


Abstract
Supported by a usage history that predates written records and the perception that “natural” ensures safety, herbal products have increasingly been incorporated into Western health care. Consumers often self-administer these products concomitantly with conventional medications without informing their health care provider(s). Such herb–drug combinations can produce untoward effects when the herbal product perturbs the activity of drug metabolizing enzymes and/or transporters. Despite increasing recognition of these types of herb–drug interactions, a standard system for interaction prediction and evaluation is nonexistent. Consequently, the mechanisms underlying herb–drug interactions remain an understudied area of pharmacotherapy. Evaluation of herbal product interaction liability is challenging due to variability in herbal product composition, uncertainty of the causative constituents, and often scant knowledge of causative constituent pharmacokinetics. These limitations are confounded further by the varying perspectives concerning herbal product regulation. Systematic evaluation of herbal product drug interaction liability, as is routine for new drugs under development, necessitates identifying individual constituents from herbal products and characterizing the interaction potential of such constituents. Integration of this information into in silico models that estimate the pharmacokinetics of individual constituents should facilitate prospective identification of herb–drug interactions. These concepts are highlighted with the exemplar herbal products milk thistle and resveratrol. Implementation of this methodology should help provide definitive information to both consumers and clinicians about the risk of adding herbal products to conventional pharmacotherapeutic regimens.
Introduction
Brief History of Natural Product Use for Medicinal Purposes.
Healing plants gracing Neanderthal tombs and in the personal belongings of Ötzi the Iceman indicate that knowledge of the pharmacologic activity of herbs and other natural products predates written records (Tyler, 2000; Goldman, 2001). Exploitation of natural products for both therapeutic and nefarious purposes during the Greek and Roman empires was well documented by Hippocrates and Galen (Forte and Raman, 2000). Perhaps the most famous early use of an herbal product for pharmacologic activity was the execution of Socrates by poison hemlock. By the early 19th century, the scientific method had advanced such that promotion of natural products for healing purposes was considered quackery (Winslow and Kroll, 1998). During the 1950s in the United States, herbal products began to regain popularity due to pharmaceutical tragedies, such as the use of thalidomide during pregnancy (Brownie, 2005). The herbal product market continued to grow in the 1960s, as consumers focused on the perceived lack of side effects and advances in scientific knowledge about natural products (Winslow and Kroll, 1998; Tyler, 2000). In 1974, the World Health Organization began encouraging developing countries to supplement modern pharmacotherapy with traditional herbal medicines to fulfill needs unmet by conventional drugs (Winslow and Kroll, 1998). Herbal product sales have continued to increase, reaching an estimated $5.6 billion in the United States in 2012 (Lindstrom et al., 2013).
Prevalence of Coadministration of Herbal Products with Conventional Medications.
An accurate estimate of the prevalence of herbal product usage and coadministration with conventional medications is difficult, because consumers of herbal products seldom inform their health care providers (Gardiner et al., 2006). Since these products usually are self-administered as a means to treat or prevent the onset of a medical condition (Winslow and Kroll, 1998), concomitant intake with conventional medications can be expected (Gardiner et al., 2006; Kennedy et al., 2008). The National Health Interview Survey provides the most comprehensive evaluation of herbal product usage rates in the United States, the most recent of which reported that approximately 20% of the population acknowledges taking herbal products (Bent, 2008). This percentage may be even greater in patients with medical conditions such as chronic gastrointestinal disorders, insomnia, liver disease, chronic pain, depression, asthma, and menopause (Gardiner et al., 2006). Of the survey responders who took an herbal product with conventional therapy, nearly 70% neglected to inform their health care providers (Gardiner et al., 2006; Kennedy et al., 2008). These practices raise concerns about increased probability of an adverse herb–drug interaction (HDI)—any alteration of the “victim” drug’s pharmacokinetics and/or pharmacodynamics perpetrated by an herbal product that may lead to drug-related toxicity or reduced efficacy. Knowledge of mechanisms underlying HDIs is critical to identify and prevent adverse interactions prospectively, as well as to modulate potentially beneficial interactions. Because most reported HDIs are of pharmacokinetic (PK) origin (Shi and Klotz, 2012), this review focuses on PK-based HDIs.
Biochemical Mechanisms Underlying Pharmacokinetic HDIs
Inhibition of Drug Metabolizing Enzymes.
Drug-mediated inhibition of drug metabolizing enzymes is the most common and well studied mechanism underlying PK drug–drug interactions (DDIs) (Wienkers and Heath, 2005). Enzyme inhibition can manifest as reversible or irreversible loss of activity, the kinetics of which can range from relatively straightforward (e.g., Michaelis–Menten) to complex (atypical) and should be considered for appropriate experimental design and data interpretation (Tracy, 2006). The proceeding concepts are predicated on the assumption that Michaelis–Menten kinetics apply.
Reversible Inhibition.
Competitive inhibition occurs when the “perpetrator” drug or other xenobiotic, including an herb, binds to the active site of the enzyme, preventing the victim drug from binding (Lin and Lu, 1998; Hollenberg, 2002) (Fig. 1). The simplest case is when two substrates for the same enzyme are administered concomitantly, albeit the perpetrator need not be a substrate for the enzyme to demonstrate competitive inhibition (Kunze et al., 1991). The functional consequence is that higher concentrations of the victim drug are needed to compete for the binding site, thereby increasing the concentration needed for half-maximal rate of metabolism (Km) while having no change in the maximal rate of metabolism (Vmax) (Lin and Lu, 1998; Hollenberg, 2002). The net result is a decrease in the intrinsic clearance (Vmax/Km or Clint) of the victim drug. Noncompetitive inhibition occurs when the perpetrator binds to a region of the enzyme that decreases the enzyme’s capacity to metabolize the victim drug (Fig. 1). Since the perpetrator does not bind to the same site on the enzyme as the victim drug, increasing victim drug concentrations cannot compensate for the decreased activity, leaving Km unchanged while decreasing Vmax (Lin and Lu, 1998; Hollenberg, 2002); the net result is a decrease in Clint. Uncompetitive inhibition occurs when the perpetrator binds to the enzyme–victim drug complex, modulating both and Km and Vmax (Lin and Lu, 1998; Hollenberg, 2002) (Fig. 1); Clint may or may not change. Regardless of the mode of reversible inhibition, return to basal enzyme activity can be achieved by removing the perpetrator from the system. Clinically, reversible inhibition, via competitive and noncompetitive modes, manifests as an increase in the systemic exposure of the victim drug due to a decrease in metabolic clearance and/or increase in bioavailability.

Biochemical mechanisms underlying metabolic HDIs. In the absence of herbal constituents, drug molecules are metabolized. Competitive inhibition by an herbal constituent prevents the drug molecule from binding to the active site of the enzyme. Noncompetitive inhibition by an herbal constituent decreases the catalytic activity of the drug metabolizing enzyme without interfering with the binding of drug molecule to the enzyme active site. Uncompetitive inhibition by an herbal constituent modulates both apparent affinity and activity by binding to the enzyme-drug molecule complex. Irreversible inhibition occurs when the herbal constituent mediates enzymatic degradation. Enzyme induction occurs when herbal constituents bind to nuclear receptors and activate mRNA expression and protein synthesis.
Irreversible Inhibition.
Inhibition by perpetrators that do not associate and dissociate rapidly from the enzyme is termed time-dependent inhibition (TDI). Mechanism-based inhibition (MBI), often observed as TDI, is characterized by irreversible or quasi-irreversible noncovalent binding of a reactive metabolite to the enzyme (Grimm et al., 2009). Such binding can impede access to the active site, target the protein for proteasomal degradation, or alkylate the heme (Silverman and Daniel, 1995; Kalgutkar et al., 2007) (Fig. 1). Comprehensive reviews detail the mechanisms and clinical implications of irreversible inhibition (Venkatakrishnan et al., 2007; Grimm et al., 2009). Due to the time-dependent nature, onset of irreversible inhibition in vivo can appear to be delayed from initial exposure to the perpetrator (Grimm et al., 2009). Like reversible inhibition, irreversible inhibition will manifest as an increase in the systemic exposure of the victim drug. Unlike reversible inhibition, the interaction can persist after removal of the perpetrator since recovery of enzyme activity depends on de novo protein synthesis (Grimm et al., 2009).
Inhibition of Protein-Mediated Flux.
Compared with metabolism-based interactions, mechanistic information about transporter-based interactions is limited, although the knowledge gap is beginning to narrow (Han, 2011). Similar to drug metabolizing enzymes, transport proteins are susceptible to competitive and noncompetitive reversible inhibition due to the perpetrator blocking the victim drug binding site or causing a conformational change that decreases transport activity, respectively (Arnaud et al., 2010; Harper and Wright, 2013). In addition to these traditional modes of inhibition, the in vitro activity of drug transporters can be modulated by the composition of the cell membrane; however, the clinical consequence remains unclear (Annaba et al., 2008; Molina et al., 2008; Kis et al., 2009; Clay and Sharom, 2013). Inhibition of transporter activity in vivo can manifest as increased or decreased systemic exposure and possibly altered tissue concentrations of the victim drug. The direction of change depends on the site of transporter expression (i.e., apical/canalicular or basolateral/sinusoidal) and direction of flux (i.e., uptake or efflux).
Induction of Drug Metabolizing Enzymes and Transporters.
In addition to inhibition, DDIs can reflect increased enzyme or transporter expression. Common mechanisms of induction include increased gene transcription or stabilization of mRNA or active protein (Okey, 1990). The predominant mechanism for enzyme and transporter induction is a receptor-mediated increase in gene transcription due to the perpetrator activating one or more nuclear receptors (Hewitt et al., 2007). Binding of the perpetrator to the ligand binding domain of a nuclear receptor causes a cascade of events leading to the activated receptor binding to the xenobiotic response element located in the promoter region of the gene (Fig. 1). This process leads to increased transcription and subsequent translation of mRNA into protein (Lin and Lu, 1998). Induction of protein function also can reflect stabilization of mRNA or protein (Novak and Woodcroft, 2000; Raucy et al., 2004; Kato et al., 2005; Ménez et al., 2012). Enzyme induction manifests clinically as increased clearance or decreased bioavailability of the victim drug, leading to a decrease in systemic exposure. Like inhibition, induction of transporters manifests as increased or decreased circulating and/or tissue concentrations of the victim drug depending on the site of transporter expression and direction of flux.
Challenges with Evaluating and Predicting PK HDIs
As aforementioned, both PK and pharmacodynamic (PD) mechanisms underlie HDIs. PD mechanisms include common receptors or signaling pathways between the herb and drug targeted for therapy and any common off-target receptors or pathways. Whereas the drug alone may not modulate an off-target mechanism significantly, it is conceivable that a concomitant herb might exacerbate off-target modulation and perpetrate an unexpected adverse event. Such mechanisms result in the biologic action of an herbal product antagonizing, enhancing, or synergizing that of the victim drug (Shi and Klotz, 2012). The most commonly reported PD-based HDIs involve antithrombotic drugs, because several herbal products have anticoagulant, antiplatelet, and/or fibrinolytic properties; for example, gingko and garlic have been implicated to increase the bleeding risk of warfarin (de Lima Toccafondo Vieira and Huang, 2012; Tsai et al., 2013). Other widely reported PD interactions include those involving central nervous system–active agents. For example, St. John’s wort can elicit a manic episode or serotonin syndrome when taken with selective serotonin-reuptake inhibitors, including sertraline, fluoxetine, paroxetine, and nefazodone; the underlying mechanism likely involves an additive effect of St. John’s wort on serotonin reuptake (de Lima Toccafondo Vieira and Huang, 2012; Shi and Klotz, 2012).
As discussed earlier, the majority of potential HDIs are of PK origin. These interactions typically involve an alteration in the victim drug’s clearance or systemic exposure due to inhibition or induction by the herbal product of drug metabolizing enzymes and transporters (de Lima Toccafondo Vieira and Huang, 2012; Shi and Klotz, 2012). The proceeding discussion focuses on common challenges when assessing such PK-based HDIs.
Variability in Herbal Product Composition.
Unlike most drug products, herbal products frequently consist of multiple constituents that vary in composition, both between manufacturers and between batches from the same manufacturer. The putative bioactive constituents in herbal products often are plant-derived secondary metabolites produced as part of normal plant metabolism or as a reaction to environmental stress (Rousseaux and Schachter, 2003). The relative concentration of each pharmacologically active compound may vary widely depending on growing conditions such as temperature and rainfall (Rousseaux and Schachter, 2003). A simple illustration of this variability is the extreme differences in wine quality and price between vineyards and vintages, even when produced from the same variety of grapes (Paine and Oberlies, 2007). Strict attention should be paid to the composition of herbal products to ensure reproducibility within studies and to enable comparisons between studies. At minimum, the brand name, manufacturer, lot number, ingredients, preparation and storage directions, manufacturing process, and origins of growth and production should be provided (Won et al., 2012).
Identification of Causative Constituents.
Modulation of drug metabolizing enzymes and transporters by herbal products can reflect interactions with one or more herbal product constituent. The net effect can result from additive, synergistic, or antagonistic interactions among multiple constituents (Efferth and Koch, 2011). Accordingly, identification of the causative constituent(s) is required to make accurate predictions of HDIs. Some herbal products, including St. John’s wort and milk thistle, are well characterized, and individual constituents have been isolated in quantities sufficient for interaction screening (Obach, 2000; Weber et al., 2004; Lee et al., 2006; Graf et al., 2007; Tatsis et al., 2007; Brantley et al., 2010, 2013). Other techniques, such as bioactivity-guided fractionation (Kim et al., 2011a; Roth et al., 2011), can be used to elucidate the causative constituents from herbal products.
Pharmacokinetics of Causative Constituents.
As with conventional DDI predictions, knowledge of the pharmacokinetics of the perpetrator herbal product is needed to make accurate predictions of HDIs. Herbal product constituents that undergo extensive presystemic (first-pass) clearance via metabolism and/or efflux in the intestine and/or liver are already marketed (e.g., milk thistle and resveratrol), whereas traditional pharmaceutical compounds with these characteristics typically are excluded from further development. This extensive elimination/low bioavailability results in low circulating concentrations of the parent herbal constituent. As a consequence, the systemic concentration of the perpetrator constituent(s), if measurable, may be a less than ideal surrogate for the concentration at the site of interaction. Moreover, upon oral dosing, high presystemic exposure of the herbal perpetrator (parent and/or metabolite) can inhibit first-pass intestinal or hepatic extraction of the victim drug. With respect to induction, concentrations of the victim drug (and all perpetrator constituents) should be monitored upon chronic exposure to the herbal product to detect time-dependent changes in systemic drug exposure.
Regulatory Perspectives on Herbal Products
Although regulatory agencies recommend full characterization of the drug interaction liability of conventional pharmaceutical agents prior to market approval, perspectives vary regarding evaluation of herbal products. Because herbal product usage is woven into cultural traditions, the ability of regulatory agencies to restrict herbal pharmacotherapy and establish regulatory precedent is limited (Rousseaux and Schachter, 2003). Various agencies have developed different guidances for addressing the balance between market availability and safety. Cultural and economic factors often dictate the final course of action. Regulatory views on herbal products in the United States, the European Union, and Canada are summarized below.
Regulation in the United States.
The Food and Drug Administration (FDA) received jurisdiction to regulate herbal products under the Dietary Supplement Health and Education Act (1994) (DSHEA) (Table 1). DSHEA provides the legal definition of dietary supplements, including herbal products, and dictates that such supplements be regulated as foods rather than drugs. Under this classification, dietary supplements are presumed to be safe “within a broad range of intake.” Herbal products marketed after passage of the DSHEA are subject to a premarket review of safety data, whereas products sold prior to the passage of the DSHEA are exempt (de Lima Toccafondo Vieira and Huang, 2012). Contrary to conventional drugs, the burden of proof is on the FDA to demonstrate that these products pose “significant or unreasonable risk” before removal from the market (Brownie, 2005). Supplement manufacturers are prohibited from making claims about the ability of their products to diagnose, mitigate, treat, cure, or prevent a specific disease or class of diseases without undergoing evaluation as conventional drugs (Dietary Supplement Health and Education Act, 1994). For herbal products with established drug interaction liability, the FDA requires mention of potential HDIs in the prescribing information of victim drugs, but not in the label of the perpetrator herbal product.
Key regulatory guidance points associated with herbal products
Refer to citations in the text for additional information.
Regulation in the European Union.
Herbal product usage varies widely among countries of the European Union, leading to differences in regulatory classifications in individual countries. Germany and France have a long history of herbal product use, reporting combined sales of $3.2 billion in 2003 (De Smet, 2005). By contrast, Portugal, Hungary, Ireland, Slovakia, Finland, and Norway have a shorter history of herbal product use, with less than $0.15 billion in combined sales in 2003 (De Smet, 2005). Initial attempts in 2002 to harmonize these disparate views generated safe lists of vitamins and minerals, but national rules for other nutrients and dietary supplements remained intact (European Parliament, 2002). With regulation of herbal products left to the agencies in each member country, at least 27 different national perspectives exist (Table 1). The second attempt in 2004 to harmonize perspectives created a category termed traditional herbal medicinal products (THMPs), providing some progress at the national level for medicinal products with traditional or historic uses (Silano et al., 2011). Authorization as a THMP requires that the product be marketed for at least 30 years, 15 of which must be in an EU member country (Silano et al., 2011). Registration under this directive requires more information than the US FDA requires for dietary supplements but less information than the US FDA or European Medicines Agency (EMA) requires for conventional drugs. Herbal product manufacturers were given until April 2011 to register a product for consideration as a THMP (Silano et al., 2011). Although market harmonization has begun, decisions as to market authorization are still left to individual EU member countries. Such incomplete harmonization creates an environment in which an herbal product can be marketed as a food supplement in one country, a THMP in a second country, and prohibited in a third country (Silano et al., 2011).
Regulation in Canada.
Herbal products in Canada are regulated by the Natural Health Product Directorate (NHPD) branch of Health Canada (Table 1). The role of the NHPD is to “ensure that Canadians have ready access to natural health products that are safe, effective and of high quality while respecting freedom of choice and philosophical and cultural diversity” (Health Canada, 2006). Unlike in the United States and European Union, herbal product manufacturers in Canada must provide evidence to support both the safety and efficacy of a product before market approval. As part of the required safety information, a summary report containing information about the interaction potential with other medicinal products, foods, or clinical laboratory tests must be provided (Health Canada, 2006). Upon approval, herbal products receive a license and identification number. All approved herbal products must meet strict labeling requirements. In addition, the process of removing an herbal product from the market is less cumbersome than in the United States. Specifically, the Canadian Health Minister can suspend sales of natural health products if a manufacturer does not provide requested safety information or if the Minister has reasonable grounds to believe that the product is not complying with other provisions of NHPD regulations (Health Canada, 2006).
HDI Predictions
Current Strategies.
Compared with qualitative descriptions of HDIs, prospective quantitative predictions of these interactions are in embryonic stages at best. Since herbal products are not regulated in the same manner as conventional drugs, at least in the United States and the European Union, rigorous assessment of HDI liability generally is not requested prior to marketing. As such, HDI studies typically are initiated upon receipt of case reports documenting a putative interaction or data from in vitro experiments highlighting a potential interaction. A prospective, systematic process would advance the mechanistic understanding of HDIs, helping to predict, mitigate, and ideally prevent adverse HDIs.
Limitations of Current Strategies.
As aforementioned, herbal products typically are mixtures of potentially bioactive constituents, any of which may interact with drug metabolizing enzymes or transporters. Information from in vitro experiments, preclinical and clinical studies, and in silico simulations can be used to assess HDI potential. Static equations usually are not amenable to complex interactions due to multiple constituents; consequently, more sophisticated approaches, such as physiologically based pharmacokinetic (PBPK) modeling and simulation, are preferable (de Lima Toccafondo Vieira and Huang, 2012; Huang, 2012). The lack of standardization of herbal products (discussed earlier), coupled with variable experimental design across laboratories (Table 2), has produced large variability in the quality of reported data, rendering application of PBPK modeling, as well as in vitro to in vivo extrapolation, approaches particularly challenging. Summarized below are current approaches for evaluating the drug interaction potential of conventional pharmaceutical compounds that can be applied to herbal products. Milk thistle and resveratrol are subsequently presented as case studies.
Milk thistle and resveratrol inhibition kinetics in various enzyme systems
Evaluation of HDIs in In Vitro Systems.
In vitro systems are fundamental tools used to estimate the contribution of drug metabolizing enzymes and transporters to the disposition of an herbal product. Results derived from in vitro experiments can be used to predict quantitatively the in vivo potential of an HDI. Systems commonly used to assess metabolism include microsomes, recombinant enzymes, and hepatocytes. Transport activity typically is determined using cell lines such as Caco-2 or MDCK cells overexpressing specific human transporters, in which bidirectional transport can be measured (Cvetkovic et al., 1999; Cui et al., 2001; Troutman and Thakker, 2003; Kindla et al., 2011; Kimoto et al., 2013; Kock et al., 2013). Sandwich-cultured hepatocytes, which mimic three-dimensional hepatic architecture, can be used to estimate biliary transport (Liu et al., 1999; Annaert et al., 2001). Continual refinement of these systems provides improved estimates of xenobiotic disposition.
Human-derived microsomes or recombinant enzymes are used to determine both the potency and mode of enzyme inhibition (Table 2). Details about the appropriate conduct of these studies are described elsewhere (Bjornsson et al., 2003; Grimm et al., 2009). Cell lines are used to determine whether the xenobiotic inhibits transport of probe substrates such as digoxin [P-glycoprotein (P-gp)] or some statins [breast cancer resistance protein (BCRP) and organic anion transporting polypeptides (OATPs)]. The likelihood of inhibition occurring in vivo can be estimated by using the in vitro–determined kinetic parameters and observed systemic concentrations of the perpetrator xenobiotic (if available), as discussed subsequently under the section on modeling and simulation approaches. A caveat is that circulating concentrations may not represent the HDI liability during first-pass extraction or may not reflect local concentrations at the site of the interaction.
Unlike inhibition experiments that can rely on cell fractions, induction experiments must rely on intact cells. Assessment of induction is dependent upon the measurement of mRNA or protein expression for both metabolic enzymes and transporters. Increased activity of the induced protein also must be demonstrated, because increased mRNA or protein expression may not always correlate with a proportional increase in activity. The induction response of immortalized cells (e.g., Caco-2 or HepG2) may not be as robust as in human hepatocytes because the immortalization process can alter expression of particular transcription factors or nuclear receptors.
Evaluation of HDIs in Preclinical Animal Models.
Appropriate animal models are critical in the drug development process. Although predictions can be made using in vitro data, several key characteristics of drug/xenobiotic disposition can only be determined in vivo, namely the relative contribution of metabolic and excretory routes to total clearance. Moreover, mass balance and the percent contribution of an enzymatic pathway to overall elimination can only be estimated using in vivo data. Without these data, the appropriateness of PBPK models cannot be assessed. Information derived from properly designed PK studies can be used to develop or refine PBPK models. Thus, in addition to helping determine bioavailability and tissue localization of a drug, animal models can provide an estimate of exposure to metabolites after administration of the parent drug. In general, in vitro data are scaled to determine drug interaction liability and whether human in vivo DDI studies are warranted. In some instances, animal models can provide mechanistic insight into a DDI using an experimental design that is not amenable to humans. A major disadvantage of animal models is differing metabolic and transport pathways compared with humans, because animals can have enzyme and transporter orthologs that differ in tissue expression or substrate specificity (Martignoni et al., 2006; Chu et al., 2013).
Human Clinical Studies.
Best practices for appropriate conduct of human clinical HDI studies closely resemble those for food–drug interaction studies as reviewed previously (Gurley, 2012; Won et al., 2012). As with food–drug interaction studies, the critical step in HDI studies is quantification of the putative perpetrator constituent(s). The Consolidated Standards of Reporting Trials checklist was updated in 2006 to include herbal medicinal products (Gagnier et al., 2006). The interventions section of this checklist was extended to highlight the importance of the name, characteristics, dosage regimen, quantitative description, and qualitative testing of the herbal product. Although this checklist is meant to enable quality reporting of trials involving herbal medicines, the major emphasis of this update is also applicable to interaction studies. Ideally, with increased awareness, HDI studies will more closely resemble those for DDIs, guidances for which have been extensively discussed (European Medicines Agency, 2012; US Food and Drug Administration, 2012).
Modeling and Simulation Approaches.
Modeling and simulation-based approaches have become useful tools for DDI predictions. The first step regarding in vitro to in vivo extrapolation is to recover robust estimates of requisite kinetic parameters (e.g., Km, Vmax, Ki, KI, kinact). Single-site Michaelis–Menten kinetics typically are assumed for the cytochromes P450 (P450); however, the possibility of atypical kinetics, including enzyme activation, biphasic kinetics, and multienzyme kinetics, should be considered, especially if multiple herbal constituents are involved (Wienkers and Heath, 2005; Tracy, 2006). Similarly, atypical kinetics such as enzyme activation and partial substrate inhibition have been reported for conjugative enzymes (Stone et al., 2003; Uchaipichat et al., 2008; Tyapochkin et al., 2011) and may complicate recovery of relevant parameters (Iwuchukwu and Nagar, 2008). Finally, the determination of unbound concentrations in incubation mixtures to estimate Km (Obach, 1999; Wienkers and Heath, 2005) is increasingly appreciated. Taken together, selecting the appropriate kinetic model is imperative for accurate recovery of parameters that are used as inputs for whole-system models, including PBPK models.
PBPK models in particular are emphasized in regulatory guidances for predicting the likelihood and magnitude of DDIs and for providing greater insight into causes of uncertainty and variability in the evaluation of DDIs (European Medicines Agency, 2012; US Food and Drug Administration, 2012). Several commercial software packages are available that facilitate model development. Differential equation solving software packages include MATLAB Simulink, Berkeley Madonna, Wolfram Mathematica, and acsIX; these programs do not contain predefined model structures or differential equations, rendering model complexity and flexibility dependent upon the ambition and coding acumen of the modeler. Alternatively, software packages such as Simcyp, PK-Sim, GastroPlus, and MATLAB SimBiology provide template model structures at the expense of full customization. Regardless of the software package selected, PBPK models generally require more parameters than other modeling approaches. Compound-independent physiologic parameters such as organ weights and blood flows can be obtained from the literature (Brown et al., 1997; Boecker, 2003). Compound-dependent parameters, such as tissue partition coefficients, absorption rate constants, and metabolic clearances, can be determined from in vitro and animal experiments or estimated from physicochemical parameters of individual constituents (Poulin and Theil, 2000; Rodgers and Rowland, 2007). PBPK models of victim and perpetrator compounds can be linked through appropriate interaction mechanisms, such as reversible or time-dependent inhibition, to simulate HDIs (European Medicines Agency, 2012; US Food and Drug Administration, 2012). Comprehensive reviews of PBPK model software packages and applications are available (Khalil and Laer, 2011; Rowland et al., 2011; Zhao et al., 2012).
Case Study: Milk Thistle
Product Identification and Usage.
Milk thistle [Silybum marianum (L.) Gaertn.] is a member of the Asteraceae plant family whose use in treating hepatic disorders was documented by Pliny the Elder (AD 23–79) (Kroll et al., 2007; Post-White et al., 2007). More recently, extracts from the plant have shown promise in preclinical studies for treatment of hepatic disorders, such as acute hepatitis, chronic hepatitis B, and hepatitis C (Wei et al., 2013). Evidence of clinical efficacy, however, is limited (Gordon et al., 2006; Rambaldi et al., 2007; Seeff et al., 2008; El-Kamary et al., 2009; Payer et al., 2010; Fried et al., 2012). In addition to treatment of liver disease, milk thistle extracts may mitigate drug-induced hepatotoxicity from chemotherapeutic agents used for childhood acute lymphoblastic leukemia (Ladas et al., 2010) and acute myelogenous leukemia (McBride et al., 2012). Milk thistle extracts and chemical derivatives are used in the treatment of fulminant liver failure caused by death cap mushroom (Amanita phalloides) poisoning (Mengs et al., 2012). Although milk thistle research remains focused on liver ailments, recent research has highlighted potential uses for treatment of obsessive compulsive disorder (Sayyah et al., 2010; Camfield et al., 2011), type II diabetes (Huseini et al., 2006), β-thalassemia major (Gharagozloo et al., 2009), influenza A (Song and Choi, 2011), and prostate cancer chemoprevention (Agarwal et al., 2006; Flaig et al., 2007; Vidlar et al., 2010). Continuous use of milk thistle products for nearly 2000 years in treating various ailments suggests putative efficacy, but again, clinical evidence remains limited.
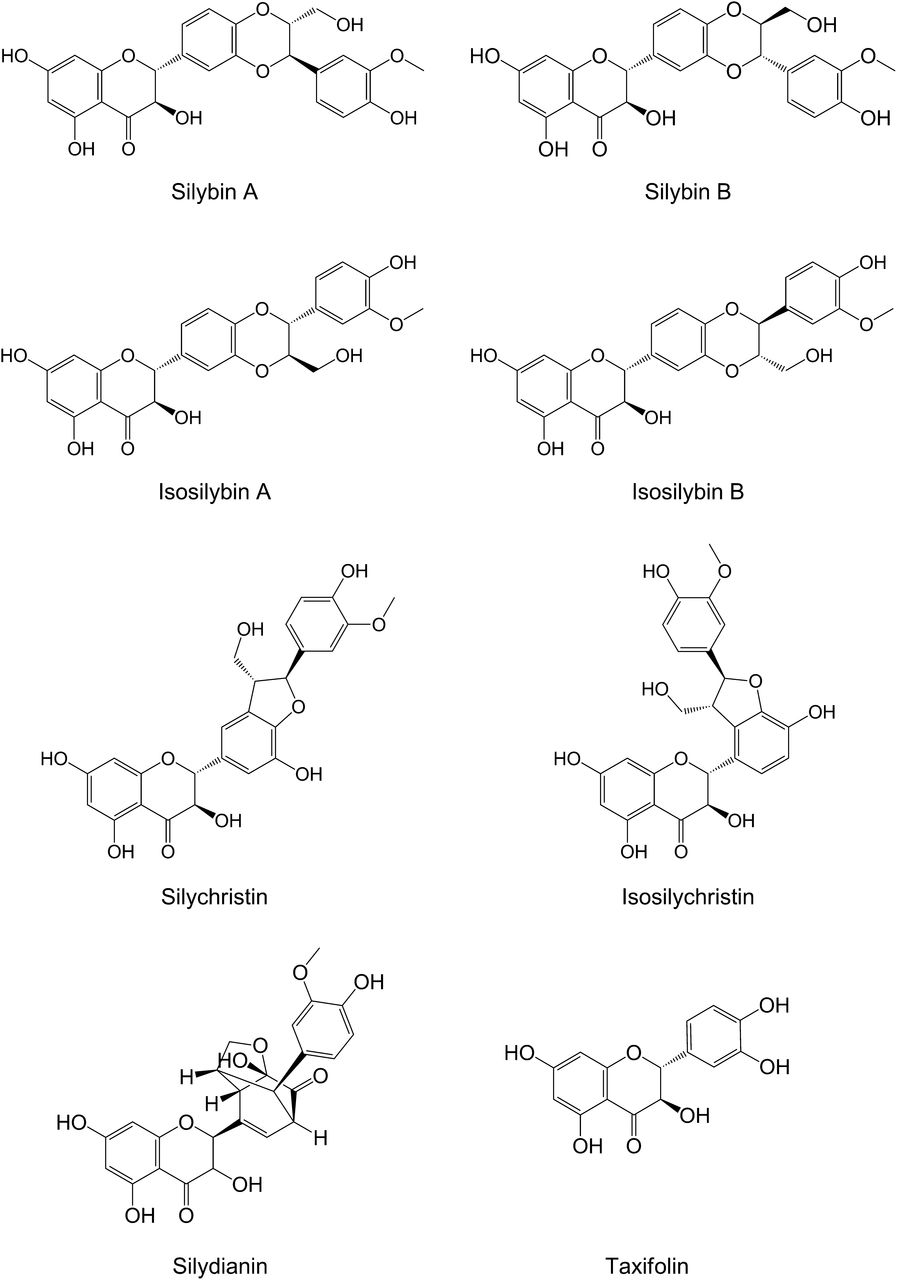
Extracts from milk thistle are commercially available with varying degrees of purification and chemical modification. Crude milk thistle extract often is standardized to contain 65%–80% silymarin and 20%–35% fatty acids (Kroll et al., 2007). Silymarin is a mixture of at least seven flavonolignans and the flavonoid taxifolin (Fig. 2). Flavonolignans are formed by conjugation of taxifolin with coniferyl alcohol to produce structural isomers with the same molecular weight, enabling rudimentary calculations of silymarin concentrations in molar units (Kim et al., 2003a; Davis-Searles et al., 2005; Graf et al., 2007). Although the abundance of flavonolignans varies among different preparations, the most prevalent flavonolignans usually are the diastereoisomer pair silybin A and silybin B (Davis-Searles et al., 2005; Wen et al., 2008). Silychristin and silidianin also are relatively abundant in most silymarin preparations. The diastereoisomeric pair isosilybin A and isosilybin B, as well as isosilychristin, are relatively scarce in most preparations. Semipurification of the crude extract yields a roughly 1:1 mixture of silybin A and silybin B, termed silibinin. The semipurified mixture of isosilybin A and isosilybin B, isosilibinin, has been used in preclinical research but is not yet available as a commercial preparation (Kroll et al., 2007). Chemical modification of silybin A and silybin B to increase water solubility for administration as an intravenous formulation led to generation of the dihemisuccinate ester derivative, Legalon SIL (Mengs et al., 2012). As aforementioned about herbal products in general, large differences exist in the relative composition of the various constituents in milk thistle products (with the exception of prescription preparations available in some countries) (Davis-Searles et al., 2005; Lee et al., 2006; Wen et al., 2008).

Structures of known milk thistle constituents.
Metabolism of Milk Thistle Constituents.
Investigations of the metabolic clearance of milk thistle products have focused on the oxidative and conjugative metabolism of silibinin. The major oxidative metabolite of silibinin is an O-demethylated product generated by CYP2C8 in human liver microsomes (Gunaratna and Zhang, 2003; Jancová et al., 2007). All milk thistle flavonolignans share the methoxy moiety (Fig. 2), located in a region of the coniferyl alcohol that does not participate in the conjugation to taxifolin. Thus, oxidation of this moiety could be similar among all flavonolignans. Relative to the O-demethyl product of silibinin, formation of the monomethylated and dimethylated products was below the limit of quantification (Gunaratna and Zhang, 2003). Milk thistle flavonolignans are conjugated extensively by uridine 5′-diphospho-glucuronosyltransferases (UGTs). Conjugation of silybin A and silybin B by human liver microsomes and hepatocytes showed preferential formation of the 7-O-glucuronide (Jančová et al., 2011). Among recombinant UGTs, UGT1A1, UGT1A3, UGT1A8, and UGT1A10 contributed to silybin A and silybin B conjugation (Jančová et al., 2011).
Human Pharmacokinetics of Milk Thistle Constituents.
After oral administration, milk thistle flavonolignans are absorbed rapidly, with maximum systemic concentrations achieved within less than 2 hours (Weyhenmeyer et al., 1992; Kim et al., 2003b; Wen et al., 2008). As with many natural products based on a flavonoid scaffold, milk thistle flavonolignan oral availability is low due to extensive presystemic conjugation by UGTs and sulfotransferases (SULTs) (Wen et al., 2008). Upon reaching the systemic circulation, parent flavonolignan clearance is rapid, with a terminal elimination half-life of less than 4 hours (Kim et al., 2003a,b; Wen et al., 2008; Zhu et al., 2013). Systemic exposure to conjugated flavonolignans is consistently higher than to parent flavonolignans. For example, exposure to conjugated silybin B, as measured by the area under the concentration-time curve (AUC), was nearly 9-fold higher than that of the unconjugated parent (converted to molar units) in healthy volunteers after a 600-mg dose of milk thistle (Wen et al., 2008). Subsequent to conjugation, flavonolignans are transported into the bile (Schandalik et al., 1992), and deconjugation in the intestine permits reabsorption and enterohepatic recirculation of flavonolignans. Renal clearance of total (unconjugated plus conjugated) silybin A and silybin B is roughly 30 ml/min, with approximately 5% of the dose eliminated in the urine as conjugates (Weyhenmeyer et al., 1992). Compared with healthy volunteers, patients with hepatitis C and patients with nonalcoholic fatty liver disease demonstrated increased exposure to milk thistle flavonolignans and conjugated flavonolignans (Schrieber et al., 2008). Patients with extrahepatic biliary obstruction showed increased systemic exposure to total, but not parent, silibinin. This observation suggests that biliary excretion is rate-limiting for the clearance of conjugated metabolites but not the parent flavonolignans (Schandalik and Perucca, 1994).
Inhibition of Drug Metabolizing Enzymes in Recombinant Enzymes and Microsomes.
The inhibitory effects of milk thistle extracts and constituents depend on the preparation as well as the enzyme system and substrate tested. Silibinin has been shown to be a mechanism-based inhibitor of CYP2C9 and CYP3A4 in reconstituted or recombinant enzymes (Sridar et al., 2004; Brantley et al., 2013) or a reversible inhibitor of CYP2C9 (Jancová et al., 2007; Brantley et al., 2010) and CYP3A4 (Zuber et al., 2002; Jancová et al., 2007) in human liver microsomes (Table 2). Based on IC50 and the caveat that the relationship to Ki is not always clear (i.e., depends on substrate concentration relative to Km and mechanism of inhibition), the inhibitory potency of silibinin toward CYP3A4 may be substrate dependent, with a higher potency toward oxidation of nifedipine (Beckmann-Knopp et al., 2000; Zuber et al., 2002) and testosterone (Jancová et al., 2007) than erythromycin (Beckmann-Knopp et al., 2000). Although silibinin constitutes nearly 50% of silymarin, silymarin extract appears to be a more potent inhibitor of CYP2C19-mediated (S)-mephenytoin 4′-hydroxylation than silibinin (Ki = 2.2 µM versus IC50 >200 µM) (Beckmann-Knopp et al., 2000). Compared with the P450s, inhibition of UGT activity by milk thistle constituents is less studied. Silibinin inhibited recombinant UGT1A1-mediated 7-hydroxy-4-trifluoromethylcoumarin metabolism with an IC50 of 1.4 µM (Sridar et al., 2004), whereas a milk thistle extract inhibited UGT1A-mediated estradiol metabolism in human liver microsomes with an IC50 of nearly 40 µM (Mohamed et al., 2010).
Modulation of Drug Metabolizing Enzymes and Transporters in Cell Systems.
As with recombinant enzymes and microsomes, the effects of milk thistle extracts on enzyme/transporter expression and activity in intact cell systems differs depending on the extract, cell system, and probe substrate tested. Silymarin was shown to decrease CYP3A4-mediated testosterone metabolism by 50% relative to vehicle control in plated human hepatocytes (Venkataramanan et al., 2000). Silibinin had no effect on cortisol metabolism in Caco-2 cells modified to express CYP3A4 (Patel et al., 2004) (Table 3). The effect of milk thistle on P-gp was more variable than on drug metabolizing enzymes. Silibinin decreased P-gp expression by nearly 70% in Caco-2 cells (Budzinski et al., 2007) but had no effect on ritonavir transport in either Caco-2 or MDCK-MDR1 cells (Patel et al., 2004). In contrast, silymarin inhibited the P-gp–mediated transport of digoxin and vinblastine in Caco-2 cells (Zhang and Morris, 2003a) and of daunomycin in MDA435/LCC6 cells (Zhang and Morris, 2003b). In addition to inhibiting efflux transporters, silymarin was shown to inhibit the uptake of estradiol-17β-glucuronide and estrone-3-sulfate mediated by OATP1B1, OATP1B3, and OATP2B1 in transfected Xenopus oocytes and HEK cells (Deng et al., 2008; Kock et al., 2013).
Milk thistle– and resveratrol–drug interaction liability in cell systems
Milk Thistle–Drug Interaction Predictions.
To the authors’ knowledge, no studies to date have investigated the drug interaction liability of milk thistle using PBPK modeling and simulation. Of the reported in vitro studies that mention HDIs with milk thistle products, the majority urge caution when such products are coadministered with sensitive victim drugs due to unknown interaction liability (Beckmann-Knopp et al., 2000; Venkataramanan et al., 2000; Nguyen et al., 2003; Sridar et al., 2004; Etheridge et al., 2007; Deng et al., 2008; Brantley et al., 2010; Mohamed et al., 2010; Doehmer et al., 2011; Mohamed and Frye, 2011). Other studies (Zuber et al., 2002; Jancová et al., 2007; Doehmer et al., 2008) dismiss interaction liability due to the low plasma concentrations of milk thistle constituents or low inhibitory potency. Regardless of interpretation, accurate predictions of milk thistle–drug interaction liability remain elusive.
Preclinical Milk Thistle–Drug Interaction Studies.
Silymarin increased risperidone AUC and maximal plasma concentration (Cmax) in rats after repeated oral doses, consistent with inhibition of P-gp (Lee et al., 2013) (Table 4). Silibinin also increased tamoxifen AUC in rats in a dose-dependent manner (Kim et al., 2010). Unlike for humans, tamoxifen disposition in the rat has not been defined. Although the mechanism for this increased exposure could not be identified, the net effect could reflect inhibition of one or more rodent orthologs of the relevant human enzymes and transporters.
Preclinical evaluation of milk thistle– and resveratrol–drug interaction liability in rodents
Clinical Milk Thistle–Drug Interaction Studies.
The clinical interaction liability of milk thistle products has been examined over the past decade (Table 5). Apart from increased exposure to losartan and talinolol (Han et al., 2009b), the majority of studies reported no clinically significant interactions. Limitations in study design and lack of information about the composition of the milk thistle preparations may have hampered detection of a significant interaction.
Clinical evaluation of milk thistle– and resveratrol–drug interaction liability
Relatively low dosages of silymarin (140 mg three times a day) inhibited the hepatic clearance of the CYP2C9/3A substrate losartan in Chinese subjects homozygous for the CYP2C9 reference allele (CYP2C9*1), leading to a doubling of losartan AUC (Han et al., 2009b). Individuals carrying a reduced activity allele (CYP2C9*3) experienced an increase in losartan Cmax without a significant increase in AUC (Han et al., 2009b). Losartan is a prodrug that is converted to the active metabolite, E-3174, by CYP2C9. Consistent with a decrease in formation clearance by CYP2C9, exposure to E-3174 was decreased after milk thistle administration. The decrease was relatively modest (approximately 15%), indicating limited clinical importance of this interaction (Han et al., 2009b). However, clinically important interactions with larger doses of milk thistle or more sensitive victim drugs known to be affected by CYP2C9 inhibition (e.g., warfarin, phenytoin, celecoxib) cannot be dismissed.
Studies of interactions between milk thistle and the HIV protease inhibitors and CYP3A substrates indinavir and ritonavir demonstrated no interaction. Long-term administration of milk thistle products (2–4 weeks) at various dosages (160–450 mg three times a day) did not alter indinavir Cmax or AUC significantly (Piscitelli et al., 2002; DiCenzo et al., 2003; Mills et al., 2005). Plasma Cmax and AUC of indinavir decreased after milk thistle administration (by 8.8% and 9.2%, respectively), which is inconsistent with inhibition of CYP3A (Piscitelli et al., 2002). Interaction studies with indinavir are not amenable to fixed sequence design because indinavir alone exhibits significantly decreased systemic exposure after long-term treatment, consistent with autoinduction of CYP3A. Compared with baseline conditions, healthy volunteers had 40% lower indinavir exposures 1 week after a 28-day cycle of indinavir (Mills et al., 2005). Indinavir is also a potent CYP3A inhibitor after acute (single dose) treatment, decreasing study sensitivity to detect mild or moderate inhibition of CYP3A. As with indinavir, milk thistle administration with ritonavir or darunavir was not associated with a significant change in drug exposure in HIV-infected patients (Moltó et al., 2012). Like indinavir, ritonavir is a potent CYP3A inhibitor after acute treatment, decreasing sensitivity to detect further enzyme inhibition. Based on the complex modulation of CYP3A by these victim drugs, these results cannot be extrapolated to milk thistle interactions with other CYP3A substrates, warranting further evaluation.
Case Study: Resveratrol
Product Identification and Usage.
Resveratrol (3,5,4′-trihydroxy-trans-stilbene) (Fig. 3) is a naturally occurring phytoalexin produced by a variety of plants, including grapes, mulberries, and peanuts (Wang et al., 2002). Resveratrol has demonstrated antioxidant (Leonard et al., 2003), lipid-lowering (Miura et al., 2003), chemopreventive (Kundu and Surh, 2008), and cardioprotective properties (Kopp, 1998). Clinical evidence regarding the cardioprotective and cancer chemopreventive effects is limited (Walle et al., 2004; Aggarwal and Shishodia, 2006). Current clinical trials with resveratrol include evaluation of effects on type II diabetes, colon cancer, Alzheimer’s disease, Friedreich’s ataxia, obesity, nonalcoholic fatty liver disease, and polycystic ovary syndrome (http://clinicaltrials.gov/ct2/results?term=resveratrol).

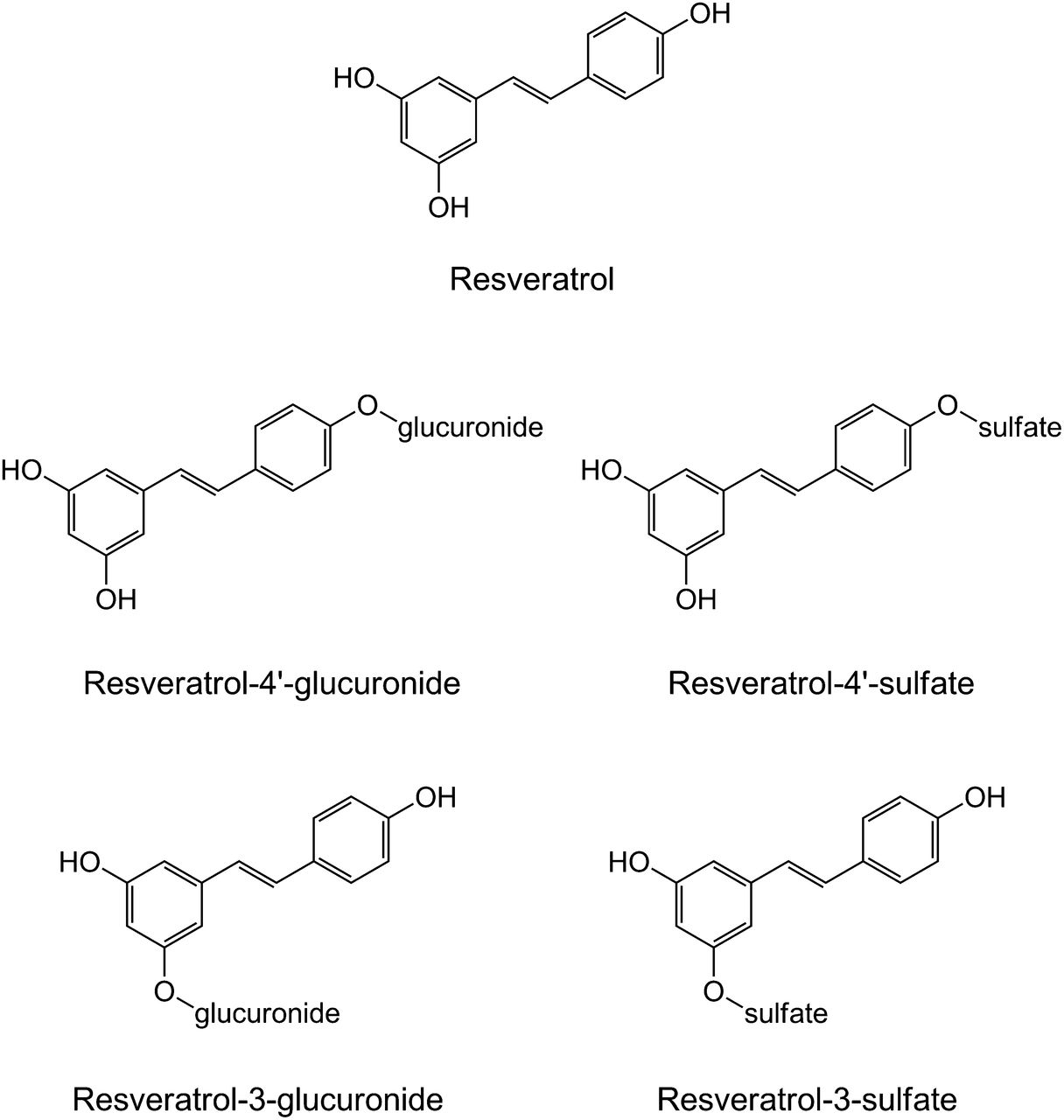
Structures of resveratrol and major conjugated metabolites.
Metabolism of Resveratrol.
Resveratrol contains three phenolic moieties (Fig. 3) that are vulnerable to conjugation by UGTs and SULTs (De Santi et al., 2000a,b; Walle et al., 2004). As with milk thistle flavonolignans, resveratrol undergoes rapid conjugation in the intestine and liver (Wenzel et al., 2005), limiting oral bioavailability (De Santi et al., 2000a,b). Both the cis- and trans-isomers are converted to resveratrol-3-O-glucuronide and resveratrol-4′-O-glucuronide in human liver microsomes and recombinant UGTs (Aumont et al., 2001). The cis-isomer is glucuronidated more rapidly than the trans-isomer, suggesting regioselective and stereoselective glucuronidation of resveratrol. Resveratrol-3-O-sulfate is the most abundant SULT-mediated metabolite (Yu et al., 2003; Boocock et al., 2007). Other metabolites include trans-resveratrol-4′-sulfate, trans-resveratrol-3,5-disulfate, trans-resveratrol-3,4′-disulfate, and trans-resveratrol-3,4′,5-trisulfate. The lack of authentic standards of some of these metabolites, coupled with the prohibitive cost of some commercially available metabolites, has precluded quantitation in biologic matrices (Wenzel et al., 2005). Nevertheless, several laboratories have published synthetic methods for resveratrol conjugates (Wang et al., 2004; Iwuchukwu et al., 2012).
Human Pharmacokinetics of Resveratrol.
After oral administration of resveratrol (0.36 mg/kg body weight or approximately 25 mg/70 kg) dissolved in white grape juice, white wine, or vegetable homogenate (V8 juice) to healthy volunteers, peak concentrations (approximately 2 μM) were detected within 30 minutes (Goldberg et al., 2003). 14C-Resveratrol (25 mg) was administered intravenously and orally to healthy volunteers to examine the absorption, metabolism, and oral bioavailability of resveratrol; similar peak concentrations were observed within 1 hour after oral administration (Walle et al., 2004). Resveratrol underwent rapid presystemic metabolism, possibly contributing to the low bioavailability (<5%). A second resveratrol peak was observed at 4–8 hours, which was attributed to enterohepatic recirculation of the conjugated metabolites (Marier et al., 2002; Walle et al., 2004). Maximum resveratrol concentrations after grape juice, white wine, or 14C-resveratrol administration were well below the EC50 values (5–100 µM) determined for various pharmacologic effects. Likewise, resveratrol doses from dietary exposure are low, leading to systemic concentrations well below pharmacologically relevant concentrations; as such, substantial supplementation may be required to attain a biologic effect (Goldberg et al., 2003).
Because resveratrol is metabolized extensively in vivo, the metabolites have been hypothesized to provide an inactive pool of resveratrol and extend pharmacologic activity beyond the short half-life of the parent compound (Walle et al., 2004; Wang et al., 2004; Wenzel et al., 2005). Resveratrol metabolites have also been hypothesized to have pharmacologic activity (Calamini et al., 2010; Hoshino et al., 2010), showing cyclooxygenase inhibition, nuclear factor-κB inhibition, and antiestrogenic effects (Aires et al., 2013; Ruotolo et al., 2013). Further studies are needed to test these hypotheses as well as to determine the extent of conversion of metabolites back to the parent compound (Goldberg et al., 2003; Walle et al., 2004; Wang et al., 2004).
Inhibition of Drug Metabolizing Enzymes by Resveratrol.
The inhibitory effects of resveratrol are enzyme system dependent (Table 2). Resveratrol was reported to inhibit the P450-mediated metabolism of 7-ethoxyresorufin in human liver microsomes and recombinant enzymes and was shown to be a mechanism-based inhibitor of CYP1A2 but a reversible, mixed-type inhibitor of CYP1A1 and CYP1B1 (Chang et al., 2001). Resveratrol suppressed CYP1A1 expression in HepG2 cells by preventing aryl hydrocarbon receptor activation and xenobiotic response element binding; these activities were hypothesized to contribute to the chemopreventive effect of resveratrol (Ciolino et al., 1998). Resveratrol was shown to be a mechanism-based inhibitor of recombinant CYP3A4 (Chan and Delucchi, 2000). High resveratrol concentrations (10–500 μM) inhibited estradiol glucuronidation in human liver microsomes (Ung and Nagar, 2009). Resveratrol inhibited SULT1E1 activity in primary human mammary epithelial cells, as measured by the formation of 17β-estradiol-3-sulfate (Otake et al., 2000).
Induction of Drug Metabolizing Enzymes by Resveratrol.
Resveratrol showed transcriptional induction of UGT1A1 in Caco-2 cells (Iwuchukwu et al., 2011). UGT1A1 mRNA increased by 3- to 5-fold and was hypothesized to occur via activation of the aryl hydrocarbon receptor and nuclear factor erythroid 2–related factor (Nrf-2) by resveratrol. A combination of resveratrol and another polyphenol, curcumin, had a synergistic effect on UGT1A1 induction, whereas a combination of resveratrol and chrysin, also a polyphenol, had an additive effect on UGT1A1 induction in Caco-2 cells (Iwuchukwu et al., 2011). In HepG2 cells treated with 10 or 30 µM resveratrol, mRNA and protein content of UGT1A1 and UGT2B7 increased after 24 and 48 hours (Lançon et al., 2007). Induction was observed in vivo in rats treated with 0.3, 1, or 3 mg/kg per day of resveratrol for 28 days. Hepatic gene expression of rat Ugt 1a1, 1a6, and 1a7 was upregulated with increasing doses of resveratrol. This increase in expression was accompanied by an increase in Ugt activity in liver microsomes prepared from these animals (Hebbar et al., 2005).
Modulation of Transporters by Resveratrol.
Resveratrol and associated monoconjugates are substrates for several transporters, including BCRP, MRP (multidrug resistance-associated protein) 2, and MRP3 (Alfaras et al., 2010; Planas et al., 2012; van de Wetering et al., 2009). Modulation of transporter activity by resveratrol has been examined using various cell lines. Resveratrol alone did not alter MDR1 expression in MCF-7 and HeLa cells, but resveratrol in combination with docetaxel or with doxorubicin reduced MDR1 expression in HeLa cells (Al-Abd et al., 2011). Resveratrol also reduced P-gp activity and significantly increased the intracellular accumulation of doxorubicin (Al-Abd et al., 2011). Resveratrol, as well as other dietary components, have been shown to modulate BCRP (Ebert et al., 2007; You and Morris, 2007). Although 50 µM resveratrol induced MRP2 activity in HepG2-C3 cells, this same concentration inhibited genistein-mediated MRP2 mRNA induction (Kim et al., 2011b). Resveratrol metabolites were reportedly weak competitive inhibitors of MRP3-mediated transport of estradiol 17-β-glucuronide (van de Wetering et al., 2009).
Few in vivo studies have assessed the effect of resveratrol on transporter activity. Biliary excretion of the Mrp2 substrate, bromosulphothalein, was inhibited competitively by resveratrol or associated glucuronide in Wistar rats (Maier-Salamon et al., 2008). Interactions between various transporters and resveratrol and other nutrients inherently are complex, as detailed in comprehensive reviews (You and Morris, 2007; Li et al., 2012).
PK Modeling of Resveratrol.
Various PK modeling approaches have been used to characterize the disposition of resveratrol and metabolites. For example, population PK modeling was used to describe resveratrol disposition (Colom et al., 2011). Traditional compartmental modeling was used to distinguish the metabolite kinetics of preformed from generated metabolites (Sharan et al., 2012). Independent models for resveratrol (three-compartment model) and two major metabolites, resveratrol-3-glucuronide (enterohepatic recirculation model) and resveratrol-3-sulfate (2-compartment model), were incorporated. The pharmacokinetics of structurally similar dietary flavonoids alone or in combination with coadministered drugs have been characterized using various methods (Moon and Morris, 2007; Wang and Morris, 2007; Lin et al., 2008; Li and Choi, 2009). Collectively, these approaches provide a framework for developing tools to predict resveratrol–drug interactions.
Clinical HDI Studies of Resveratrol.
The potential for resveratrol to modulate drug metabolizing enzyme activity was examined in healthy subjects (Chow et al., 2010). Resveratrol had no effect on UGT1A1 or glutathione S-transferase activity, but in general, P450 activity was inhibited (Table 5). Comprehensive reviews of ongoing clinical trials involving resveratrol have been published (Patel et al., 2011; Smoliga et al., 2011). Additional human data regarding drug interaction liability are forthcoming.
Summary
Herbal product usage likely will continue to increase, in part due to attempts by consumers to decrease medical costs through self-diagnosis and treatment. In parallel, the prevalence of concomitant administration of herbal products with conventional medications will increase. Despite the mounting likelihood of HDIs, a standard system for evaluating the drug interaction liability of herbal products remains elusive. The high compositional variability inherent to herbal products, multiple perpetrator constituents, scant knowledge of perpetrator constituent pharmacokinetics, and differing regulatory perspectives render prospective assessment of HDIs more challenging than DDIs. Taken together, an unprecedented opportunity exists to develop a framework for improving HDI predictions. Strategies to evaluate conventional DDIs, such as integrating in vitro parameters with the pharmacokinetics of individual herbal product constituents into PBPK interaction models, could be applied to herbal products, helping prioritize them for clinical evaluation. Adoption of these strategies should streamline safety assessment of herbal products, assist in the management of HDIs, and ultimately promote the safe use of herbal products.
Acknowledgments
The authors thank Dr. Peng Hsiao for artistic assistance with Figure 1 and Dr. Brandon Gufford for assistance with manuscript formatting. M.F.P. dedicates this article to Dr. David P. Paine.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Brantley, Argikar, Lin, Nagar, Paine.
Footnotes
- Received October 4, 2013.
- Accepted December 11, 2013.
This research was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant R01-GM077482] and the National Institutes of Health National Cancer Institute [Grant R03-CA159389]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AUC
- area under the concentration-time curve
- BCRP
- breast cancer resistance protein
- DDI
- drug–drug interaction
- EMA
- European Medicines Agency
- FDA
- Food and Drug Administration
- HDI
- herb–drug interaction
- MBI
- mechanism-based inhibition
- MRP
- multidrug resistance-associated protein
- OATP
- organic anion-transporting polypeptide
- P450
- cytochrome P450
- PBPK
- physiologically based pharmacokinetic
- PD
- pharmacodynamic
- P-gp
- P-glycoprotein
- PK
- pharmacokinetic
- SULT
- sulfotransferase
- TDI
- time-dependent inhibition
- THMP
- traditional herbal medicinal product
- UGT
- uridine 5′-diphospho-glucuronosyltransferase
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Biochemical Mechanisms Underlying Pharmacokinetic HDIs
- Challenges with Evaluating and Predicting PK HDIs
- Regulatory Perspectives on Herbal Products
- HDI Predictions
- Case Study: Milk Thistle
- Case Study: Resveratrol
- Summary
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters