver (PXB mice) can be a suitable animal model for investigating the PXR-mediated induction of CYP2C subfamily, together with CYP3A4. We evaluated the inductive effect of rifampicin (RIF), a typical human PXR ligand, on the plasma exposure to the four P450 substrate drugs (triazolam/CYP3A4, pioglitazone/CYP2C8, (S)-warfarin/CYP2C9, and (S)-(−)-mephenytoin/CYP2C19) by cassette dosing in PXB mice. The induction of several drug-metabolizing enzymes and transporters in the liver was also examined by measuring the enzyme activity and mRNA expression levels. Significant reductions in the exposure to triazolam, pioglitazone, and (S)-(−)-mephenytoin, but not to (S)-warfarin, were observed. In contrast to the in vivo results, all the four P450 isoforms, including CYP2C9, were elevated by RIF treatment. The discrepancy in the (S)-warfarin results between in vivo and in vitro studies may be attributed to the relatively small contribution of CYP2C9 to (S)-warfarin elimination in the PXB mice used in this study. In summary, PXB mice are a useful animal model to examine DDIs caused by PXR-mediated induction of CYP2C and CYP3A4.
Introduction
The induction of cytochrome P450 (P450) enzymes is one of the risk factors for drug-drug interactions (DDIs) (Niemi et al., 2003; Luo et al., 2004). The human pregnane X receptor (PXR) is a key nuclear receptor principally responsible for the induction of several P450 enzymes, including CYP3A4, -2C8, -2C9, -2C19, -2A6, and -2B6 (Niemi et al., 2003; Sinz et al., 2008; Chen and Goldstein, 2009). In addition to P450 enzymes, the expression of several drug transporters, such as multidrug resistance gene (MDR) 1, multidrug resistance-associated protein (MRP) 2, organic anion-transporting polypeptides, and phase II metabolic enzymes, including UDP-glucuronosyltransferase (UGT), sulfotransferase, and glutathione S-transferase, are also regulated by human PXR (Dixit et al., 2007; Nishimura et al., 2008a,b; Sinz et al., 2008).
In humans, CYP3A4 plays a major role in drug metabolism because of its abundant expression in the liver and intestine and its broad substrate specificity. In fact, CYP3A4 contributes to the oxidative metabolism of more than 50% of all currently used drugs (de Wildt et al., 1999; Luo et al., 2004). Therefore, CYP3A4-related DDIs have a major clinical impact. CYP3A4 is the most studied isoform among the P450s in terms of DDIs caused by PXR-related induction of drug-metabolizing enzymes. Both in vitro and in vivo experimental models for CYP3A4 induction have been reported by several pharmaceutical companies (Cui et al., 2008; Kanebratt and Andersson, 2008; Kim et al., 2008, 2010; Kamiguchi et al., 2010).
The human CYP2C subfamily has four members: CYP2C8, CYP2C9, CYP2C18, and CYP2C19 (Läpple et al., 2003; Chen and Goldstein, 2009). Of these, CYP2C8, CYP2C9, and CYP2C19 are of clinical importance and are collectively responsible for the metabolism of ∼20% of clinically used drugs (Chen and Goldstein, 2009). The substrate specificity of CYP3A4 and CYP2C enzymes sometimes overlaps. In that case, the overall contribution of PXR-regulated P450 enzymes in the drug elimination process is relatively large. These observations suggest that new investigations should focus on the DDIs between PXR ligands and CYP2C substrate drugs. The quantitative polymerase chain reaction (PCR) analyses using human hepatocytes have demonstrated that the magnitude of CYP3A4 induction by PXR ligand is the largest followed by CYP2C enzymes, including CYP2C8, CYP2C9, and CYP2C19 (Raucy et al., 2002; Niemi et al., 2003). In fact, DDIs caused by induction of CYP2C enzymes have been also reported (Chen and Goldstein, 2009). However, there has been no systematic in vivo analysis focusing on the differences in the degree of the inductive effects of PXR ligands on the each of these P450 enzymes.
It has been reported that there is a large species difference in ligand recognition by the PXR between rodents and humans (Jones et al., 2000; LeCluyse, 2001). For example, RIF is more selective for human PXR, whereas the synthetic C21 steroid pregnenolone-16[propto]-carbonitrile, is a weak ligand for the human PXR but a potent ligand for rodents (Jones et al., 2000; LeCluyse, 2001). In fact, the expression of mouse Cyp3a is not influenced by the administration of RIF (Ma et al., 2007). The species differences in the ligand recognition of the PXR limit the utility of animal models to predict PXR-related DDIs in humans. In addition, it is known that there are species differences in metabolic patterns, as well as in the contribution of each P450 isoform to drug elimination (Shin et al., 2009; Kamimura et al., 2010). These species differences make it hard to predict human pharmacokinetics from animal data. Recently, several groups, including our own, have generated the humanized mouse models of the PXR and CYP3A4 by gene knockout and transgenic techniques (Xie et al., 2000; Ma et al., 2007; Kim et al., 2008; Scheer et al., 2008; Hasegawa et al., 2011). These models are useful for investigating CYP3A4 induction by human PXR ligands. However, the effect of the human PXR ligand on drug-metabolizing enzymes other than CYP3A4 cannot be examined using these mouse models.
The chimeric mouse with a humanized liver is an alternative mouse model (Strom et al., 2010). This mouse model, designated as the “PXB mouse,” has been established by the transplantation of human hepatocytes into urokinase-type plasminogen activator-transgenic severe combined immunodeficient mice (Tateno et al., 2004). The livers of the PXB mice are replaced with more than 70% human hepatocytes, although the remaining 30% are mouse hepatocytes (Strom et al., 2010). It has been reported that the mRNA expression of several P450 enzymes in the PXB mouse liver is induced by RIF treatment and that PXB mice also show similar drug-metabolizing profiles of CYP3A4 and CYP2C substrate drugs to humans (Katoh et al., 2005a,b; Kamimura et al., 2010). The PXB mouse is expected to provide the opportunity to examine the inductive effect of PXR ligands on the plasma profiles of not only CYP3A4 but also CYP2C substrate drugs. This study will provide important information on DDIs caused by CYP2C induction in addition to CYP3A4.
In the present study, we evaluated the inductive effect of three different doses of RIF on the plasma exposure of PXB mice to the substrate drugs of CYP3A4, CYP2C8, CYP2C9, and CYP2C19, which have been reported to have DDIs with RIF in humans. Furthermore, the induction of several drug-metabolizing enzymes and transporters in the liver was also examined by measuring the enzyme activities and mRNA expression levels.
Materials and Methods
Materials.
RIF, rosiglitazone (ROS), (S)-warfarin (WAR), dextromethorphan hydrobromide monohydrate, and propranolol hydrochloride were purchased from Wako Pure Chemical Industries (Osaka, Japan). Triazolam (TRZ), phenacetin, bupropion, and diclofenac were purchased from Sigma-Aldrich (St. Louis, MO). (S)-(−)-Mephenytoin (MEP) was purchased from Toronto Research Chemicals (North York, Canada). NADP+, d-glucose 6-phosphate (G-6-P), and G-6-P dehydrogenase (G-6-P-DH) were purchased from Oriental Yeast (Tokyo, Japan). All other chemicals and solvents were of analytical grade otherwise noted.
Generation of Chimeric Mice with a Humanized Liver.
All animal studies were conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals, and the experimental protocol used in this study was approved by the Committee for Animal Experiments of PhoenixBio Co., Ltd., and Kyowa Hakko Kirin Co., Ltd. The PXB mice were generated as described previously (Tateno et al., 2004). All PXB mice used in the present study were derived from the same donor cryopreserved hepatocytes (BD85, from a 5-year-old black male; BD Biosciences, Franklin Lakes, NJ). The blood concentration of human albumin in the PXB mice was measured according to a previous report (Tateno et al., 2004) to predict the replacement index of human hepatocytes that had repopulated in the host mouse liver. The actual values of the replacement index in PXB mice used in this study ranged from 82 to 94%.
Pharmacokinetic DDI Study in PXB Mice.
Male PXB mice (11–12-weeks-old, 18–23 g) were used in this study. The suspensions of RIF prepared with corn oil were given intraperitoneally at doses of 2, 10, and 50 mg/kg to the PXB mice once daily for 4 days. After the 4 days of treatment of RIF, the PXB mice received a mixture of CYP3A4, CYP2C8, CYP2C9, and CYP2C19 substrate drugs orally via cassette dosing. The dosing mixture was prepared by adding a dimethyl sulfoxide solution of each drug to a 0.5% methylcellulose aqueous solution. The dose of each substrate drug was as follows: TRZ (CYP3A4 substrate), 5 mg/kg; ROS (CYP2C8 substrate), 1 mg/kg; WAR (CYP2C9 substrate), 0.1 mg/kg; and MEP (CYP2C19 substrate), 5 mg/kg. Blood samples were collected at 2 h after administration on days 1 and 4 and 0.5, 1, 2, 4, and 7 h on day 5. The blood samples were centrifuged, and the plasma samples obtained were stored at −80°C until the analysis. After blood sampling on day 5, the mice were euthanized, and a piece of the liver was collected and preserved in RNAlater solution (Invitrogen, Carlsbad, CA) to stabilize the RNA. The remaining liver tissue was frozen in liquid nitrogen and stored at −80°C until microsomal preparation.
RNA Isolation and Quantitative Reverse Transcription-PCR.
Total RNA was extracted from the liver using an RNeasy Plus mini kit (QIAGEN, Hilden, Germany) and was reverse-transcribed to obtain cDNA using a PrimeScript RT reagent kit (Takara Bio, Shiga, Japan) according to the manufacturer's instructions. SYBR-PCR was performed using an ABI PRISM 7900HT (Invitrogen) with SYBR Premix Ex Taq (Takara Bio). The PCR conditions were as follows: after initial denaturation at 94°C for 5 min, the amplification was performed by denaturation at 94°C for 30 s, annealing at 65°C for 30 s, and extension at 72°C for 30 s for 45 cycles. In all cases, the input cDNA concentrations were normalized to those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH; ΔCt). The relative mRNA expression was determined by a 2−ΔΔCt calculation. The primer sequences used in the present study are summarized in Table 1. We confirmed that these primers were capable of amplifying human but not mouse genes.
The sequences of the primers for SYBR-PCR
Preparation of Liver Microsomes and Metabolic Assay.
Liver microsomes were prepared from the frozen liver tissues as described previously (Sugihara et al., 2001). The reaction conditions for the enzyme activity of each P450 isoform are summarized in Table 2. The optimized substrate concentrations, the microsomal concentrations, and the reaction times were used to determine metabolic activity precisely. A reaction mixture (50 μl) consisted of 100 mM phosphate buffer, pH 7.4, 3 mM magnesium chloride, 8 mM G-6-P, 1 U/ml G-6-P-DH, 0.8 mM NADP+, microsomal protein, and substrate. The reaction was initiated by the addition of an NADPH-generating system (a mixture of magnesium chloride, G-6-P, G-6-P-DH, and NADP+) after preincubation of the mixture without the NADPH-generating system for 5 min at 37°C. The reaction was terminated at the designated time by the addition of ice-cold methanol containing propranolol as an internal standard. The sample was centrifuged, and the supernatant was diluted with water. The metabolite of each substrate was analyzed using a liquid chromatography-tandem mass spectrometry (LC/MS/MS) system.
The reaction conditions for P450 enzyme assay using the liver microsomes
Pretreatment of Plasma.
Two microliters of plasma sample, 2 μl of dimethyl sulfoxide, and 30 μl of the ice-cold methanol containing the internal standard were mixed and centrifuged. The calibration standards were prepared in the same manner as the plasma samples. The supernatant was mixed with 10 mM ammonium acetate, and the mixture was injected into the LC/MS/MS system.
LC/MS/MS Analysis.
The concentrations of the substrate drugs (in plasma samples), and metabolites (in microsomal samples) were measured using the LC/MS/MS system consisting of an ACQUITY UPLC (Waters, Inc., Bedford, MA) connected to a 4000 QTRAP mass spectrometer (AB Sciex, Foster City, CA).
For the plasma samples, chromatographic separation was performed on a CAPCELL PAK C18 MGII column (3 μm, 3 mm inner diameter × 35 mm; Shiseido, Tokyo, Japan) using an injection volume of 10 μl (ROS and WAR) or 25 μl (TRZ and MEP) and a run time of 4 min. The elution was conducted at a flow rate of 0.8 ml/min by a linear gradient with the mobile phase, which consisted of 10 mM ammonium acetate in water (A) and methanol (B). The gradient condition of B (%) was as follows: at 0, 0.2, 2.2, 2.21, 3, and 3.01 min, the B% was 80, 80, 25, 10, 10, and 80%, respectively. The mass spectrometry detection was performed by positive ionization electrospray. The multiple reaction monitoring mode was used, and the monitor ions (m/z precursor ion > product ion) were as follows: ROS (358.1 > 153.3), WAR (309.6 > 163.5), TRZ (343.4 > 308.1), and MEP (219.6 > 134.4). The plasma concentration ranges of quantification were as follows: ROS (1.07–3570 ng/ml), WAR (0.925–9250 ng/ml), TRZ (0.343–3430 ng/ml), and MEP (2.18–6550 ng/ml).
For the microsomal samples, chromatographic separation was performed on an ACQUITY UPLC BEH C18 column (1.7 μm, 2.1 mm inner diameter × 50 mm; Waters, Milford, MA) using an injection volume of 7.5 μl and a run time of 2.5 min. The elution was conducted at a flow rate of 0.5 ml/min by a linear gradient with the mobile phase, which consisted of 10 mM ammonium acetate in water (A for CYP1A2, CYP2B6, CYP2C9 (7-hydroxywarfarin), CYP2C19, and CYP2D6 assays) or 0.05% formic acid in water (A for CYP2C8, CYP2C9 (4′-hydroxydiclofenac), and CYP3A4 assays) and methanol (B). The gradient condition of B (%) was as follows: at 0, 0.2, 1.5, 2 and 2.01 min, the B% was 95, 95, 5, 5, and 95%, respectively. The mass spectrometry detection was performed by positive ionization electrospray. The multiple reaction monitoring mode was used, and the monitor ions (m/z precursor ion > product ion) were as follows: CYP1A2 (acetaminophen, 152.0 > 110.0), CYP2B6 (hydroxybupropion, 256.1 > 238.1), CYP2C8 (N-demethyl rosiglitazone, 344.1 > 121.1; 5-hydroxyrosiglitazone, 374.1 > 151.1), CYP2C9 (4′-hydroxydiclofenac, 312.05 > 230.45; 7-hydroxywarfarin, 325.6 > 163.5), CYP2C19 (4′-hydroxymephenytoin, 235.0 > 150.15), CYP2D6 (dextrorphan, 258.1 > 157.1), and CYP3A4 (1′-hydroxytriazolam, 359.1 > 176.1; 4-hydroxytriazolam, 359.1 > 314.1). Although the metabolites concentrations in the microsomal samples were not quantified, we have confirmed the linearity of signal intensities and no signals in the blank samples.
Pharmacokinetic Analysis.
The pharmacokinetic parameters for TRZ, ROS, WAR, and MEP were obtained by a noncompartmental analysis. The log-transformed plasma concentrations were plotted against time. The slope of the elimination phase (λz) was estimated by linear regression. The maximal plasma concentration (Cmax) and time to Cmax (tmax) were obtained directly from the observed values. The apparent t1/2 was obtained as ln2/λz. The area under the plasma concentration-time curve (AUC) from time 0 to the last data point (AUC0-t) was calculated using the linear trapezoidal method. The AUC after the last data point (AUCλz) was estimated by extrapolating with λz. The sum of AUC0-t and AUCλz was regarded as AUC0-∞.
Statistical Analysis.
A one-way analysis of variance with a Dunnett's test was performed to assess for significant differences in the pharmacokinetics, metabolic activity in the liver microsomes, and mRNA expression in the liver between vehicle- and RIF-treated groups. The statistical analyses were performed using the SAS software program (SAS Institute, Cary, NC). The criterion for statistical significance was P < 0.05.
Results
The Effect of RIF Treatment on the Pharmacokinetics of CYP3A4 and CYP2C Substrate Drugs.
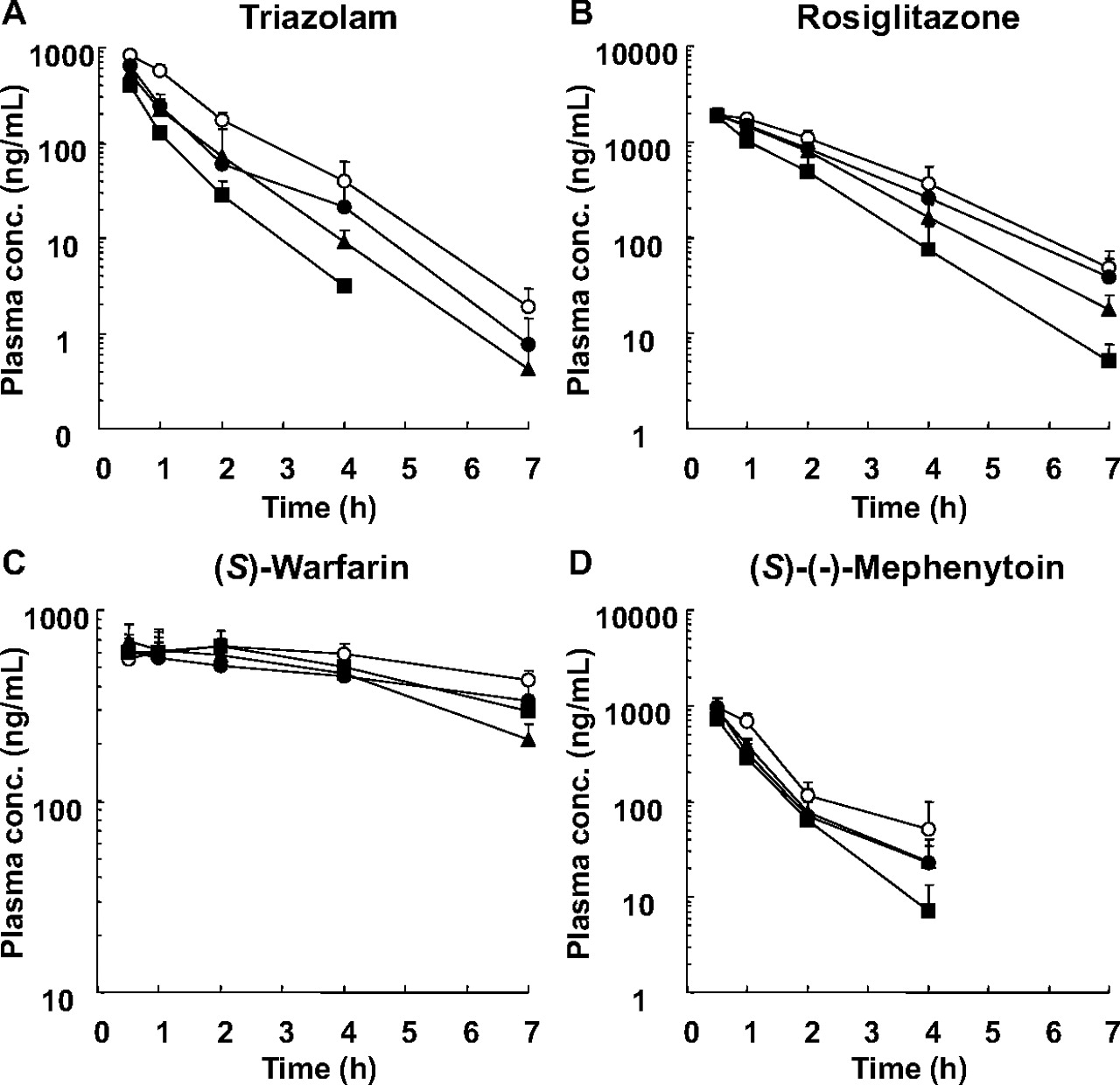
The pharmacokinetics of CYP3A4 (TRZ) and CYP2C substrate drugs (ROS, MEP, and WAR) was evaluated after repeated intraperitoneal administration of RIF (2, 10, and 50 mg/kg daily for 4 days) or vehicle to the PXB mice. The plasma concentration-time profiles of the substrate drugs are shown in Fig. 1, and the pharmacokinetic parameters and the AUC decrease (percentage) of substrate drugs are summarized in Table 3. The plasma exposure to TRZ was decreased with increased doses of RIF (Fig. 1A). The TRZ AUC was significantly decreased by 46 (2 mg/kg), 54 (10 mg/kg), and 71% (50 mg/kg) compared with the vehicle control (Table 3). RIF treatment also resulted in an AUC decrease of ROS and MEP with increased doses of RIF, and the statistically significance in the AUC decrease was observed only at the dose of 50 mg/kg RIF as 47% for ROS and 46% for MEP (Fig. 1, B and D; Table 3). Treatment with RIF had no effect on the pharmacokinetics of WAR (Fig. 1C; Table 3).

The plasma concentration-time profiles of TRZ (A), ROS (B), WAR (C), and MEP (D) after repeated intraperitoneal administration of RIF once daily for 4 days to male PXB mice. The PXB mice were given oral doses of 5 mg/kg TRZ (CYP3A4 substrate), 1 mg/kg ROS (CYP2C8 substrate), 0.1 mg/kg WAR (CYP2C9 substrate), and 5 mg/kg MEP (CYP2C19 substrate) via cassette dosing after 4 days of treatment with 2 (●), 10 (▴), or 50 (■) mg/kg RIF or vehicle (○). Each point represents the mean ± S.D. of three mice.
The pharmacokinetic parameters of TRZ, ROS, WAR, and MEP administered orally in cassette dosing after repeated intraperitoneal administration of RIF once daily for 4 days to the male PXB mice
Each value was determined from the data shown in Fig. 1. Data represent the mean ± S.D. of three mice.
The Metabolic Activities of Human P450 Enzymes in the Liver Microsomes.
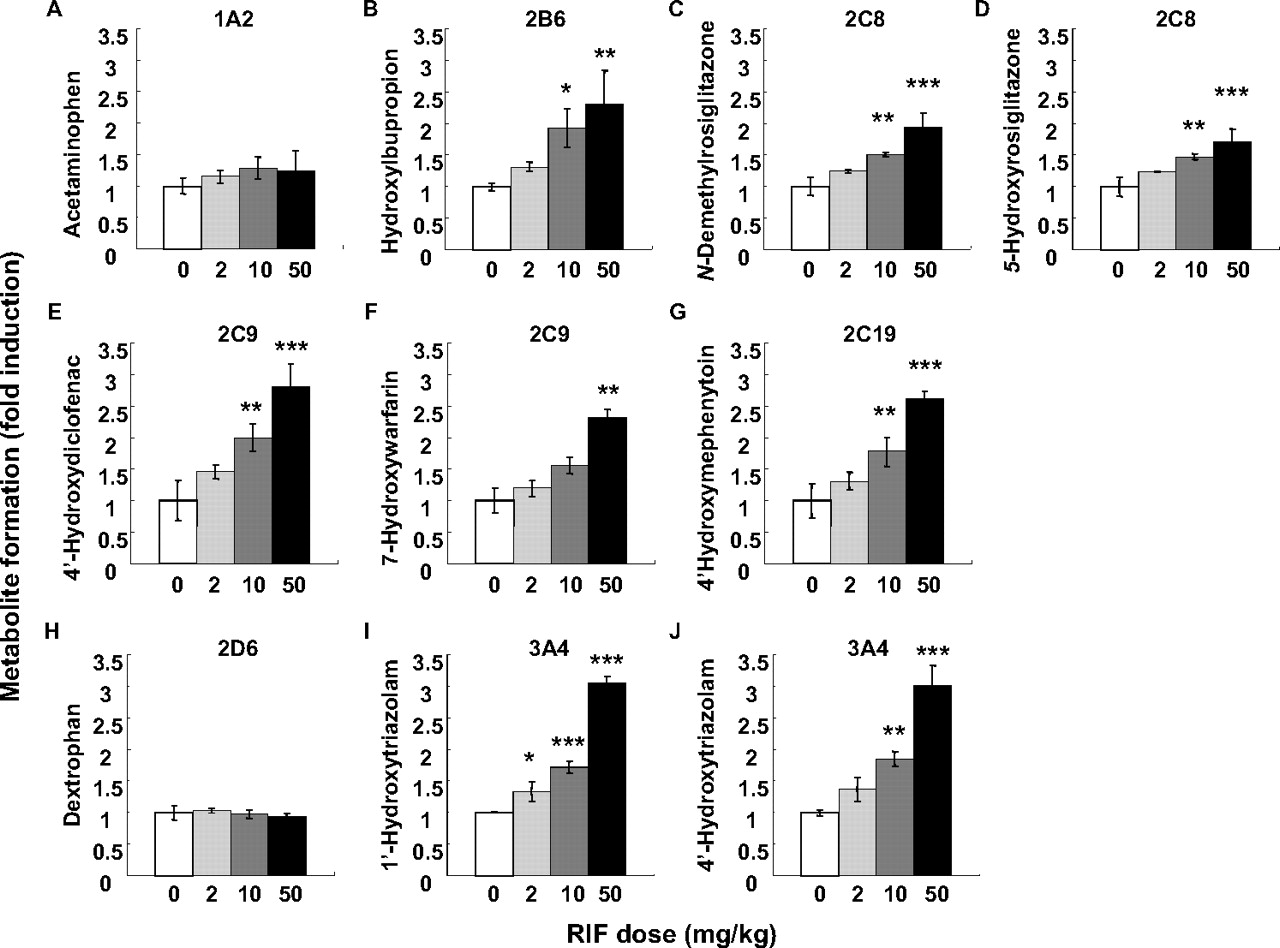
The metabolic activities of seven human P450 enzymes were determined in the liver microsomes prepared from PXB mice treated with RIF. The fold-induction of enzyme activity for each P450 isoform in the RIF-treated group is shown in Fig. 2. The metabolic activities of CYP3A4 (1′- and 4-hydroxy-TRZ), CYP2C8 (5-hydroxy- and N-demethyl-ROS), and CYP2C19 (4-hydroxy-MEP), whose induction was detected in the in vivo study, were significantly increased with increased doses of RIF. Although CYP2C9 induction was not detected in the in vivo study, the metabolic activity of CYP2C9 (7-hydroxy-WAR and 4′-hydroxydiclofenac) was significantly increased by RIF treatment. In addition to CYP3A4 and CYP2C enzymes, the metabolic activities of the CYP1A2, CYP2B6, and CYP2D6 enzymes were also examined. The metabolic activity of CYP2B6 (hydroxybupropion) but not CYP1A2 (acetaminophen) or CYP2D6 (dextrorphan) was increased in a dose-dependent manner by RIF.

The metabolic activities of P450 enzymes in the liver microsomes prepared from PXB mice. The liver microsomes were prepared from RIF- or vehicle-treated PXB mice. The activities of P450 enzymes, CYP1A2 (A), CYP2B6 (B), CYP2C8 (C and D), CYP2C9 (E and F), CYP2C19 (G), CYP2D6 (H), and CYP3A4 (I and J), were determined. The experimental conditions are summarized in Table 2. The fold induction in the RIF-treated group compared with vehicle-treated group was calculated. The metabolic activity of each of the mouse liver microsomes was determined from the means of duplicate assay. Each bar represents the mean ± S.D. of three mice. Statistically significant from the vehicle-treated group; *, P < 0.05, **, P < 0.01, and ***, P < 0.001.
The mRNA Expression of Human P450 Enzymes and Transporters.
The mRNA expression of the seven human P450 enzymes, UGT1A1, and transporters, including MDR1 and MRP2, was evaluated in the livers of the PXB mice treated with RIF. The fold induction of mRNA expression of enzymes and transporters in the RIF-treated group is shown in Fig. 3. The magnitude of CYP3A4 induction was the largest among the P450 enzymes, followed by CYP2C8 and CYP2B6. Although the enzyme activities of CYP2C9 and CYP2C19 were increased by RIF treatment, the increase in mRNA expression was too slight to detect significant difference. No changes in the mRNA expression of CYP1A2 and CYP2D6 were observed. RIF treatment significantly increased the mRNA expression of UGT1A1, but not MDR1 and MRP2.
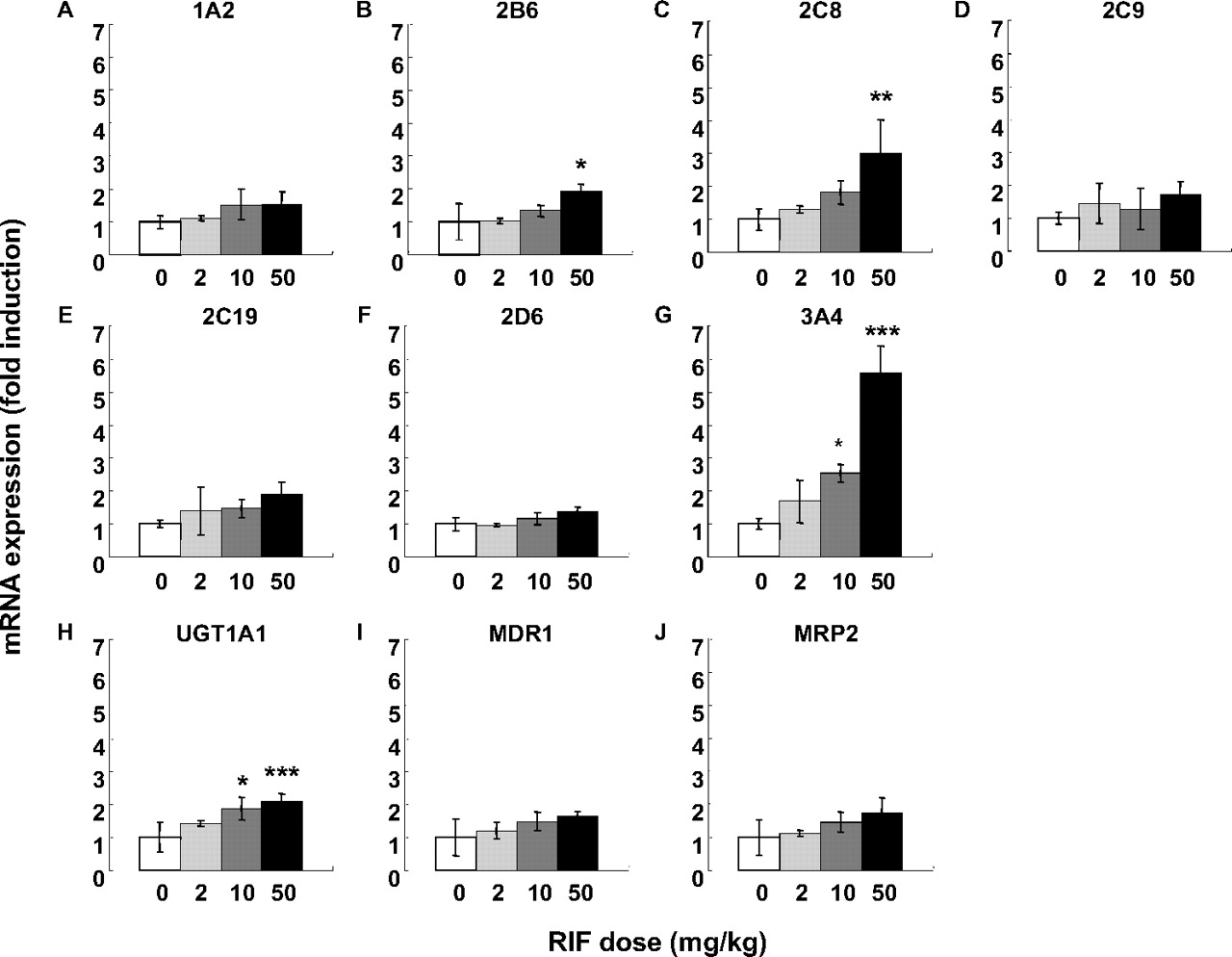
The mRNA expression levels of P450 enzymes and drug transporters in the liver of PXB mice. The mRNA expression levels of CYP1A2 (A), CYP2B6 (B), CYP2C8 (C), CYP2C9 (D), CYP2C19 (E), CYP2D6 (F), CYP3A4 (G), UGT1A1 (H), MDR1 (I), and MRP2 (J) were determined by SYBR-PCR. The mRNA expression level of each gene was normalized to that of GAPDH. The fold induction in the RIF-treated group compared with vehicle-treated group was calculated. Each bar represents the mean ± S.D. of three mice. Statistically significant from the vehicle-treated group; *, P < 0.05, **, P < 0.01, and ***, P < 0.001.
Discussion
In the present study, in vivo study using PXB mice, we simultaneously investigated the inductive effect of RIF on CYP3A4 and CYP2C enzymes. We demonstrated that concomitant use of RIF affects the pharmacokinetics of both CYP3A4 and CYP2C substrate drugs and that the inductive effect of RIF on CYP3A4 is greater than that on CYP2C enzymes. In addition, in vitro studies using the liver samples after RIF treatment were also carried out to examine the enzyme activities of human P450s and the mRNA expression levels of human P450s, UGT, and transporters. The induction by RIF was observed in the genes whose expression levels were known to be regulated by the human PXR, but no change was observed in the genes not regulated by the PXR (CYP2D6).
To compare the pharmacokinetic data between studies conducted in humans and this study, we selected substrate drugs that have previously been reported to have DDIs with RIF in humans. In a clinical study, the AUC decreases in the P450 substrate drug with concomitant use of RIF (600 mg daily) were 46 to 78% (ROS), 57 to 85% (WAR), and 95% (TRZ) (Villikka et al., 1997; Niemi et al., 2003, 2004; Park et al., 2004). The DDI information on CYP2C19 substrate drugs is very limited, and we could only find information about the urinary excretion data for 4′-hydroxymephenytoin, the main metabolite of MEP (Zhou et al., 1990; Feng et al., 1998). The concomitant use of RIF (600 mg daily) increased the urinary excretion of 4′-hydroxymephenytoin from 1.4- to 2.8-fold (Zhou et al., 1990). Assuming that the urinary excretion amount of the metabolite reflects the metabolic clearance of MEP, the decrease in the AUC would be between 29 to 64% as a result of RIF treatment. These reported clinical data suggest that CYP3A4 is the most susceptible to induction by RIF treatment and that the magnitude of induction of CYP2C8, CYP2C9, and CYP2C19 by RIF seems to be relatively weak compared with CYP3A4. In this study using PXB mice, RIF treatment resulted in the largest AUC decrease in TRZ, followed by ROS and MEP (Fig. 1; Table 3). The response to RIF treatment observed in the PXB mice is therefore similar to that in humans.
It was unexpected that the exposure to WAR was not affected by RIF treatment, despite CYP2C9 induction in the liver microsomes (Figs. 1C and 2, E and F; Table 3). The elimination pathways of WAR other than metabolism by CYP2C9 might have made it harder to detect CYP2C9 induction in the in vivo study. In humans, WAR is mainly metabolized to the 7-hydroxyl metabolite by CYP2C9, although WAR is also metabolized to other hydroxyl metabolites by other P450 isoforms (Inoue et al., 2009). Given that the PXB mice used in the present study were derived from the hepatocytes of a single donor, the contribution of CYP2C9 to WAR metabolism in these PXB mice might not be uniformly typical of humans in general. The availability of murine P450 isoforms remaining in the liver of PXB mice could also potentially have affected the overall metabolism of WAR in the in vivo study.
We measured the metabolic activities of WAR in the liver microsomes of SCID (severe combined immunodeficiency) mice (the background strain of the PXB mice) and in pooled human liver microsomes to compare them with PXB mice (supplemental figure). The three types of hydroxyl metabolites of WAR, including 7-hydroxy-WAR, were detected in all of the microsomes. However, the 7-hydroxylation activity in the liver microsomes of PXB mice was only one fifth of that in the human liver microsomes. Therefore, the contribution of CYP2C9 to WAR metabolism in the PXB mice may have been smaller than expected. In fact, the formation of another hydroxyl WAR (M2) in the liver microsomes of SCID mice was greater than that in the human liver microsomes, although the absolute metabolic clearance was not determined (supplemental figure). The metabolic activity of murine P450s remaining in the liver could possibly make it difficult to examine CYP2C9 induction by examining the pharmacokinetics of WAR.
To investigate whether a genetic polymorphism could explain the lower CYP2C9 activity in PXB mice, the CYP2C9 genomic polymorphism was determined by Invader assay (BML, Inc., Tokyo, Tokyo) for the cryopreserved human hepatocytes (lot BD85) used in this study. Although no variant sequence was detected in the CYP2C9 gene (data not shown), real-time quantitative reverse transcription-PCR analysis revealed that the mRNA expression level of CYP2C9 in the hepatocytes was relatively low compared with that in other donor hepatocytes (data not shown). In fact, the plasma elimination of WAR in this study seems to be slower than that in the previous study using PXB mice transplanted with a different lot of human hepatocytes (Inoue et al., 2008). Therefore, the main reason for the failure to detect of CYP2C9 induction in the in vivo study was probably the low hepatic expression of CYP2C9 in the PXB mice used in this study.
In humans, a therapeutic dose of RIF (600 mg) resulted in an AUC0-∞ of 22,400 to 35,300 ng · h/ml (Polk et al., 2001), which was a similar range of plasma exposure as the PXB mouse receiving the 10 mg/kg RIF (M. Kakuni, unpublished data). Considering the AUC decrease of the substrate drugs in PXB mice and humans caused by RIF treatment, the inductive response in PXB mice seems to be relatively weaker than that in humans. It has been reported that CYP3A4, CYP2C8, CYP2C9, and CYP2C19 are expressed in the human intestine (Kolars et al., 1992; Läpple et al., 2003; van de Kerkhof et al., 2008). In addition, it is well known that drug metabolism by intestinal CYP3A4 affects the pharmacokinetics of orally administered drugs (Kato et al., 2003). RIF was previously reported to induce CYP3A4 not only in the liver but also in the intestine in humans (Kolars et al., 1992; van de Kerkhof et al., 2008). Therefore, the decrease in the AUC by concomitant use of RIF in the clinic is accounted for by induction of P450 enzymes both in the liver and in the intestine. In PXB mice, only the liver, but not the intestine, is humanized. Therefore, the intestinal P450 enzymes in PXB mice cannot be induced by RIF, which is a specific human PXR ligand. As a result of the lack of induction in the intestinal P450 enzymes in PXB mice, the reduction of the AUC in the PXB mice would be predicted to be smaller than that in humans. In addition, the hepatic exposure of PXB mice to RIF might be smaller than the expected level, because RIF was administered intraperitoneally, not orally, to PXB mice in this study. The relatively low exposure of the liver to RIF may have result in a weaker induction in the PXB mice.
The induction of CYP3A4 and CYP2C8, CYP2C9, and CYP2C19 by RIF in PXB mice was also demonstrated by examining the enzyme activities using typical substrates for each P450 isoform (Fig. 2). The induction of CYP2B6 in the liver microsomes was also detected (Fig. 2B). This result is consistent with the fact that the expression of CYP2B6 is regulated by the human PXR (Sinz et al., 2008). Next, we determined the mRNA expression levels of other genes, including UGT1A1, MDR1, and MRP2, whose expression levels are also under the regulation of the human PXR (Fig. 3) (Nakata et al., 2006). It was previously demonstrated that RIF led to a small increase in the mRNA expression of these genes using human hepatocytes (Nishimura et al., 2008a,b). In this study, the mRNA expression levels seemed to be slightly increased by RIF in a dose-dependent manner. Statistically significant increase was observed in the mRNA expression levels of UGT1A1 but not in those of MDR1 or MRP2 expression (Fig. 3, H–J). These results might be also attributed to the use of single donor hepatocytes as discussed above.
In the present study, we have performed a DDI study focusing on the human PXR-related induction of CYP3A4 and CYP2C enzymes simultaneously by using the cassette dosing of substrate drugs in PXB mice. We have demonstrated that the PXB mice show a similar response to humans in terms of human PXR-related P450 induction by RIF. Because the PXB mice used in the present study were derived from the hepatocytes of a single donor, further studies are needed to generalize the present findings by performing DDI studies using PXB mice derived from the different hepatocyte donors.
Considering the magnitude of induction and its contribution to the drug metabolism in clinical situations, CYP3A4 is the most important enzyme to examine during the preclinical development of a new drug candidate. Several groups have established PXR and/or CYP3A4 humanized mice using gene knockout and transgenic techniques (Xie et al., 2000; Ma et al., 2007; Kim et al., 2008; Scheer et al., 2008; Hasegawa et al., 2011). On the other hand, the previous and present DDI studies have demonstrated that the induction of CYP2C enzymes also has a large impact on the pharmacokinetics of CYP2C substrate drugs (Niemi et al., 2003). At present, chimeric mice with a humanized liver, including the PXB mice, are only animal model available to investigate DDIs caused by the induction of CYP2C together with CYP3A4. Furthermore, several groups have reported that the drug-metabolizing profiles in PXB mice are similar to those in humans (Kamimura et al., 2010). Therefore, PXB mice seem to be a suitable animal model to examine the enzyme induction by a drug and its metabolite(s) if these are ligands for the human PXR. In conclusion, PXB mice will provide the opportunity to examine potential DDIs caused by PXR-related enzyme induction in a situation similar to that observed in humans.
Authorship Contributions
Participated in research design: Hasegawa and Tahara.
Conducted experiments: Hasegawa, Tahara, Inoue, Kakuni, and Tateno.
Performed data analysis: Hasegawa.
Wrote or contributed to the writing of the manuscript: Hasegawa, Tahara, and Ushiki.
Acknowledgments
We thank Tatsuya Matsumi and Dr. Saburo Sugai for supporting this research and reviewing the manuscript.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- P450
- cytochrome P450
- DDI
- drug-drug interaction
- PXR
- pregnane X receptor
- MDR
- multidrug resistance gene
- MRP
- multidrug resistance-associated protein
- UGT
- UDP-glucuronosyltransferase
- RIF
- rifampicin
- ROS
- rosiglitazone
- WAR
- (S)-warfarin
- MEP
- (S)-(−)-mephenytoin
- TRZ
- triazolam
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- PCR
- polymerase chain reaction
- G-6-P
- d-glucose 6-phosphate
- G-6-P-DH
- G-6-P dehydrogenase
- LC/MS/MS
- liquid chromatography-tandem mass spectrometry
- AUC
- area under the plasma concentration-time curve.

- Received September 14, 2011.
- Accepted November 29, 2011.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}