


Abstract
Lipophilic (logP > 1) and amphiphilic drugs (also known as cationic amphiphilic drugs) with ionizable amines (pKa > 6) can accumulate in lysosomes, a process known as lysosomal trapping. This process contributes to presystemic extraction by lysosome-rich organs (such as liver and lung), which, together with the binding of lipophilic amines to phospholipids, contributes to the large volume of distribution characteristic of numerous cardiovascular and central nervous system drugs. Accumulation of lipophilic amines in lysosomes has been implicated as a cause of phospholipidosis. Furthermore, elevated levels of lipophilic amines in lysosomes can lead to high organ-to-blood ratios of drugs that can be mistaken for active drug transport. In the present study, we describe an in vitro fluorescence-based method (using the lysosome-specific probe LysoTracker Red) to identify lysosomotropic agents in immortalized hepatocytes (Fa2N-4 cells). A diverse set of compounds with various physicochemical properties were tested, such as acids, bases, and zwitterions. In addition, the partitioning of the nonlysosomotropic atorvastatin (an anion) and the lysosomotropics propranolol and imipramine (cations) were quantified in Fa2N-4 cells in the presence or absence of various lysosomotropic or nonlysosomotropic agents and inhibitors of lysosomal sequestration (NH4Cl, nigericin, and monensin). Cellular partitioning of propranolol and imipramine was markedly reduced (by at least 40%) by NH4Cl, nigericin, or monensin. Lysosomotropic drugs also inhibited the partitioning of propranolol by at least 50%, with imipramine partitioning affected to a lesser degree. This study demonstrates the usefulness of immortalized hepatocytes (Fa2N-4 cells) for determining the lysosomal sequestration of lipophilic amines.
Introduction
Lysosomes are acidic organelles (pH 4–5) that play a key role in various metabolic processes, such as the turnover of phospholipids, the breakdown of endogenous waste products (including bacteria and viruses), autophagy, and apoptosis. First described by de Duve et al. (1974), lysosomes are also able to sequester drugs by a physicochemical (nonenzymatic, non–transporter-mediated) process known as lysosomal trapping. The targets of sequestration are typically lipophilic and amphiphilic drugs [also known as cationic amphiphilic drugs (CADs)], and their propensity to be sequestered (trapped) in lysosomes is dictated by their physicochemical properties. Many central nervous system and cardiovascular drugs are lipophilic amines (logP > 1) with ionizable amine groups (pKa > 6). Such drugs have a neutral fraction at physiologic pH (7.2–7.4) and can readily diffuse across cell membranes by passive diffusion. When such drugs diffuse into lysosomes, they become protonated (positively charged) because of the acidic environment of the lysosomes, which restricts diffusion of drug molecules back across the lysosomal membrane into the cytosolic space (Fig. 1). Of note, compared with the pH of blood (7.4), the pH of cytosol is slightly acidic (7.0–7.2) and this slight difference may play a significant role in the disposition of drugs (Berezhkovskiy, 2011; Hallifax and Houston, 2012; Poulin et al., 2012). The pH partitioning of lipophilic amines into acidic compartments is the mechanistic basis by which these drugs become highly sequestered in lysosomes. Together with their binding to phospholipids throughout the cell, lysosomal trapping explains why many basic (cationic) drugs exhibit high liver-to-blood ratios and large volumes of distribution. Active transport by OCTs (organic cation transporters) may play a role in the cellular uptake of certain lipophilic amines at plasma concentrations of drug at or below the Km value for the specific OCT (Aki et al., 2012); however, at higher concentrations, cellular uptake largely involves passive diffusion. For acidic (anionic) drugs, such as some statins, high liver-to-blood ratios are largely attributable to active uptake by sinusoidal transporters, such as hepatic OATP1B1/1B3 (organic anion transporting polypeptide) or renal OAT1/3 (Neuvonen et al., 2006). However, for lipophilic amines, such as antidepressants, rapid passive diffusion coupled with lysosomal trapping can also yield high liver-to-blood ratios that are not attributable to active uptake processes, such as hepatic or renal transport, which is an important consideration when evaluating the mechanism of hepatic or renal accumulation of drugs.
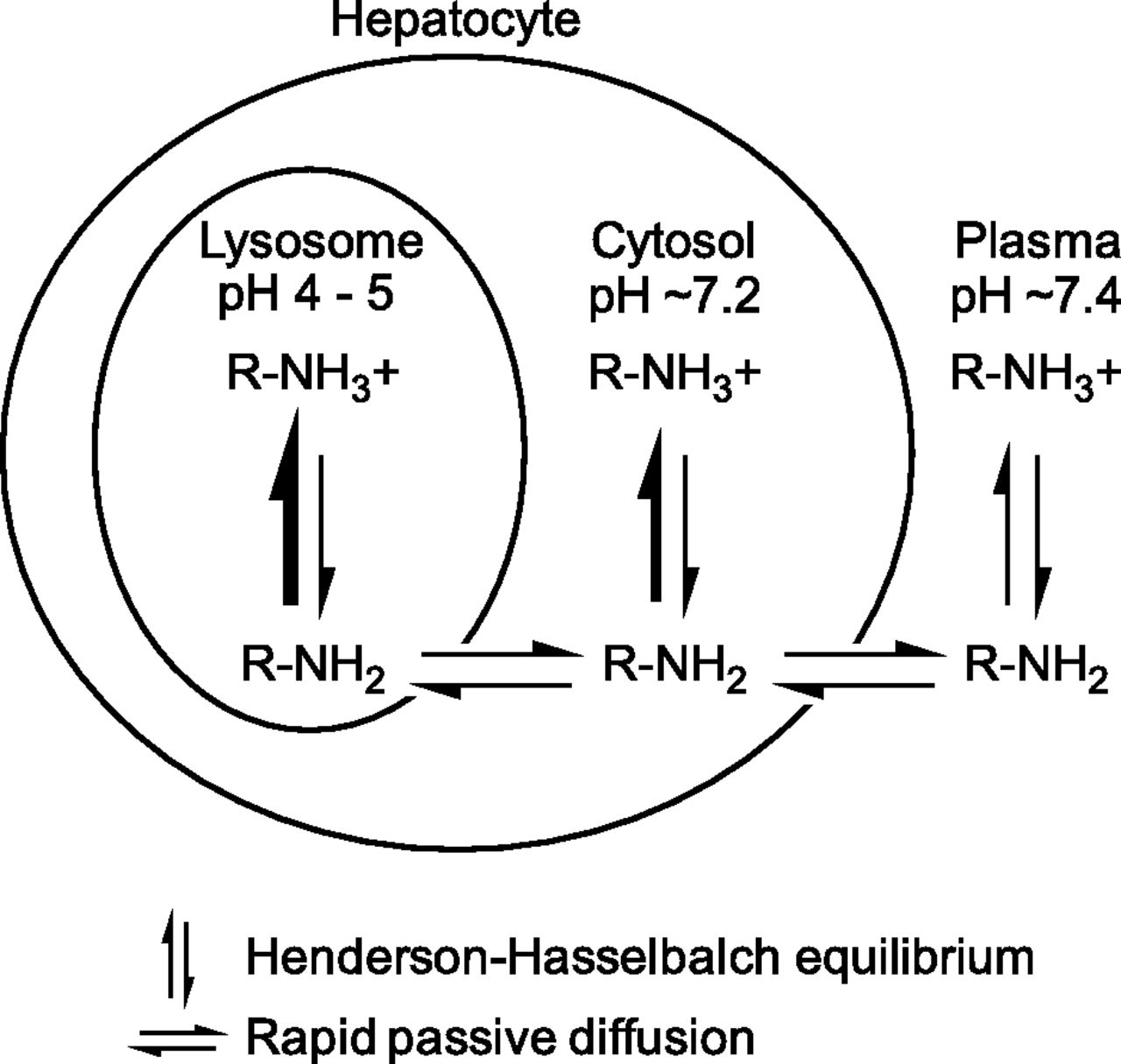
The basis of pH partitioning of lipophilic amines into lysosomes. The diagram illustrates the mechanism by which lipophilic amines (i.e., CADs) accumulate in lysosomes. From plasma (pH 7.4) and cytosol (∼7.2), a lipophilic amine (logP > 1, pKa > 6.5) will readily diffuse across membranes in its unionized form (RNH2) while maintaining Henderson-Hasselbach equilibrium with its ionized form (RNH3+, which cannot readily diffuse across membranes). After diffusion into the acidic environment of the lysosome (pH 4–5), the equilibrium between charged and uncharged species shifts in favor of the ionized form of the lipophilic amine, limiting diffusion of the drug back into the cytosol and, in effect, trapping the drug in lysosomes. For highly permeable lipophilic amines, the concentration of unionized drug (RNH2) at equilibrium is assumed to be the same in all three compartments (lysosomes, cytosol, and plasma). The figure is not to scale; lysosomes make up about 1% of the hepatocyte volume.
Competition for lysosomal trapping has been the subject of some speculation as a potential mechanism of drug-drug interactions (DDIs) (Daniel and Wojcikowski, 1999; Chadwick et al., 2005; Funk and Krise, 2012; Logan et al., 2012). Because many central nervous system and cardiovascular drugs are lysosomotropics (drugs that undergo lysosomal sequestration), there is the possibility that concomitant administration of lysosomotropics could lead to elevated drug exposure levels as competition for lysosomal sequestration increases or lysosomal pH is elevated by amine accumulation (Kornhauser et al., 1980; Vestal et al., 1980; Logan et al., 2012). Furthermore, prolonged accumulation of CADs in lysosomes, which impairs lysosomal function, has been implicated as the major cause of drug-induced phospholipidosis, characterized by an excessive accumulation of phospholipids in various tissues because of decreased phospholipid catabolism (Hanumegowda et al., 2010).
The sequestration of lipophilic amines into lysosomes can be evaluated in vitro by assessing the accumulation of fluorescent lipophilic amine probes. The probe Red DND-99 (also known as LysoTracker Red), a lipophilic amine with logP 2.10 and pKa 7.5 (Duvvuri et al., 2004), has been shown to be highly specific for lysosomal accumulation and has been used in numerous nonhepatic cell lines (Lemieux et al., 2004; Duvvuri and Krise, 2005; Nadanaciva et al., 2011). However, because the presystemic extraction of lipophilic amines into lysosome-rich organs (such as the liver) would be expected to play a key role for CADs, the potential for lysosome-based DDIs may be better evaluated in a more suitable test system, such as human hepatocytes or a human hepatic-derived cell line with functional lysosomes. The Fa2N-4 cells are SV40 virus large T antigen–transformed human hepatocytes that do not express constitutive androstane receptor and do not retain significant transporter activity (Hariparsad et al., 2008). However, the Fa2N-4 cells propagate in culture and readily attach to collagen-coated plates, circumventing some of the limitations of using cryopreserved human hepatocytes.
In the present study, we describe an in vitro method to screen compounds for lysosomal trapping potential in immortalized hepatocytes (Fa2N-4 cells) with use of the lysosome-specific fluorescent probe LysoTracker Red. Over two dozen compounds were screened for their propensity to inhibit LysoTracker Red fluorescence signal in Fa2N-4 cells. Studies were conducted to quantify the partitioning of the lysosomotropics propranolol and imipramine in Fa2N-4 cells and to evaluate whether such partitioning could be inhibited by other lysosomotropics.
Materials and Methods
Chemicals and Reagents.
Acetaminophen, amitriptyline, ammonium chloride, astemizole, chloroquine, d5-atorvastatin, dextromethorphan, diclofenac, fluoxetine, imipramine, ketoprofen, labetalol, monensin, nifedipine, nigericin, paroxetine, pravastatin, propranolol, and raclopride were purchased from Sigma-Aldrich (St. Louis, MO); atorvastatin, cetirizine, desipramine, erlotinib, fluconazole, fluvastatin, gefitinib, ketorolac, lapatinib, rosuvastatin, and tenoxicam were purchased from Toronto Research Chemicals Inc. (North York, ON, Canada); amodiaquine was purchased from US Pharmacopeia (Rockville, MD); d7-propranolol was purchased from CDN isotopes (Pointe-Claire, QC, Canada); and LysoTracker Red was purchased from Invitrogen (Eugene, OR). The sources of the other reagents used in this study have been described elsewhere (Paris et al., 2009; Parkinson et al., 2011; Funk and Krise, 2012).
Test System and Cell Culture.
Fa2N-4 cells (hepatocytes that have been stably transformed with the SV-40 large T antigen), multifunction enhancing (MFE) plating media containing 10% newborn calf serum (Hyclone, Logan, UT), MFE support media, and human hepatocytes from nontransplantable livers were prepared at XenoTech, LLC (Lenexa, KS). Cryopreserved immortalized Fa2N-4 cells were thawed and cultured to confluency in T-150 flasks (Corning Incorporated, Corning, NY) in serum-free MFE medium (∼30 ml), which was replaced every other day. When the cells reached confluency (every ∼3–4 days), as determined by light microscopy, the cells were rinsed with ∼30 ml of prewarmed (37°C) 1 × phosphate-buffered saline (PBS). The PBS was then aspirated and replaced with 5–8 ml of prewarmed (37°C) trypsin-EDTA (Gibco, Grand Island, NY). The flasks were incubated in a humidified environment (37°C with 95% humidity and 5% CO2) until the cells began to detach from the flask (3–15 minutes). The detached cells were subsequently transferred to a 50-ml conical tube, and the volume was diluted to 50 ml with MFE plating medium. The conical tube was centrifuged at 120 ± 10 relative centrifugal force for 5 ± 1 minutes at room temperature. The resulting supernatant fraction was aspirated and the pellet resuspended with 1–3 ml of MFE plating medium. Trypan Blue exclusion analysis determined the concentration of cells, which were diluted to the desired concentration for plating on either fresh flasks or 96-well plates. Before experiments, Fa2N-4 cells were plated on collagen-coated 96-well plates from Nunc (Rochester, NY) or collagen-coated black-clear bottom 96-well microtiter plates (Corning Incorporated) in MFE medium containing 10% newborn calf serum at a concentration of 50,000 cells/well (100 µl incubation volume). Cells were maintained at 37°C with 95% humidity and 5% CO2. After the cell attachment period (3–20 hours), media were replaced with fresh, serum-free medium, and experiments were performed 36–48 hours after plating.
Fluorescence Microscopy.
Epifluorescence microscopy was conducted as described previously (Duvvuri et al., 2004; Lemieux et al., 2004; Funk and Krise, 2012). In brief, cryopreserved human hepatocytes or Fa2N-4 cells were thawed and diluted to 1 × 106 cells/ml. Cells (0.5 ml) were added to 8-chamber slides and allowed to adhere overnight. LysoTracker Red (200 nM) was added to each well, except for background wells, for two hours. Controls included incubations with nigericin (10 μM) and monensin (20 μM), which are ionophores that uncouple the proton gradient present in lysosomes and ammonium chloride (10 mM), which raises lysosomal pH through buffering. The cells were subsequently washed three times with D-PBS and immediately mounted with a coverslip for imaging. Samples were visualized using a Nikon Eclipse 80i epifluorescence microscope (Nikon Instruments Inc., Melville, NY) equipped with a 40× (1.30 NA) oil-immersion objective at the excitation/emission wavelengths for LysoTracker Red (λex 530 nm, λem 590 nm). Images were captured using an ORCA ER camera (Hamamatsu, Hamamatsu City, Japan) and analyzed using Metamorph, version 7.0 (Molecular Devices, Sunnyvale, CA). Background images of nonfluorescently labeled cells were acquired to correct for auto-fluorescence. Images were scaled identically to allow for comparison.
Assessment of Lysosomal Trapping in Fa2N-4 Cells with LysoTracker Red.
Fa2N-4 cells (50,000 cells/well) were plated in collagen-coated black-clear–bottom 96-well microtiter plates (Corning Incorporated), as described above. Before the assay, the MFE plating medium was removed and the wells were rinsed twice with 100 μl prewarmed (37°C) 1 × PBS. Stock solutions of each test drug were initially prepared in methanol and diluted with supplemented modified Chee’s medium such that the final incubation concentrations of each drug were 1, 5, 10, 50, 100, and 500 μM. LysoTracker Red was added to each test drug substock solution such that the final incubation concentration of LysoTracker Red was 50 nM. Incubations with the cells were performed at a final volume of 100 μl in triplicate for 30 minutes at 37°C with 95% humidity and 5% CO2. The incubation matrix was then aspirated (and kept for analysis of lactate dehydrogenase), and the plate was rinsed twice with 200 μl 1 × PBS (at room temperature) using a Tecan Genesis EVO automated liquid handling system (Tecan, Morrisville, NC) before solubilizing the cells with 100 μl of acetonitrile. Samples were then analyzed for LysoTracker Red fluorescence (λex 530 nm, λem 590 nm) with a BioTek Synergy plate reader (BioTek, Winooski, VT).
Cytotoxicity Assessment.
The incubation medium from plated Fa2N-4 cells used to assess lysosomal trapping was analyzed for lactate dehydrogenase (LDH) release, a marker of drug-induced cytotoxicity, using a cytotoxicity detection kit (LDH) purchased from Roche Diagnostics (Indianapolis, IN). In brief, 80 μl of the incubation medium was transferred to a Costar 96-well block plate (Fisher Scientific, Pittsburgh, PA) and diluted with 300 μl of supplemented modified Chee’s medium to have adequate sample volume for the assay. A single well was treated with 1% Triton X-100 (Sigma-Aldrich) as a positive control for the assay (100% LDH release).
Assessment of Cellular Partitioning Lysosomal Trapping.
Fa2N-4 cells were cultured and plated as described above. Substrates (1 μM) were incubated with plated Fa2N-4 cells in serum-free MFE medium at 37±1°C in the presence and absence of competing drugs, ammonium chloride, nigericin, and monensin. Before the incubations were started, the cells were washed once with fresh, serum-free MFE media. Cells and the substrate solution were preincubated separately (5 minutes), and incubations were initiated by the addition of the substrate solution with or without inhibitor to the cells. After 5 minutes, incubations were terminated by the addition of ice-cold PBS (100 μl). The cells were then washed two times with ice-cold PBS (100 μl) and were subsequently lysed with acetonitrile containing internal standard (amitriptyline for imipramine, d5-atorvastatin for atorvastatin, and d7-propranolol for propranolol; 150 μl). An aliquot (100 μl) of the cell lysate was transferred to a Costar 96-well analytical plate (Fisher Scientific). The final volume was adjusted to 200 μl with water. The 96-well analytical plate was vortexed, and residual protein was precipitated by centrifugation (920 relative centrifugal force for 10 minutes at 10°C). The amount of substrate in the cells was determined by liquid chromatography-tandem mass spectrometry.
Analytical Methods.
Atorvastatin, propranolol, and imipramine in the cells were quantified using high-performance liquid chromatography–tandem mass spectrometry against calibration curves constructed over the range 0.002–2.0 µM. Atorvastatin was analyzed using a Shimadzu Nexera LC system comprising LC-20AD pumps, a CBM-20A controller, a SIL-20AC autosampler, and a DGU-14 solvent degasser (Shimadzu, Kyoto, Japan) interfaced by electrospray ionization to an AB Sciex API4000 QTrap mass spectrometer (Foster City, CA) operated in negative multiple reaction monitoring (MRM) mode. Mobile phases were 0.1 mM ammonium acetate in water:methanol 95:5 v/v (A) and 0.1 mM ammonium acetate in methanol (B). A linear gradient ramping from 50% to 85% B over 1.9 minute was applied to a Waters Atlantis dC18, 5 μm, 100 × 2.1 mm column (Milford, MA), preceded by a direct connection Phenomenex Luna C8, 4 × 2 mm guard cartridge (Torrance, CA), for separation. MRM transitions for detection of atorvastatin and d5-atorvastatin internal standard were 557.3/397.2 and 562.3/402.2, respectively. Atorvastatin and d5-atorvastatin eluted at approximately 1.8 minutes. Propranolol and imipramine were analyzed using a Shimadzu LC system comprising LC-10ADVP pumps, a SIL-HTA autosampler, and a DGU-14A solvent degasser (Shimadzu) interfaced by electrospray ionization to an AB Sciex API2000 triple quadrupole mass spectrometer (Foster City, CA) operated in positive MRM mode. Mobile phases were 0.2% formic acid in water (A) and 0.2% formic acid in methanol (B). A linear gradient ramping from 40 to 98% B over 1.65 minutes was applied to a Waters Atlantis dC18, 5 μm, 50 × 2.1 mm column, preceded by a direct connection Phenomenex Luna C8, 4 × 2 mm guard cartridge, for separation. MRM transitions for detection of propranolol and d7-propranolol internal standards were 260.1/116.0 and 267.1/116.0, respectively and propranolol and d7-propranolol eluted at approximately 1.8 minutes. MRM transitions for detection of imipramine and amitriptyline internal standard were 281.2/86.0 and 278.1/232.9, respectively. Imipramine and amitriptyline were eluted at approximately 1.9 minutes.
Data Analysis.
Physicochemical information (Table 1) for each compound was predicted using MarvinSketch 5.9.0 (ChemAxon, Cambridge, MA). Each compound tested was classified as an acid, base, neutral, or zwitterion based on the major molecular species present at pH of 7.4. LysoTracker Red IC50 values were calculated using GraFit 4.0.21 (Erithacus Software Ltd., Horley, Surrey, UK). All other data were processed and graphed using Microsoft Excel 2007 (Microsoft, Redmond, WA).
Predicted physicochemical properties and experimentally determined LysoTracker Red IC50 values for 27 compounds
All physicochemical properties were calculated with MarvinSketch 5.9.0 as described in the Materials and Methods; compounds that showed >25% LysoTracker Red inhibition at the highest tested concentration but did not yield an IC50 value or exhibited concentration-dependent cytotoxicity were classified as possible for undergoing lysosomal sequestration.
Results
Epifluorescence Microscopy of Human Hepatocytes and Fa2N-4 Cells.
Initially cryopreserved human hepatocytes were evaluated by epifluorescence microscopy with LysoTracker Red to determine the distribution of functional lysosomes in these cells. As shown in Fig. 2A, LysoTracker Red fluorescence was localized to lysosomes, and after treatment of the cells with nigericin and monensin (ionophores that uncouple the proton gradient present in lysosomes), the localized (vesicular) fluorescent signal was lost (Fig. 2B), indicating that, as expected, LysoTracker Red localized to acidic lysosomes in cryopreserved human hepatocytes. Cryopreserved human hepatocytes were also treated with 10 mM NH4Cl with similar results (data not shown). To determine whether immortalized hepatocytes (Fa2N-4 cells) also expressed functional lysosomes, the cells were incubated with LysoTracker Red and examined by epifluorescence microscopy. The Fa2N-4 cells also localized LysoTracker Red into vesicle-like compartments, similar to the distribution of lysosomes observed in cryopreserved hepatocytes (Fig. 2C). To confirm that these vesicle compartments had functional proton gradients indicative of lysosomes, the immortalized hepatocytes were treated with 10 mM NH4Cl (which partitions into acidic compartments and neutralizes intracellular pH levels). After treatment with NH4Cl, LysoTracker Red florescence signal was markedly decreased (Fig. 2D), indicating the presence of functional lysosomes in Fa2N-4 cells.

Epifluorescence microscopy of cryopreserved human hepatocytes and immortalized hepatocytes (Fa2N-4 cells) treated with LysoTracker Red. Cryopreserved human hepatocytes and immortalized hepatocytes (Fa2N-4 cells) were diluted to 1 × 106 cells and incubated with 200 nM of LysoTracker Red for two hours, as described in Materials and Methods. Cryopreserved human hepatocytes (A) showed punctate fluorescent staining characteristic of lysosomes, which was attenuated when treated with the ionophores nigericin and monensin (B). Fa2N-4 cells showed a similar punctate staining pattern (C), which was also mitigated with an inhibitor of lysosomal sequestration, ammonium chloride (D)
Compound Screen for Inhibition of LysoTracker Red Fluorescence.
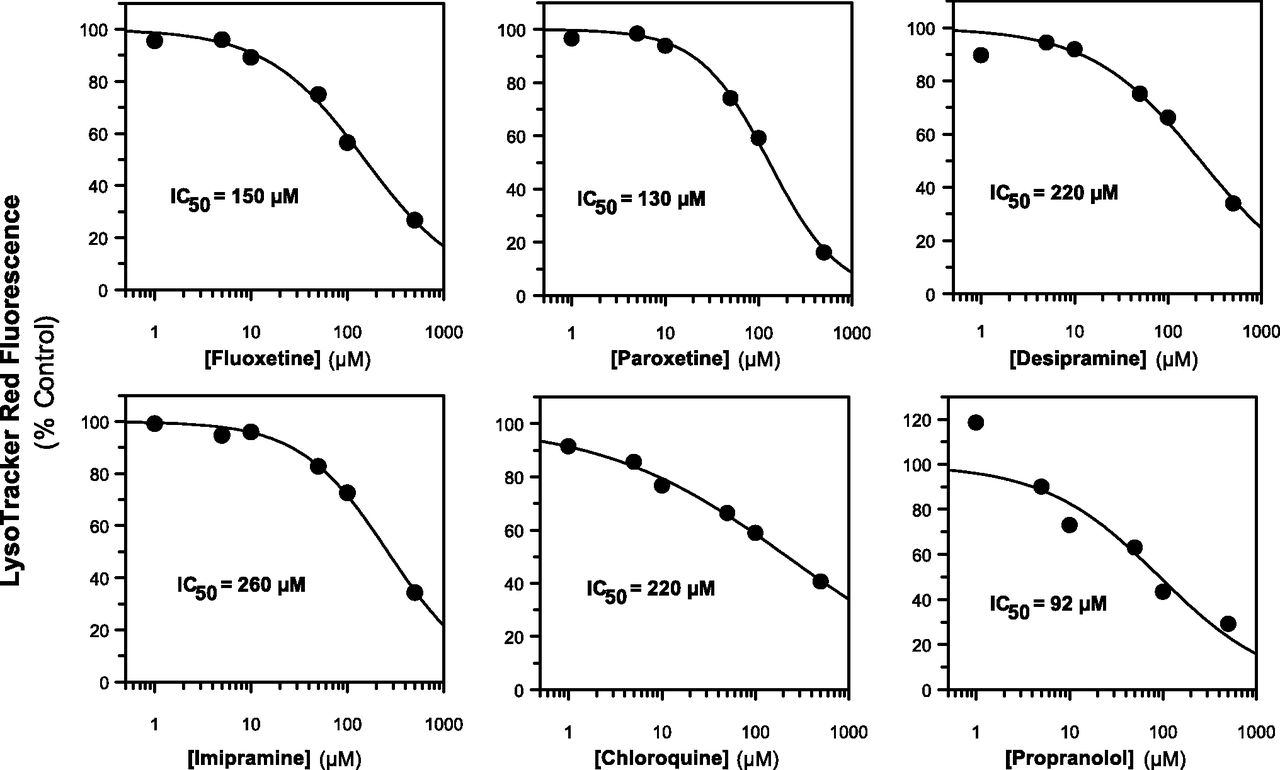
To ascertain the propensity for the Fa2N-4 cells to trap lysosomotropics or nonlysosomotropics, 27 compounds spanning various physicochemical properties and diverse therapeutic indications were tested for their ability to inhibit LysoTracker Red fluorescence in a concentration-dependent manner, including acids, lipophilic amines, neutrals, and zwitterions (Table 1). Of the compounds tested, those that were acids (diclofenac, ibuprofen, ketoprofen, ketorolac, tenoxicam, atorvastatin, fluvastatin, pravastatin, and rosuvastatin) did not inhibit LysoTracker Red fluorescence, demonstrating little or no lysosomal trapping of these drugs. Most drugs that were bases and known lysosomotropics (fluoxetine, paroxetine, desipramine, imipramine, chloroquine, and propranolol) blocked LysoTracker Red partitioning. IC50 values are shown in Fig. 3. The basic drugs amodiaquine, dextromethorphan, and labetalol did not give a measurable LysoTracker Red IC50 value in the concentration range tested; however, weak inhibition was observed at the highest concentration tested (> 25% inhibition; unpublished data). Of the drugs that are neutral at plasma pH, gefitinib inhibited LysoTracker Red fluorescence with an IC50 value of 77 μM. None of the zwitterions tested (cetirizine and raclopride) inhibited LysoTracker Red fluorescence. Both astemizole (base) and lapatinib (neutral) also inhibited LysoTracker Red signal; however, these compounds also exhibited marked cytotoxicity (as measured by LDH release) at the same concentrations. Thus their propensity to inhibit lysosomal trapping of LysoTracker Red could not be determined.

The effect of known lysosomotropics on LysoTracker Red fluorescence in immortalized hepatocytes (Fa2N-4 cells). The lipophilic amines fluoxetine, paroxetine, desipramine, imipramine, chloroquine, and propranolol were incubated in Fa2N-4 cells at 1, 5, 10, 50, 100, and 500 μM for 30 minutes in the presence of 50 nM LysoTracker Red, as described in Materials and Methods. The data are summarized in Table 1.
Quantification of Lysosomotropic and Nonlysosomotropic Partitioning.
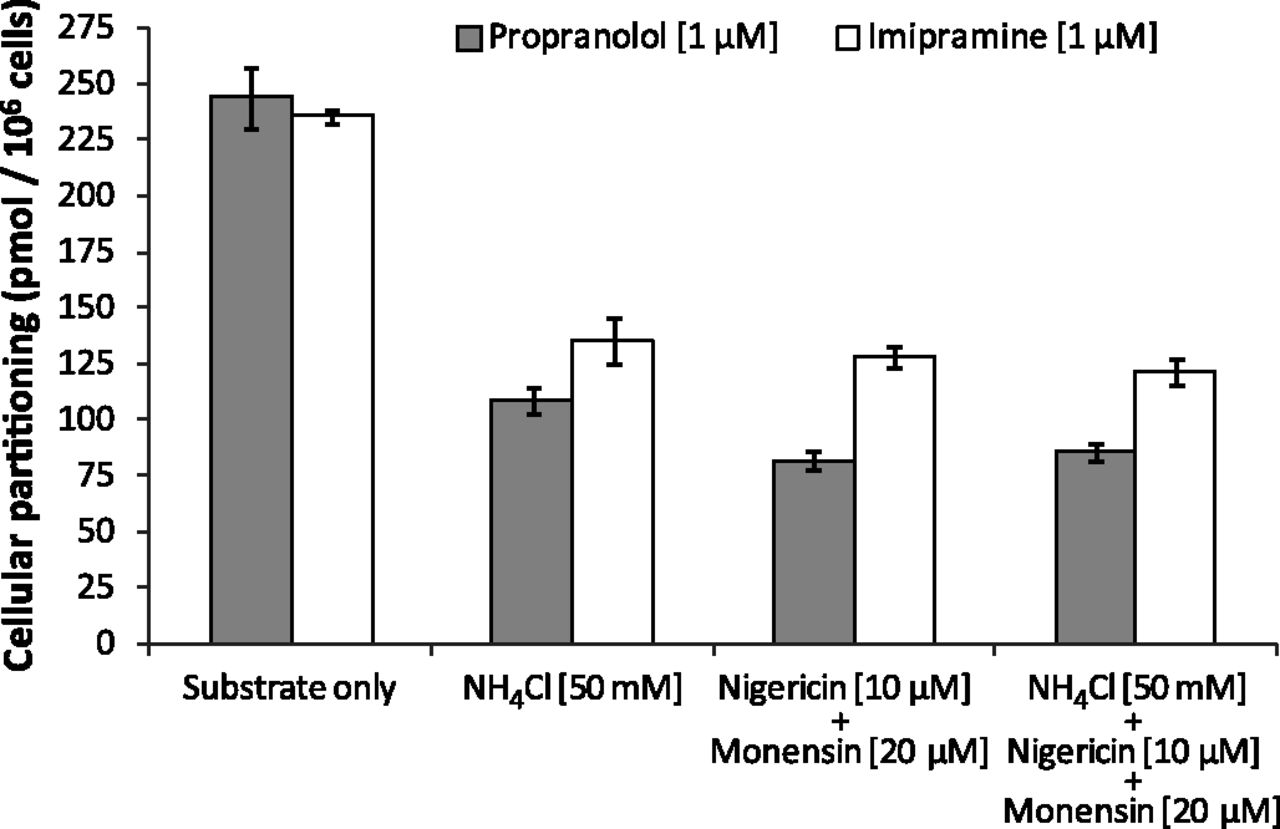
Although the aforementioned studies qualitatively evaluated the propensity of immortalized hepatocytes to lysosomally trap drugs, a quantitative approach was used to ascertain changes in cellular partitioning of the lysosomotropics propranolol and imipramine and the nonlysosomotropic atorvastatin in the presence of ammonium chloride. Propranolol, imipramine, or atorvastatin (1 μM) was incubated for 5 minutes with immortalized hepatocytes with or without 50 mM ammonium chloride, as described in Materials and Methods. As shown in Fig. 4, both propranolol and imipramine showed marked partitioning in Fa2N-4 cells with cellular partitioning values of 189 and 320 pmol/106 cells, respectively, whereas atorvastatin showed little to no partitioning. In the presence of ammonium chloride, the partitioning of propranolol and imipramine was reduced by approximately 50%. A time course of propranolol and imipramine partitioning was examined and was shown to be linear up to 10 minutes (Fig. 5). In the presence of ammonium chloride, the partitioning of propranolol and imipramine was reduced by approximately 60 and 45%, respectively, at 10 minutes. The effect of inhibitors of lysosomal trapping (ammonium chloride and chloroquine) on the partitioning of propranolol in Fa2N-4 cells was examined, specifically the effect of (1) coincubation of both substrate and inhibitor, (2) preincubation of the inhibitor followed by coincubation of both substrate and inhibitor, and (3) preincubation of the inhibitor followed by removal of media and replacement with media containing substrate only. As shown in Fig. 6, similar results (∼50% inhibition) were obtained when ammonium chloride or chloroquine was coincubated with propranolol or preincubated with the cells followed by coincubation with propranolol. The mitigation of lysosomal trapping by ammonium chloride was reversed with a short wash-out period before incubation with propranolol. In contrast, the inhibition of propranolol partitioning by chloroquine persisted despite removal of the medium before propranolol addition. The additive effects of multiple inhibitors of lysosomal trapping (ammonium chloride, nigericin, and monensin) on the partitioning of propranolol and imipramine in Fa2N-4 cells was examined. No difference in the inhibition of lysosomal trapping was observed between ammonium chloride (50 mM) and the ionophores (10 μM nigericin plus 20 μM monensin), and no additive effects were observed when all 3 inhibitors of lysosomal trapping were combined (Fig. 7).

A comparison of the partitioning of propranolol, imipramine, and atorvastatin in Fa2N-4 cells with and without ammonium chloride. The lipophilic amines propranolol and imipramine and the OATP substrate atorvastatin were incubated at 1 μM in the presence or absence of 50 mM ammonium chloride for 5 minutes in Fa2N-4 cells, and the partitioning of each compound was determined by liquid chromatography-tandem mass spectrometry, as described in Materials and Methods.

Time course of the partitioning of propranolol and imipramine in Fa2N-4 cells with and without ammonium chloride. The lipophilic amines propranolol (A) and imipramine (B) (1 μM) were incubated in the presence or absence of 50 mM ammonium chloride in Fa2N-4 cells for 1, 3, 5, 7, 10, 15, 20, and 30 minutes, and the cellular partitioning of each lipophilic amine was determined by liquid chromatography-tandem mass spectrometry, as described in Materials and Methods.

The effect of order of addition of substrate and inhibitor on the partitioning of propranolol (substrate) in Fa2N-4 cells. Propranolol (1 μM) partitioning into Fa2N-4 cells was assessed for 5 minutes with and without inhibitor [50 mM ammonium chloride (A) or 200 μM chloroquine (B)] under three conditions: (1) the substrate and inhibitor were added simultaneously (coincubation), (2) the inhibitor was added 5 minutes before substrate but was not removed (preincubation and coincubation), and (3) the inhibitor was added 5 minutes before the substrate, but the cells were washed before adding the substrate (preincubation), as described in Materials and Methods.

The effect of inhibitors of lysosomal trapping on the partitioning of propranolol and imipramine in Fa2N-4 cells. Propranolol and imipramine partitioning into Fa2N-4 cells was assessed for 5 minutes in the presence of (1) 50 mM ammonium chloride alone, (2) the ionophores nigericin (10 μM) and monensin (20 μM) together, or (3) a combination of all three inhibitors, as described in Materials and Methods.
Lysosomal Trapping with Concomitant Lysosomotropic or Nonlysosomotropic Drugs.
The propensity for lysosomotropics and nonlysosomotropic drugs to affect the partitioning of propranolol, imipramine, and atorvastatin was examined, and the results are shown in Fig. 8. The partitioning of both propranolol and imipramine was inhibited by the lysosomotropics astemizole, dextromethorphan, chloroquine, ammonium chloride, as well as imipramine and propranolol, respectively. Propranolol partitioning was inhibited to a greater degree than was imipramine partitioning in Fa2N-4 cells. Nonlysosomotropics, such as diclofenac, rifampin, or atorvastatin, had little to no effect on the partitioning of propranolol or imipramine. In comparison with propranolol and imipramine, very little atorvastatin partitioning was observed (< 11 pmol/106 cells), consistent with the observation that the Fa2N-4 cells retain little to no transporter activity (Hariparsad et al., 2008). Of interest, atorvastatin partitioning in Fa2N-4 cells increased marginally, up to a maximum of approximately 34 pmol/106 cells, in the presence of either lysosomotropic or nonlysosomotropic drugs. However, the mechanism of this activation of partitioning was not further investigated.

An assessment of propranolol, imipramine, and atorvastatin partitioning in Fa2N-4 cells in the presence of lysosomotropic (basic) and nonlysosomotropic (acidic) drugs. Propranolol (1 μM), imipramine (1 μM), and atorvastatin (1 μM) partitioning was determined in Fa2N-4 cells (5-minute incubation) in the presence of nonlysosomotropic drugs (rifampin, diclofenac, and atorvastatin) and lysosomotropic compounds (astemizole, dextromethorphan, chloroquine, ammonium chloride, imipramine, or propranolol), as described in Materials and Methods.
Discussion
Lysosomal trapping involves the partitioning of the neutral form of lipophilic amines (R-NH2) into cells, generally by passive diffusion (with possible involvement of OCTs) and the subsequent accumulation of the protonated and less permeable form of the drug (R-NH3+) in the acidic environment of lysosomes (pH 4–5) (Fig. 1). Macintyre and Cutler (1988) calculated that, if the volume of lysosomes equaled the volume of cytosol, a strongly basic compound (pKa > 11) would have an accumulation ratio of 400. A strong dibasic compound, where diffusion from the acidic lysosome to the cytosol requires 2 simultaneous deprotonation events to form the permeable neutral compound, has an accumulation ratio of 160,000. The volume of lysosomes is about 0.7% of hepatocyte volume (MacIntyre and Cutler, 1988); thus, the actual accumulation ratios for mono- and dibasic drugs are about 3 and 1000 times liver volume, respectively. The rapid accumulation of lipophilic amines in lysosome-rich organs, such as liver and lung, can contribute (together with phospholipid and protein binding) to first-pass extraction of orally administered drugs and the rapid distribution of intravenously administered drugs into tissues, which can lead to high liver-to-blood ratios and large volumes of distribution (MacIntyre and Cutler, 1988; Hallifax and Houston, 2007). The dibasic drug chloroquine has a volume of distribution at steady state (Vdss) of 140 l/kg, second only to hydroxychloroquine (Vdss = 700 l/kg). In the pharmacokinetic database of 670 drugs compiled by Obach et al. (2008), all of the drugs with a Vdss of 25 l/kg or more are mono-, di-, or tri-basic compounds.
The term trapping is widely used to describe lysosomal accumulation of drugs. but the term is misleading. The ionized form of the drug that is trapped in lysosomes is in equilibrium with the neutral drug that, in most cases, can rapidly cross the lysosomal membrane by passive diffusion when the cytosolic concentration of drug decreases as the result of metabolism, diffusion back into plasma, or diffusion/active transport into the bile. In essence, lysosomes function as a drug reservoir and do not indefinitely trap xenobiotics. The rapid reversibility of lysosomal trapping of a lipophilic amine is shown by the effects of asphyxiating rats with carbon dioxide, which slightly acidifies the blood and causes a slight decrease in tissue levels and an increase in plasma drug levels (Angus et al., 2008). Although passive diffusion is one mechanism by which lipophilic amines can exit lysosomes, there are other mechanisms by which this can occur, namely mechanisms more relevant for the recycling of phospholipid-bound drugs (Goldman et al., 2009).
In the present study, Fa2N-4 cells were examined for their ability to accumulate the lysosome-specific fluorescent probe LysoTracker Red. Epifluorescence microscopy established that the immortalized hepatocytes resembled cryopreserved primary human hepatocytes in their ability to accumulate the pH-sensitive probe LysoTracker Red with a punctate signal pattern characteristic of localization in acidic vesicles (Fig. 2). These vesicles were identified as lysosomes based on the ability of ammonium chloride to block vesicular localization of LysoTracker fluorescence. The use of Fa2N-4 cells for studies of the lysosomal sequestration of drugs has several advantages over the use of primary cells. Lysosomal sequestration is an active process (energy is constantly expended by the lysosomal V-ATPase to maintain the pH gradient), as is the transporter-mediated uptake of drugs. In primary cells, the mechanism for active accumulation of drugs must be investigated to distinguish active uptake by drug transporters from active accumulation by lysosomal trapping. Fa2N-4 cells lack significant transporter expression (Hariparsad et al., 2008); thus, the accumulation of drugs in Fa2N-4 cells is predominantly by lysosomal trapping. The ease of culturing and attaching Fa2N-4 cells to collagen substrata contribute to the usefulness of these cells as an in vitro system to examine lysosomal trapping.
To validate the lysosomal trapping of xenobiotics in immortalized hepatocytes, a selection of 27 compounds spanning diverse physicochemical properties was investigated for their ability to inhibit the accumulation of LysoTracker Red in cultured Fa2N-4 cells. However, of note, drugs can inhibit the accumulation of LysoTracker Red by four mechanisms: (1) the drug can compete with LysoTracker Red for lysosomal accumulation, which is the basis of inhibiting lysosomal trapping with ammonium chloride; (2) the drug can permeabilize the lysosomal membrane to protons, which is the mechanism by which the ionophores nigericin and monensin raise lysosomal pH; (3) the drug can inhibit the lysosomal V-ATPase that maintains the acidic environment of the lysosome, which has been documented for the antibiotic bafilomycin A1 (Bowman et al., 1988; Yoshimori et al., 1991); and (4) the drug can cause cell toxicity. In the present study, the latter possibility was assessed on the basis of LDH release into the cell culture medium. Of the 27 compounds screened in this study (Table 1), only the lipophilic amines caused concentration-dependent inhibition of LysoTracker Red accumulation in Fa2N-4 cells (Fig. 3). Certain lipophilic amines, such as amodiaquine, dextromethorphan, and labetalol, inhibited LysoTracker Red accumulation by >25% at only the highest concentration tested, despite being known lysosomotropics (Pappu et al., 1985; Hallifax and Houston, 2007; Hayeshi et al., 2008). Gefitinib caused marked attenuation of LysoTracker Red fluorescence, indicating it is sequestered in lysosomes, as reported previously (Nadanaciva et al., 2011). Astemizole and lapatinib both inhibited LysoTracker Red signal but were also cytotoxic to the Fa2N-4 cells; however, both have been previously shown to be lysosomotropic (Nadanaciva et al., 2011). In general, the physicochemical properties of the compounds that undergo trapping suggest that lysosomotropic drugs are those that have a logP > 2 and a basic pKa of 6.5–11, which is consistent with findings in the literature (Nadanaciva et al., 2011).
In direct studies of drug partitioning into Fa2N-4 cells, the lipophilic amines propranolol and imipramine showed marked cellular partitioning, with lysosomal sequestration contributing approximately 50% to their accumulation based on the degree of inhibition by ammonium chloride. Because Fa2N-4 cells lack OATPs, acidic compounds (charged drugs at pH 7.4) with low intrinsic permeability would not be expected to enter Fa2N-4 cells at an appreciable rate. Atorvastatin is an acidic drug with low intrinsic permeability at pH 7.4 and whose uptake into hepatocytes is largely mediated by OATP (Jigorel and Houston, 2012). As expected, atorvastatin showed little to no partitioning by Fa2N-4 cells (Fig. 4). Partitioning of propranolol and imipramine was linear up to 10 minutes. Ammonium chloride reduced but did not eliminate the accumulation of propranolol and imipramine in Fa2N-4 cells (Fig. 5), suggesting that accumulation of these lipophilic amines is partly attributable to lysosomal trapping and partly attributable to binding to phospholipids and cellular proteins. Ammonium chloride, chloroquine, and the ionophores nigericin and monensin blocked the partitioning of propranolol to about the same extent (∼50%). The effects of these inhibitors were not additive (Fig. 7). Adding chloroquine 5 minutes before the addition of propranolol did not enhance its ability to block propranolol partitioning (Fig. 6). A wash-out period between treating the cells with chloroquine or ammonium chloride and adding propranolol reversed the inhibitory effect of ammonium chloride but not that of chloroquine, a dibasic lipophilic amine that leaves the lysosome very slowly (of note, the plasma half-life of chloroquine is 570 hours) (Obach et al., 2008). Overall, these results support that immortalized hepatocytes (Fa2N-4 cells) are an appropriate system to evaluate the lysosomal sequestration of lipophilic amines, because both the qualitative and quantitative approaches confirmed that immortalized hepatocytes maintain functional lysosomes with the propensity to trap lipophilic amines.
The potential for DDIs involving lysosomes is a topic of speculation (Daniel and Wojcikowski, 1999; Chadwick et al., 2005; Funk and Krise, 2012; Logan et al., 2012). Although coadministration-related subtle changes in organ distribution may occur, there are no known clinical examples of lysosomal-related DDI in which changes in drug clearance have been causally attributed to competition among lysosomotropic agents. In the present study, we examined the partitioning of propranolol, imipramine, and atorvastatin with lysosomotropics and nonlysosomotropics. All lysosomotropic drugs decreased the partitioning of propranolol and imipramine, whereas nonlysosomotropics did not (Fig. 8). There are limited examples in the literature of similar findings, such as the inhibition (40–70%) of the in vitro partitioning of imipramine by other lipophilic amines in isolated rat hepatocytes (Hallifax and Houston, 2006; Hallifax and Houston, 2007) and ion-trapping in lysosomes accounting for approximately 50% of propranolol hepatic sequestration during in situ perfusions of isolated rat livers (Siebert et al., 2004), consistent with data in this study (∼50% propranolol partitioning). More recently, the P-glycoprotein inhibitor tariquidar has been shown to significantly decrease the in vitro and in vivo accumulation of the positron emission tomography imaging agent [11C]-N-desmethyl-loperamide in lysosome-rich tissues through competition for lysosomal trapping (Kannan et al., 2011). However, the clinical significance of lysosomal trapping–based DDIs is not well understood and requires further study.
Lysosomal trapping of drugs has also been associated with drug-induced phospholipidosis, an iatrogenic lysosomal storage disorder characterized by the accumulation of phospholipids in tissues (Daniel and Wojcikowski, 1999; Anderson and Borlak, 2006; Hanumegowda et al., 2010; Shayman and Abe, 2013). At least 385 drugs that cause phospholipidosis have been identified in the Food and Drug Administration database (Advisory Committee for Pharmaceutical Science, April 2010; http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/advisorycommitteeforpharmaceuticalscienceandclinicalpharmacology/ucm210798.pdf), all of which have been identified as CADs. The development of drug-induced phospholipidosis in nonclinical species usually requires prolonged treatment with high doses of CADs, which are lipophilic amines that cannot only increase lysosomal pH but can bind tightly to phospholipids. This leads to the impediment of lysosomal-based catabolism of phospholipids through inhibition of acidic hydrolases and the endo- and exocytosis of endobiotics in and out of lysosomes as well as impeding lysosomal cycling (Daniel and Wojcikowski, 1999; Hanumegowda et al., 2010; Muehlbacher et al., 2012). Because of the strong association between lysosomal trapping of CADs and drug-induced phospholipidosis, screening approaches to identify lysosomotropic compounds will help to identify early in drug discovery those compounds with the potential to cause phospholipidosis.
In conclusion, we have shown the usefulness of the Fa2N-4 immortalized hepatocyte cell line to study the lysosomal sequestration of lipophilic amines. Transporter-mediated uptake by OATPs is a likely mechanism for the rapid uptake and accumulation of acidic drugs in hepatocytes, especially acidic drugs with low intrinsic permeability. Although the uptake of lipophilic amines into hepatocytes can be also be mediated by transporters, such as OCT, in many cases, the accumulation of basic drugs in hepatocytes is not attributable to active transport but to passive diffusion and lysosomal sequestration. For drugs that undergo lysosomal trapping, toxicity associated with phospholipidosis and in vivo interactions related to competition at the lysosomal level are not well understood.
Acknowledgments
The authors thank Dr. Jeffrey Krise at the University of Kansas (Lawrence, KS) for valuable discussions and the use of instrumentation at his laboratory; Phyllis Yerino, Paul Bolliger, Steve Otradovec, Robert Grbac, and the analytical chemistry department at XenoTech, LLC (Lenexa, KS) for technical assistance; Dr. Joanna Barbara for manuscript assistance; Dr. Maciej Czerwinski for cell culture assistance and manuscript review; Kammie Settle for assistance with figure preparation; and Brian Ogilvie for manuscript review.
Authorship Contributions
Participated in research design: Kazmi, Hensley, Loewen, Funk, Buckley, Parkinson.
Conducted experiments: Kazmi, Hensley, Pope, Funk.
Contributed new reagents or analytic tools: Funk.
Performed data analysis: Kazmi, Hensley, Loewen, Buckley, Parkinson.
Wrote or contributed to the writing of the manuscript: Kazmi, Buckley, Parkinson.
Footnotes
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant T32-GM008359] (to R.S.F.).
Abbreviations
- CAD
- cationic amphiphilic drug
- DDI
- drug-drug interaction
- LDH
- lactate dehydrogenase
- MFE
- multifunction enhancing
- MRM
- multiple reaction monitoring
- OAT
- organic anion transporter
- OATP
- organic anion transporting polypeptide
- OCT
- organic cation transporter
- PBS
- phosphate-buffered saline
- VDss
- volume of distribution at steady state
- Received November 29, 2012.
- Accepted February 1, 2013.
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}