


Abstract
Recent analyses demonstrated that metabolites are unlikely to contribute significantly to clinical inhibition of cytochrome P450 (P450)–mediated drug metabolism, and that only ∼2% of this type of drug interaction could not be predicted from the parent drug alone. Due to generally increased polarity and decreased permeability, metabolites are less likely to interact with P450s, but their disposition is instead more likely to involve transporters. This commentary presents case studies illustrating the potential importance of transporters as determinants of metabolite disposition, and as sites of drug interactions, which may alter drug efficacy and safety. Many of these examples are hydrophilic phase II conjugates involved in enterohepatic cycling, where modulation of transporter-dependent disposition may alter pharmacokinetics/pharmacodynamics. The case studies suggest that characterization of metabolite disposition, toxicology, and pharmacology should not focus solely on metabolites with appreciable systemic exposure, but should take into consideration major excretory metabolites. A more thorough understanding of metabolite (phase I and II; circulating and excreted) transport properties during drug development may provide an improved understanding of complex drug-drug interactions (DDIs) that can alter drug and/or metabolite systemic and intracellular exposure. Knowledge and capability gaps remain in clinical translation of in vitro and animal data regarding metabolite disposition. To this end, useful experimental and modeling approaches are highlighted. Application of these tools may lead to a better understanding of metabolite victim and perpetrator DDI potential, and ultimately the establishment of approaches for prediction of pharmacodynamic and toxicodynamic consequences of metabolite transport modulation.
Introduction
Preface.
There has been a longstanding concern that metabolites may contribute to drug-drug interactions (DDIs) or toxicity by modulating drug-metabolizing enzymes and/or transporters (perpetrator DDIs) or through comedications altering metabolite disposition (victim DDIs). Recent reviews of public domain data concluded that metabolites rarely perpetrate clinical DDIs via inhibition of cytochrome P450 (P450) enzymes (Yeung et al., 2011). A review of 129 clinical P450-inhibitor drugs reported that only 2% (amiodarone, bupropion, and sertraline) were identified as perpetrators of clinical DDIs that could not be predicted without consideration of metabolites (Yeung et al., 2011). Metabolites are unlikely to cause clinical P450-mediated DDIs generally due to increased polarity and metabolic stability. However, these properties, together with reduced passive permeability, render metabolites more susceptible to interactions with drug transporters (Zamek-Gliszczynski et al., 2006a; Benet, 2010; Smith and Dalvie, 2012; Loi et al., 2013) (Fig. 1; Table 1). The concern of metabolites as both victims and perpetrators of transporter-based DDIs has been highlighted in recent DDI guidance and the International Transporter Consortium white paper (Zamek-Gliszczynski et al., 2013b). This commentary presents a case for a greater understanding of metabolite transport properties considering the increased likelihood of transporter-based relative to P450-based metabolite interactions.
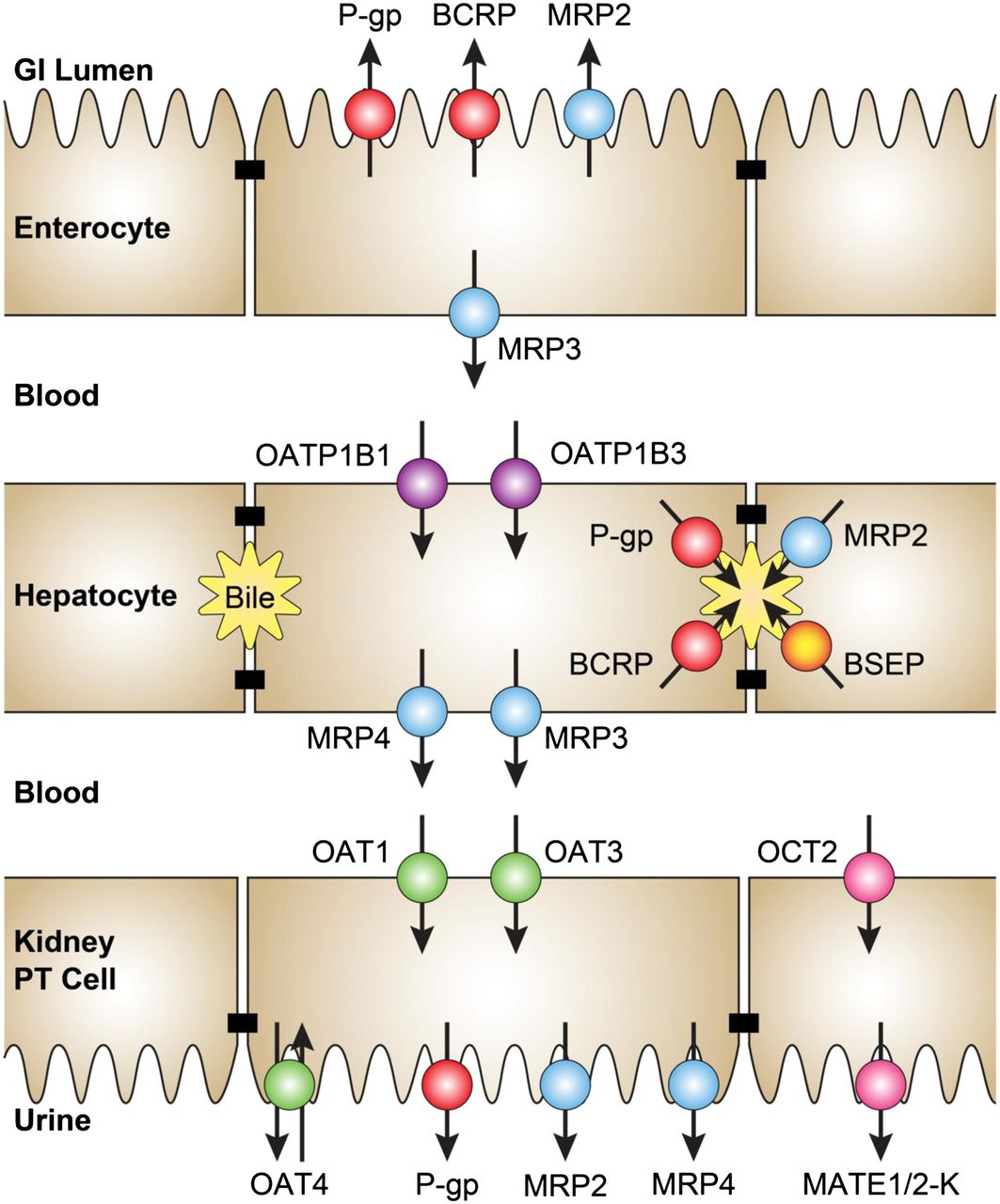
Key transporters interacting with drug metabolites in the intestine, liver, and kidney. GI, gastrointestinal; PT, proximal tubule.
Summary of metabolite-transporter DDIs and associated changes in efficacy and toxicity
A summary of current regulatory positions on metabolite characterization is provided in the initial section, as a starting point for drug development teams to create project-specific strategies, and is followed by an overview of key transporters interacting with drug metabolites. A series of diverse case studies follows, highlighting scenarios when alteration of transporter-mediated disposition of a metabolite (victim) or by a metabolite (perpetrator) can be clinically relevant. A subsequent section on experimental approaches describes useful in vitro, preclinical, and clinical study designs for the assessment of metabolite-transporter interactions. In addition, application of modeling and simulation as well as several newer technologies that can facilitate investigation of metabolite disposition are highlighted. The final section provides recommendations for more thorough characterization of metabolite transport properties.
Regulatory Expectations Regarding Metabolite Disposition.
Current regulatory framework considers metabolites as potential contributors to toxicity, efficacy, and DDIs based on relative systemic exposure. For example, the U.S. Food and Drug Administration 2012 DDI Draft Guidance recommends evaluation of metabolites for effects on P450 activity when systemic exposure exceeds 25% of parent drug exposure and suggests mechanistic understanding of the disposition of these metabolites, especially when pharmacologic or toxicologic activity is evident (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf). Nonclinical toxicology coverage originally was suggested for metabolites that account for more than 10% of human parent drug exposure (http://www.fda.gov/downloads/Drugs/GuidelineComplianceRegulatoryInformation/Guidelines/ucm079266.pdf), but this threshold was later relaxed to 10% of total drug-related radioactivity by the International Conference on Harmonization Guideline M3 (R2) (http://www.ema.europa.eu). The European Medicines Agency 2012 DDI Guidance recommends an understanding of phase I metabolite perpetrator interactions with common drug-metabolizing enzymes when exposure exceeds both 25% of parent and 10% of total radioactivity exposure; in addition, the enzymes involved in the formation and elimination pathways of metabolites contributing more than 50% to overall pharmacologic activity based on unbound concentrations should be identified (http://www.ema.europa.eu). Notably, phase II conjugates are not highlighted based on the view that they are generally inactive, a position refuted in this commentary.
The only regulatory opinion on evaluation of noncirculating metabolites was reported as a footnote to the “Metabolites in Safety Testing” guidance, which stated that, in addition to the systemic exposure criteria, “urinary and fecal metabolites accounting for more than 10% of the bioavailable dose may necessitate further characterization” (http://www.fda.gov/downloads/Drugs/GuidelineComplianceRegulatoryInformation/Guidelines/ucm079266.pdf). This safety characterization ideally entails demonstration of urinary or fecal safety coverage in nonclinical toxicology studies, or if coverage cannot be established, in vitro characterization of the metabolite’s toxicologic properties. Major metabolites in excreta that do not meet the aforementioned circulation criteria may still warrant mechanistic understanding of their transport properties and consideration of victim and perpetrator drug interaction potential. Although not specifically recommended in the current regulatory framework, the evaluation of major excretory metabolites, even if they do not meet the systemic exposure criteria or are altogether not detectable in circulation, has been conducted on a case-by-case basis to inform drug disposition, interactions, efficacy, and/or safety; recent examples include faldaprevir, LY2090314 (3-[9-fluoro-2-(piperidin-1-ylcarbonyl)-1,2,3,4-tetrahydro[1,4]diazepino[6,7,1-hi]indol-7-yl]-4-imidazo[1,2-a]pyridin-3-yl-1H-pyrrole-2,5-dione), and eltrombopag (Deng et al., 2011; Li, 2013; Zamek-Gliszczynski et al., 2013a).
Overview of Metabolite-Transporter Interactions.
The intestinal wall contains metabolic activity of a number of drug-metabolizing enzymes, including P450s (primarily CYP3A), UDP glucuronosyltransferase (UGTs), sulfotransferases, and carboxylesterases (Paine et al., 2006; Galetin et al., 2008). Metabolites formed in enterocytes can be excreted back into the gut lumen by P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and to a lesser extent multidrug resistance-associated protein 2 (MRP2) (Enokizono et al., 2007b; van de Wetering et al., 2009) (Fig. 1). MRP3 has demonstrated importance in efflux of anionic metabolites from the enterocytes into blood (van de Wetering et al., 2009). Finally, evidence exists for apical organic anion transporting polypeptide (OATP) uptake into enterocytes, but its relevance to metabolite transport and interactions in vivo is unclear at present.
The liver generally has the highest expression levels of P450s, UGTs, sulfotransferases, glutathione S-transferases, and other drug-metabolizing enzymes. The liver is very efficient in excreting polar metabolites, which otherwise would have limited ability to diffuse passively out of hepatocytes. BCRP, P-gp, and MRP2 are the three main transport proteins on the bile canalicular membrane responsible for biliary excretion of drug metabolites (Zamek-Gliszczynski et al., 2006a; Giacomini et al., 2010) (Fig. 1). Although bile salt export pump (BSEP) is not known to transport metabolites directly, metabolites can inhibit BSEP-mediated transport of unconjugated bile acids, leading to cholestasis and potential liver injury (Zamek-Gliszczynski et al., 2012). MRP3 and MRP4 on the basolateral membrane have been documented extensively as critical to sinusoidal excretion of anionic metabolites, such as glucuronide and sulfate conjugates (Zamek-Gliszczynski et al., 2006b; Hirouchi et al., 2009). The glucuronides can subsequently be taken up from the blood back into hepatocytes by OATP1B1 or OATP1B3 expressed on the same membrane. Sinusoidal liver-to-blood shuttling or “hepatocyte hopping” of bilirubin glucuronide via MRP3 and OATPs is important to the overall bilirubin detoxification process (Iusuf et al., 2012).
Renal clearance often is the main pathway of elimination for circulating metabolites, which can occur by glomerular filtration and transport-mediated secretion into the proximal tubule. In addition, drug metabolism may occur in the kidney, which contains a range of metabolic enzymes, including P450s, carboxylesterases, UGTs, sulfotransferases, and glutathione S-transferases (Lohr et al., 1998; Knights et al., 2013). However, the kidney is most commonly an excretory organ for circulating metabolites formed in the liver and/or intestine. Uptake of drugs and metabolites into the renal proximal tubule is mediated by organic anion transporter 1 (OAT1), OAT3, and organic cation transporter 2 (OCT2) (Fig. 1). Secretion from proximal tubular cells into urine occurs via OAT4, MRP2, MRP4, multidrug and toxin extrusion protein 1 (MATE1), and MATE2-K.
Case Studies
The following series of case studies highlight scenarios when alteration of transporter-mediated disposition of a metabolite (victim) or by a metabolite (perpetrator) can be clinically relevant; a summary of these examples is provided in Table 1.
Potential for Altered Efficacy in Metabolite-Transporter Interactions
Mycophenolic Acid.
Mycophenolic acid is an active metabolite of the immunosuppressant mycophenolate mofetil. Mycophenolic acid–cyclosporine DDI observed during combination immunosuppression therapy in organ transplant patients is one of the most compelling cases for the importance of prospective understanding of DDI potential at the level of metabolite transport (Pou et al., 2001). When mycophenolate mofetil was coadministered with cyclosporine to organ transplant patients, mycophenolic acid exposure decreased by 44%. This unexpected DDI was puzzling because inhibition of CYP3A, P-gp, or hepatic OATPs by cyclosporine would be expected to increase, rather than decrease, victim drug exposure. Although the interaction could be consistent with P450 induction, cyclosporine does not induce P450 enzymes (http://www.pharma.us.novartis.com/product/pi/pdf/sandimmune.pdf).
Mycophenolic acid undergoes enterohepatic cycling involving the glucuronide conjugate, with recycled drug estimated to account for 10–60% of total systemic exposure (Bullingham et al., 1998). Cyclosporine disrupts this process, elevating circulating glucuronide concentrations and reducing mycophenolic acid availability for enterohepatic cycling, which in turn decreases overall mycophenolic acid systemic exposure (Kobayashi et al., 2004; Picard et al., 2010; Lloberas et al., 2011). Cyclosporine is a known potent inhibitor of hepatic uptake via both OATP1B3 and OATP1B1, as well as canalicular secretion via MRP2 (Kobayashi et al., 2004; Gertz et al., 2013). Recent studies suggest that the clinical mycophenolic acid glucuronide–cyclosporine interaction may occur primarily at the level of hepatic OATP uptake, rather than MRP2 biliary excretion (Patel et al., 2013; Picard, 2013). Nonetheless, biliary excretion of mycophenolic acid glucuronide was negligible in Mrp2-deficient rats, resulting in increased circulating glucuronide concentrations and ∼2-fold decreased mycophenolic acid exposure; analogous effects were observed during cyclosporine coadministration to normal rats (Kobayashi et al., 2004). Furthermore, clinical pharmacogenetic studies demonstrated that naïve patients (e.g., not coadministered with cyclosporine or other drugs that inhibit transporters) carrying OATP1B3 or MRP2 variant alleles exhibited decreased mycophenolic acid exposure and elevated circulating glucuronide concentrations relative to noncarriers. However, during cyclosporine coadministration, when mycophenolic acid glucuronide hepatic transporters are inhibited, these genotypic differences were no longer apparent (Picard et al., 2010; Lloberas et al., 2011).
Collectively, the nonclinical and clinical data suggest that the cyclosporine–mycophenolic acid interaction reflects inhibition of both hepatic uptake and likely biliary excretion. This DDI is clinically important due to the narrow therapeutic index of immunosuppressants (http://www.pharma.us.novartis.com/product/pi/pdf/myfortic.pdf). In contrast, mycophenolate mofetil coadministered with tacrolimus does not alter mycophenolic acid pharmacokinetics (Pou et al., 2001). Although tacrolimus inhibits OATP1B1 and OATP1B3 in vitro, circulating (unbound) concentrations are insufficient to perpetrate clinically relevant inhibition. In addition, tacrolimus does not inhibit MRP2 (Oswald et al., 2011), and therefore, unlike cyclosporine, tacrolimus does not disrupt mycophenolic acid enterohepatic cycling.
Notably, this metabolite transport DDI was observed empirically in clinical practice and is unlikely to be identified without thorough evaluation of the disposition and interaction mechanisms of mycophenolic acid glucuronide, despite it being pharmacologically inactive. Understanding of mycophenolic acid glucuronide transport properties would enable more rational selection of add-on immunosuppressants (e.g., tacrolimus versus cyclosporine) and/or an up-front dose adjustment in the case of cyclosporine combinations.
Ezetimibe.
Ezetimibe is an inhibitor of intestinal cholesterol absorption (http://www.merck.com/product/usa/pi_circulars/z/zetia/zetia_pi.pdf). Ezetimibe undergoes extensive glucuronidation in the gut wall, primarily by UGT1A1, with the glucuronide accounting for >95% of portal drug-related exposure (van Heek et al., 2000). The glucuronide is active and is a major contributor to in vivo efficacy (Garcia-Calvo et al., 2005; Kosoglou et al., 2005). In addition, this metabolite is subject to transporter-mediated enterohepatic cycling, which is critical to extend the residence time in the gut, the site for efficacy. Ezetimibe glucuronide formed in enterocytes is transported into portal blood by the basolateral efflux transporter MRP3. Both the metabolite and parent can also be effluxed back into the gut lumen by MRP2 and P-gp (Oswald et al., 2006b; de Waart et al., 2009). Once in portal blood, the glucuronide is taken up into the liver by OATP1B1 and subsequently excreted into the bile primarily by MRP2 and to a lesser extent by BCRP. The glucuronide also can undergo hepatic sinusoidal efflux back to blood via MRP3 (Oswald et al., 2008; de Waart et al., 2009).
Involvement of transporters in ezetimibe glucuronide disposition and their impact on the efficacy of ezetimibe was demonstrated in animal models. Ezetimibe enterohepatic cycling was disrupted in Mrp2-deficient (TR−) rats, which also exhibit decreased Bcrp and increased Mrp3 hepatic expression (Oswald et al., 2006c). These animals exhibited a 9-fold increase in glucuronide systemic exposure, 2-fold decrease in parent drug exposure, 3-fold decrease in fecal recovery, and significantly diminished efficacy. Likewise, inhibition of P-gp in mice resulted in increased circulating glucuronide concentrations, decreased parent drug concentrations, and attenuated efficacy (Oswald et al., 2010a).
Pharmacokinetic/pharmacodynamic implications of clinical DDIs involving ezetimibe glucuronide are complex (Table 1). A single dose of rifampin increased ezetimibe glucuronide exposure 2-fold but had no impact on parent exposure or overall efficacy, albeit parent Cmax increased 2.6-fold and the onset of sterol-lowering effects was accelerated (Oswald et al., 2006a). This DDI is consistent with reduced enterohepatic cycling due to inhibition of ezetimibe glucuronide hepatic transport by OATP1B1 and possibly MRP2, as well as inhibition of intestinal efflux of both parent and glucuronide by P-gp and MRP2. In contrast, chronic rifampin treatment decreased systemic exposure to ezetimibe and the glucuronide 2- and 3-fold, respectively, and nearly abolished the sterol-lowering effects (Oswald et al., 2006b). These changes are consistent with induction of intestinal P-gp, MRP2, and UGT1A1.
Cyclosporine is known to increase total ezetimibe (parent + glucuronide) systemic exposure 2- to 12-fold (Koshman et al., 2005; Bergman et al., 2006; http://www.merck.com/product/usa/pi_circulars/z/zetia/zetia_pi.pdf). Although the mechanisms of this DDI remain to be characterized, these observations are consistent with clinically relevant inhibition of intestinal P-gp and MRP2, as well as hepatic OATP and MRP2, by cyclosporine. In contrast, sirolimus and tacrolimus are not inhibitors of OATP1B1 and MRP2 at clinically relevant concentrations, resulting in no significant changes in ezetimibe and glucuronide pharmacokinetics or drug efficacy (Oswald et al., 2010b, 2011).
Ezetimibe glucuronide provides an example where knowledge of transporter-mediated metabolite disposition is critical for understanding overall drug pharmacokinetics, explaining observed drug interactions and their possible implications for drug efficacy.
Ethinylestradiol.
Oral contraceptive DDI studies are conducted during development of drugs likely to be taken by women of child-bearing age. The estrogen component of combination contraceptives usually is ethinylestradiol, whose pharmacokinetics are evaluated along with its major circulating sulfate conjugate (Zhang et al., 2007). Although this metabolite is inactive, it can be deconjugated readily to active drug at key sites of pharmacologic activity, and circulating plasma concentrations are several-fold higher than parent concentrations. Inhibition of ethinylestradiol sulfation via sulfotransferase 1E1 is extensively documented to result in decreased ethinylestradiol sulfate exposure, concurrent with elevated parent drug exposure (Zhang et al., 2007).
Recent evidence has established that altered transporter-mediated disposition of ethinylestradiol sulfate can also elicit reduced exposure to this metabolite (Zamek-Gliszczynski et al., 2011). Specifically, impaired intestinal and hepatic excretion of ethinylestradiol sulfate increases the organ residence time of the metabolite, providing opportunity for increased sulfatase-mediated desulfation. In rodent models lacking Mrp2 or Bcrp, sulfate conjugate exposure was decreased up to 83%, with minimal impact on parent drug pharmacokinetics. Perturbed ethinylestradiol sulfate excretion is the hypothesized mechanism of a clinical drug interaction, in which ethinylestradiol sulfate systemic exposure was 71% decreased, whereas parent drug exposure was modestly increased 25% (http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022332s000_ClinPharmR.pdf).
Since contraception is a binary pharmacodynamic response, and because parent drug exposure was not decreased in this scenario of diminished ethinylestradiol sulfate exposure, there is no reason to expect altered drug efficacy. Nonetheless, when interpreting ethinylestradiol sulfate interactions, it is important to consider that both sulfation and efflux transport inhibition can decrease systemic exposure of this sulfate conjugate.
Morphine.
The aforementioned three case studies pertain to metabolite transport interactions at the level of tissue uptake and/or apical efflux into excreta, reflecting current regulatory and drug development focus (Zamek-Gliszczynski et al., 2012). However, polar and poorly permeable metabolites can also return to the systemic circulation via active efflux from the sites of formation (Iusuf et al., 2012). For example, following glucuronidation in the gut wall, ezetimibe glucuronide is transported into blood by MRP3 located on the basolateral membrane of enterocytes (de Waart et al., 2009), whereas a number of other glucuronide and sulfate metabolites formed in the liver rely on MRP3 and MRP4 for efflux into sinusoidal blood (Zelcer et al., 2005; Zamek-Gliszczynski et al., 2006b). Clear cases of clinically relevant DDIs at the level of basolateral efflux of metabolites by transporters such as MRP3 and MRP4 are currently lacking. However, as illustrated by the subsequent morphine glucuronide example, this lack of cases is not because a DDI potential does not exist at these basolateral transporters, nor is it due to the absence of pharmacologic consequences, but a reflection of the knowledge gap in understanding the clinical relevance of basolateral efflux transporters.
Following morphine administration to Mrp3-knockout mice, appearance of the inactive morphine-3-glucuronide in blood was decreased 50-fold, shifting the route of excretion of this metabolite from urinary to biliary/fecal (Zelcer et al., 2005). Since rodents do not form the active morphine-6-glucuronide conjugate produced by humans, potential pharmacokinetic and analgesic alterations in Mrp3-knockout mice were determined following parenteral administration of this active metabolite. In the absence of Mrp3, morphine-6-glucuronide concentration was 2-fold lower at the only time point available (60 minutes post 24 mg/kg i.p.), and a significant decrease in the antinociceptive effect was observed over 9 hours post dose (10 mg/kg i.p.). Currently, the translation of these findings from knockout animals to humans is unknown. Further investigation of clinical drug interaction potential at the level of basolateral efflux transporters, such as MRP3 and MRP4, is needed. This research may ultimately provide mechanistic explanations concerning why some metabolites circulate at high concentrations instead of being excreted.
Potential for Altered Toxicity in Metabolite-Transporter Interactions
Irinotecan.
Irinotecan is a topoisomerase I inhibitor with potent and broad anticancer activity (http://labeling.pfizer.com/ShowLabeling.aspx?id=533). Clinical use of irinotecan is limited by myelotoxicity and severe diarrhea, which exhibit large interindividual variability (Kweekel et al., 2008). Irinotecan disposition is complex and involves multiple enzymes and transporters (Smith et al., 2006). Irinotecan is a prodrug, which is converted to the active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), by carboxylesterases. SN-38 is actively taken up into the liver by OATP1B1 (Nozawa et al., 2005), where it undergoes metabolic inactivation via glucuronidation and P450 oxidation. SN-38 and the glucuronide are excreted into bile by MRP2 (Chu et al., 1997, 1998); the glucuronide is deconjugated to SN-38 in the gut by bacterial β-glucuronidases. In addition, irinotecan is eliminated into feces by P-gp–mediated intestinal and biliary excretion (Chu et al., 1999; Bansal et al., 2008).
Irinotecan and SN-38 pharmacokinetics exhibit high interindividual variability, which has been associated with genetic polymorphisms of the relevant enzymes and transporters. SN-38 exposure is correlated positively with neutropenia (Kweekel et al., 2008). Carriers of the UGT1A1*28 allele(s) (*28B- rs4124874, rs10929302, rs887829, rs8175347; *28C- rs4124874, rs10929302, rs887829, rs8175347, rs35350960; *28D- rs887829, rs8175347) exhibit reduced SN-38 glucuronidation, higher SN-38 systemic exposure, and a greater degree of myelosuppression (Kweekel et al., 2008). Carriers of the OATP1B1*5 (rs4149056) and *15 allele(s) (rs2306283, rs4149056), who have reduced hepatic uptake activity, exhibited higher SN-38 systemic exposure, which is associated with higher risk of neutropenia (Han et al., 2008; Takane et al., 2009).
Irinotecan-induced diarrhea is hypothesized to be associated with intestinal exposure to SN-38. Biliary excretion of SN-38 and glucuronide via MRP2, with subsequent enterohepatic cycling, contributes to enterocyte SN-38 exposure. Clinical studies indicated that patients with the variant ABCC2*2 (rs2273697) haplotype had a significantly lower risk of severe diarrhea (de Jong et al., 2007). Probenecid coadministration in rats elicited a 2-fold decrease in biliary excretion of SN-38 and glucuronide, with a concomitant reduction in diarrhea incidence (Horikawa et al., 2002a). Consistent with inhibition of both Oatp hepatic uptake and Mrp2 biliary excretion, probenecid increased SN-38 systemic exposure 2-fold; when the irinotecan dose was halved, systemic SN-38 was maintained at therapeutic concentrations, whereas diarrhea incidence was significantly reduced (Horikawa et al., 2002a). This case example demonstrates how an understanding of metabolite transport not only uncovers DDI liabilities, but also presents opportunities for improved treatment outcomes by enabling design of safer drug regimens and pharmacogenetic tailoring.
Troglitazone.
Troglitazone is a thiazolidinedione antidiabetic agent, which was withdrawn from the market due to idiosyncratic hepatotoxicity. Various factors are likely contributors to the observed liver injury, including reactive metabolites, mitochondrial permeability alteration, and cholestasis (Masubuchi, 2006). The mechanism underlying cholestasis is BSEP inhibition, resulting in reduced biliary excretion of unconjugated bile acids, which can be toxic upon accumulation in hepatocytes.
As troglitazone sulfate is a BSEP inhibitor that is an order of magnitude more potent than parent drug, and because hepatic metabolite concentrations are 20-fold higher, the sulfate conjugate is considered as the primary precipitant of in vivo bile salt increases in plasma (Funk et al., 2001; Masubuchi, 2006). Troglitazone sulfate has low passive permeability, and hepatobiliary disposition is transporter-mediated (Masubuchi, 2006). OATP1B1 mediates hepatic uptake, whereas BCRP is primarily responsible for biliary excretion, with perhaps some contribution from MRP2 (Kostrubsky et al., 2001; Nozawa et al., 2004; Enokizono et al., 2007a). When hepatic efflux of troglitazone sulfate is impaired, the sulfate may accumulate in hepatocytes, increasing the risk of cholestasis. In TR− rats, which lack Mrp2 and exhibit decreased hepatic Bcrp expression, troglitazone sulfate systemic exposure was 2-fold increased and elicited a commensurate increase in serum conjugated bilirubin (Kostrubsky et al., 2001). Direct clinical translation of increased propensity for liver injury when troglitazone sulfate hepatic excretion is impaired has not been established. In addition, other mechanisms besides transporter inhibition may contribute to the toxicity of troglitazone (Kassahun et al., 2001). Nevertheless, BSEP inhibition has been considered as one of the mechanisms associated with drug-induced liver toxicity in humans.
Trabectedin.
Trabectedin is an intravenous anticancer agent for which liver injury is a common dose-limiting toxicity (http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000773/WC500045832.pdf). Trabectedin elimination occurs by extensive metabolism primarily via CYP3A, with numerous metabolites accounting for 92% of total drug-related exposure (Beumer et al., 2005, 2007). Preclinical studies suggested that hepatotoxicity may be related to hepatic exposure to trabectedin metabolites, which can be attenuated by hepatic efflux via P-gp, MRP2, and MRP3 (van Waterschoot et al., 2009). Unexpectedly, P-gp, Mrp2, or Mrp3 single-gene knockout mice elicited only modest increases in liver injury markers. However, combined genetic ablation of both Mrp2 and Mrp3 or Mrp2 and P-gp elicited severe hepatotoxicity, which was diminished markedly by the additional knockout of Cyp3a in transporter double-knockout mice. These studies indicated that trabectedin hepatotoxicity was mediated primarily by metabolites formed by CYP3A, which are then excreted by multiple transporters that protect the liver. Indeed, when hepatic excretion of the metabolites was blocked, as in the transporter double-knockout animals, the metabolites accumulated in the liver, resulting in hepatotoxicity (van Waterschoot et al., 2009). A recent clinical case report supports the importance of drug transporters in the hepatotoxicity of trabectedin, where a patient with multiple MRP2 polymorphisms, including a rare variant (rs717620) associated with reduced function, developed chronic hepatobiliary toxicity 3 weeks after the second round of treatment (Laurenty et al., 2013).
Trabectedin hepatotoxicity due to impairment of multiple hepatic excretory routes for metabolites is too complex for direct clinical translation. However, this example illustrates the importance of the “fraction excreted” concept, in which the increase in hepatic exposure is governed by the relationship 1/[1 – fe,Total], where fe,Total describes the change in total hepatic excretory clearance (Zamek-Gliszczynski et al., 2009). Hepatic metabolite exposure increases ≤2-fold when hepatic excretory clearance is decreased ≤50%; however, exposure increases exponentially when hepatic excretory clearance is impaired in excess of 50% (Fig. 2). This kinetic principle is consistent with the markedly greater trabectedin hepatotoxicity when the function of two transporters was knocked out relative to genetic ablation of a single transporter.

Metabolite hepatic exposure as a function of total hepatic excretory capacity. Fold-change in hepatic metabolite exposure as a function of total hepatic excretory capacity (fe,Total). The solid line represents the theoretical relationship (fold-change in hepatic exposure = 1/[1 – fe,Total]) ± 50% represented by dashed lines. Data points represent observed changes in hepatic exposure of metabolites in knockout rodents lacking various efflux transporters reported by Zamek-Gliszczynski et al. (2009). This relationship provides a kinetic explanation for the pronounced increase in trabectedin hepatotoxicity when the function of two transporters was knocked out relative to genetic ablation of a single transporter. When only a single excretion pathway is knocked out, i.e., total hepatic excretory clearance is decreased ≤50%, hepatic metabolite exposure increases ≤2-fold. In contrast, liver exposure increases exponentially when hepatic excretory clearance is impaired in excess of 50%.
Potential for Altered DDI Profile Due to Metabolite-Transporter Interactions
Gemfibrozil.
Gemfibrozil is a fibrate commonly used to treat dyslipidemia in the prevention of cardiovascular disease. Gemfibrozil coadministration has resulted in significant increases in the systemic exposure of numerous drugs, most notably repaglinide and cerivastatin, the latter of which was withdrawn from the market due to high incidence of rhabdomyolysis and kidney failure when administered with gemfibrozil (Backman et al., 2002; Niemi et al., 2003). Subsequent in vitro studies have demonstrated that the major gemfibrozil metabolite, an acyl-glucuronide, is an irreversible inhibitor of CYP2C8 following hydroxylation at a site distal to the glucuronide moiety (Ogilvie et al., 2006; VandenBrink et al., 2011). In contrast, the parent drug is a weak reversible inhibitor of CYP2C8. In addition, both gemfibrozil and its glucuronide are inhibitors of hepatic uptake via OATP1B1, with the glucuronide being more potent than the parent (Shitara et al., 2004; Hinton et al., 2008). Thus, the apparent DDI potential of gemfibrozil toward either CYP2C8 or OATP1B1 cannot be understood without consideration of the glucuronide, as well as systemic and intracellular drug and metabolite exposure.
Gemfibrozil glucuronide is excreted into the bile via MRP2 and is subject to basolateral efflux back to blood by MRP3, and to a lesser extent, by MRP4 (Hirouchi et al., 2009). Plasma glucuronide concentrations in Mrp3 knockout mice were 3.5-fold lower than those in wild-type mice. Mrp2 deficiency and Mrp3 upregulation in TR− rats resulted in impaired biliary excretion and compensatory urinary excretion of gemfibrozil glucuronide (Kim et al., 2003). In humans, gemfibrozil glucuronide is present in the circulation with systemic exposure representing 47–73% of the parent following gemfibrozil doses of 30–900 mg (Tornio et al., 2008) (Table 1). Currently, there is no in vitro evidence to suggest that the glucuronide in the portal blood is taken back up into hepatocytes via OATPs, as was observed with ezetimibe glucuronide. However, total gemfibrozil glucuronide concentrations in rat liver have been reported to be 42-fold higher than in the perfusate, suggesting the presence of active uptake into the liver (Sabordo et al., 1999). Whether these preclinical observations translate to humans remains to be determined.
Gemfibrozil glucuronide further highlights the need for more consistent characterization of the DDI potential of glucuronide metabolites in a prospective manner to avoid empirical DDI discovery in clinical practice. In addition, this phase II conjugate represents a clear example where understanding of the impact of transporters on the intracellular concentrations is crucial for DDI predictions, particularly when the victim drug also has a complex hepatic disposition as observed with repaglinide (Menochet et al., 2012b).
Cyclosporine.
Cyclosporine is an immunosuppressant that is associated with numerous DDIs (Gertz et al., 2013). Cyclosporine is metabolized extensively by CYP3A, and blood concentrations of the main metabolite, AM1, exceed those of the parent after both single and multiple doses (Wang et al., 1988; Aweeka et al., 1994; Bauer et al., 2003) (Table 1). Like cyclosporine, AM1 distributes extensively into red blood cells (Awni et al., 1989), but any potential nonlinearity in blood distribution, as observed with the parent drug, is currently unknown. In addition to AM1, other metabolites (AM9, AM4N) exceed 25% of steady-state parent exposure (Gertz et al., 2013). Despite extensive data on the systemic exposure of cyclosporine metabolites, minimal information was available until recently on their inhibition potency and the potential contribution to cyclosporine-perpetrated DDIs. In addition, there are currently no in vitro data on potential transport-mediated disposition of these metabolites.
In vitro studies with human embryonic kidney (HEK)–OATP1B1 cells have shown that AM1 is a potent inhibitor of OATP1B1. Despite a 5-fold lower potency relative to the parent, the IC50 was in the nanomolar range (Table 1), and combined with a high systemic exposure, highlighted the need to assess the potential role of AM1 in cyclosporine-drug interactions (Gertz et al., 2013). With respect to OATP1B3, AM1 had comparable inhibition potency to the parent drug (IC50 < 0.05 µM). These in vitro findings also highlighted the importance of the preincubation step in OATP inhibition studies, which resulted in a “time-dependent” increase in AM1 and cyclosporine inhibition potency against OATP1B1 and OATP1B3 following preincubation.
The contribution of AM1 to the cyclosporine OATP-mediated DDI was subsequently investigated using reported blood concentration-time data after single and multiple oral doses of cyclosporine and in vitro transporter inhibition data. Metabolite concentration data were combined with a physiologically based pharmacokinetic (PBPK) model for cyclosporine, allowing consideration of the time course of the perpetrator at the relevant site of interaction and assessment of DDI potential in a dynamic manner (Gertz et al., 2013). Despite high total blood AM1 concentrations after multiple dosing, the predicted maximal reduction of OATP1B1 activity by cyclosporine at steady state did not change (<5%) when AM1 was considered. Extensive distribution of AM1 into red blood cells, leading to lower unbound plasma concentration and metabolite/parent differences in potencies against OATP1B1, explain the lack of effect of AM1 in vivo despite potent inhibition in vitro. OATP1B1 inhibition by AM1 was more apparent during the terminal phase of the dosing interval because of the longer terminal half-life of AM1 relative to parent, resulting in protracted inhibition of hepatic OATP uptake. Assigning the same OATP1B1 IC50 to the other cyclosporine metabolites with comparable structure to AM1 (AM9, AM4N), for which standards were not available commercially, predicted negligible contribution of these metabolites to the magnitude of DDIs. This example emphasizes the need for prospective characterization of metabolite disposition and interaction potential, as well as integration of resultant data into PBPK models to predict the contribution of metabolite(s) to clinical DDIs in a mechanistic manner.
Mycophenolic Acid.
Potential changes in the DDI risk profile are an important consideration for special populations. For example, renal impairment can change exposure of metabolites cleared by renal excretion and/or by hepatic mechanisms, whose function is impaired secondary to kidney damage (Reyes and Benet, 2011; Rowland Yeo et al., 2011; Zhao et al., 2012; Hsu et al., 2013). For instance, both mycophenolic acid and its glucuronide metabolite are inhibitors of the renal uptake transporters OAT1 and OAT3 (Uwai et al., 2007). This DDI concern generally has not translated to the clinic due to insufficient unbound drug and metabolite concentrations. However, mycophenolic acid glucuronide exposure can increase up to 8-fold in renally impaired patients (http://www.pharma.us.novartis.com/product/pi/pdf/myfortic.pdf), raising the possibility of extensive OAT3 inhibition (unbound inhibitor concentration/IC50 > 10). This example illustrates how the DDI potential of metabolites may change in special populations.
Approaches to Understanding Metabolite Transport
Use of Cell Systems and Knockout Animal Models to Understand Metabolite Disposition.
Cell lines transfected with single (e.g., HEK-OATP1B1) or multiple (e.g., HEK-OATP1B1/MRP2/MRP3) transporters and hepatocyte systems (e.g., suspension, sandwich culture, 3D) can be used to assess the impact of uptake and/or efflux transporters on drug or metabolite intracellular concentration in vitro. These systems have been crucial in evaluating the importance of transporters in metabolite disposition and interactions (Hirouchi et al., 2009). In addition, coexpression of metabolic enzymes (e.g., OATP1B1-UGT1A1-MRP2) enables investigation of the functional interplay between uptake, phase II metabolism, and efflux, as illustrated with ezetimibe and etoposide (Fahrmayr et al., 2012). Studies in hepatocytes can delineate more complex metabolite-transporter interactions, such as the impact of canalicular efflux impairment on intracellular exposure to troglitazone metabolites, or the multifaceted alterations in hepatobiliary drug disposition during inhibition of multiple hepatic uptake and efflux transporters by sulindac metabolites (Lee et al., 2010a,b). Recently, mechanistic compartmental models have been developed for dynamic characterization of transport-metabolism interplay in different cellular systems (Lee et al., 2010a; Maeng et al., 2012; Menochet et al., 2012a,b). These complex compartmental models enable simultaneous assessment of active uptake/efflux and metabolism in addition to passive permeation and intracellular binding. Although increasing the complexity of the data analysis enables a more mechanistic description of the processes in vitro, rich data input and monitoring of both parent and metabolite(s) are generally required (Sun et al., 2008; Menochet et al., 2012a).
Studies in knockout animals are useful for investigating the effect of deficient transporter activity on the systemic and tissue exposure of the parent and metabolite, as well as the pharmacodynamic and toxic consequences of these pharmacokinetic alterations, as highlighted throughout this review (see ezetimibe, mycophenolic acid, trabectedin, morphine). These studies are of particular relevance in the case of differential effects on parent and metabolite, as illustrated recently for sorafenib and its glucuronide metabolite (Zimmerman et al., 2013). Oatp1b2-knockout mice exhibited >8-fold increased systemic sorafenib glucuronide exposure with ∼25% decreased parent drug exposure; knockin of human OATP1B1 or OATP1B3 into Oatp1b2-knockout mice partially restored sorafenib glucuronide clearance. Despite the proven utility of knockout models in elucidating metabolite-transporter interactions, caution is warranted in direct human translation, which requires confirmation of similar drug disposition between the knockout species and humans. Furthermore, pharmacokinetic alterations in knockout models, which represent 100% “inhibition” of the knocked-out pathway, are likely to be markedly larger in magnitude than clinical changes during partial inhibition of the same pathway by coadministered drugs (Zamek-Gliszczynski et al., 2009, 2013a).
In Vitro–In Vivo Extrapolation of the Disposition of Major Excretory Human Metabolites.
A recent example of an in vitro–in vivo extrapolation strategy to understand the importance of major noncirculating metabolites is Boehringer-Ingelheim’s investigational hepatitis C drug, faldaprevir (Li et al., 2013). Following administration of a radiolabeled oral dose to humans, parent drug accounted for >98% of plasma radioactivity. However, two hydroxylated metabolites accounting for 41% of the dose were detected in human feces. Per the “Metabolites in Safety Testing” recommendation of toxicology coverage for metabolites accounting for >10% of a bioavailable dose in human excreta, adequate safety coverage was established for both fecal hydroxyl metabolites in monkeys. The disposition, DDI potential, and pharmacologic relevance of the two metabolites were characterized prospectively using an in vitro–in vivo extrapolation approach.
Using recombinant P450s and intestinal and hepatic microsomes, the hydroxyl metabolites were shown to be formed predominantly by hepatic CYP3A4. In vitro transport studies in transporter-transfected cells and hepatocytes suggested that, once formed in the liver, these two metabolites were excreted into bile by BCRP and P-gp. The absence of the hydroxylated metabolites in the circulation is consistent with predominant hepatic formation and biliary excretion, as well as active hepatic uptake via OATP. The two hydroxyl metabolites are ∼3-fold more pharmacologically potent than parent drug and may contribute to drug efficacy since the liver is the target organ for hepatitis C treatment. However, these metabolites are not expected to contribute to DDIs as perpetrators based on overall lower in vitro inhibition potencies than parent and insufficient systemic and hepatic exposure to perpetrate clinical interactions. Interaction for the metabolites as victims at the level of hepatic canalicular transport is theoretically possible, but such an interaction would result in enhanced hepatic exposure to the active metabolites, and would thus be expected not to diminish efficacy.
Faldaprevir represents an example of the value of characterizing metabolite disposition. This additional understanding ensured toxicologic evaluation of the two major metabolites (i.e., confirmation of safety), dismissal of perpetrator drug interaction potential related to the metabolites, as well as demonstration that victim drug interaction potential at the level of metabolite transport will not result in diminished efficacy.
Preclinical Characterization of the Disposition of Noncirculating Excretory Metabolites.
LY2090314 is an intravenous glycogen synthase kinase-3 inhibitor in clinical studies for the treatment of cancer (Zamek-Gliszczynski et al., 2013a). Parent drug exhibited perfusion-limited clearance by extensive metabolism in humans and toxicology species; despite these clearance properties, metabolites were not present in the circulation, with the exception of a dog-specific direct glucuronide conjugate. Instead, numerous metabolites were extensively and rapidly eliminated into feces via biliary excretion, with no apparent intestinal reabsorption. Since current regulatory criteria for metabolite characterization are based upon circulating exposure, there is no expectation to understand potential victim drug interaction liabilities with respect to hepatic canalicular transport. However, biliary excretion of metabolites conceptually was consistent with an active process and was investigated further in transporter knockout rats (Zamek-Gliszczynski et al., 2013a). The tested hypothesis was that, in single-gene knockout rats, lacking one of the three key hepatic canalicular transporters (P-gp, Bcrp, or Mrp2), biliary excretion of LY2090314 metabolites may be impaired, resulting in excretion of these metabolites across the sinusoidal membrane into the circulation (Zamek-Gliszczynski et al., 2012). Unexpectedly, in all three single-gene knockout rats, metabolites did not appear in the circulation, and their urinary recovery was not enhanced. Thus, the hypothesized impairment of metabolite biliary excretion leading to increased systemic exposures and urinary excretion did not materialize. Metabolite biliary excretion either was maintained by the other efflux transporters (e.g., P-gp and Bcrp in the Mrp2 knockout) or the metabolites were metabolized further into more polar species, which were then excreted into the bile. Thus, even complete inhibition of individual canalicular efflux transporters may not significantly alter LY2090314 metabolite disposition, i.e., shunt excretion into the circulation or accumulation in the liver. As such, these preclinical studies dismissed the risk of victim drug interactions at the level of biliary excretion of LY2090314 metabolites. Specifically, coadministration with canalicular transporter inhibitor drugs does not present a risk of systemic exposure to or hepatic accumulation of LY2090314 metabolites.
Entero-Test for Clinical Identification of Biliary Metabolites.
Unlike for parent drugs, biliary excretion is often a major route of elimination of metabolites. However, biliary excretion of metabolites poses a major challenge to understanding drug disposition, because this biophase is not easily accessible in humans. Recently, Guiney and colleagues (2011) described a novel, simple, and noninvasive approach to collect small samples of duodenal bile during clinical studies using Entero-Test, a commercially available device. In brief, a subject swallows a gelatin capsule containing 140 cm of highly absorbent nylon string, which can absorb 15 μl of fluid per centimeter of string. The string is weighed at the distal end and is allowed to pass into the duodenum. Approximately 4–5 hours after swallowing the capsule, a food stimulus (e.g., sausage or orange zest) is administered to contract the gallbladder, and the string is withdrawn 1 hour later. The string can then be analyzed for drug-related material.
Application of the Entero-Test has been reported by Bloomer et al. (2013), who used this approach to study the metabolism of GSK1325756 (N-(4-chloro-2-hydroxy-3-(3-piperidinylsulfonyl)phenyl)-N′-(3-fluoro-2-methylphenyl)urea), a drug undergoing development for chronic obstructive pulmonary disease. Preliminary in vitro and preclinical studies suggested that the drug underwent direct conjugation, with this conjugate eliminated in bile, as well as additional drug clearance by oxidative metabolism. Therefore, to gain a more complete understanding of GSK1325756 metabolism in humans, and to assess for drug interaction liability, Entero-Test was incorporated into a clinical study to collect bile for metabolite profiling. Of the 16 subjects tested, 9 had visual evidence of the string being soaked in duodenal bile. No drug-related metabolites were detected in control strings (i.e., collected before drug administration). Individual bile samples were processed and analyzed for GSK1325756 and metabolites. A total of 13 metabolites were detected by LC-MS and 19F-NMR, including oxidized, cyclized, and glucuronide-conjugated metabolites. The study confirmed that the O-glucuronide was the major metabolite in bile, accounting for 80% of total drug-related material. This case study illustrates the feasibility of collecting human bile as a method to further understand metabolite disposition and transport. This relatively noninvasive approach also can be used in larger animals (e.g., dogs, monkeys), enabling correlations between nonrodent species and humans.
Mass Spectrometry Imaging of Drugs and Metabolites.
Understanding the tissue distribution of a drug and metabolite(s) can provide critical insights into drug disposition, efficacy, and safety. Historically, tissue distribution and/or concentrations were determined by isolating and homogenizing tissues, quantitative whole-body autoradiography, or microradiography of tissue sections. However, these approaches fail either to maintain the anatomic distribution or to separate parent drug from metabolites (Chu et al., 2013). Recent advances in mass spectrometry imaging (MSI) have rendered determination of drug and metabolites in tissues more quantitative while preserving the overall spatial relationship of the drug and metabolites in the tissue (Castellino et al., 2011; Prideaux and Stoeckli, 2012). Application of MSI to understand both parent and metabolite distribution has increased significantly in the past few years, with dozens of drugs studied in a variety of tissues (Prideaux and Stoeckli, 2012). One recent example using MSI was to elucidate the mechanism of toxicity in subjects administered an investigational non-nucleoside reverse transcriptase inhibitor (fosdevirine) undergoing development for the treatment of human immunodeficiency virus (Castellino et al., 2013). In a phase IIb clinical trial involving treatment-experienced human immunodeficiency virus–infected patients, five of the 20 subjects enrolled in the fosdevirine treatment arm experienced seizures after completion of at least 4 weeks of treatment. The high incidence of seizures could not be explained by background seizure incidence or risk due to concomitant medications.
Matrix-assisted laser desorption/ionization MSI was used to investigate fosdevirine central nervous system (CNS) disposition and metabolism in brain tissue from rabbits, minipigs, and monkeys (Castellino et al., 2013). LC-MS was used to characterize and estimate fosdevirine and metabolite concentrations in cerebrospinal fluid from seizure patients, rabbits, minipigs, and monkeys. A cysteine conjugate of fosdevirine was the predominant drug-related component in cerebrospinal fluid from seizure patients, rabbits, and minipigs, as well as brain tissue from rabbits and minipigs. This metabolite persisted in the CNS for an extended period of time after the last fosdevirine dose and was associated with the white matter. In contrast, in monkeys, a species without any CNS adverse events, fosdevirine was the predominant drug-related component and was associated with the gray matter. This approach provided insights into species differences in CNS toxicity/seizures. Fosdevirine provides an example of the future potential of imaging approaches such as MSI to gain an improved understanding of metabolite disposition.
PBPK Modeling and Simulation of Metabolite Disposition and DDI Potential.
PBPK models incorporating transporter-metabolite interplay in the liver, intestine, and kidney have been reported (Pang et al., 2008; Fan et al., 2010; Gertz et al., 2013), accounting also for metabolite undergoing futile cycle (Sun et al., 2010) and emphasizing the need for modeling of both parent and metabolite data. Recently, PBPK modeling approaches have increasingly been used to predict enzyme-transporter-mediated pharmacokinetics and associated complex DDIs (Gertz et al., 2013; Zamek-Gliszczynski et al., 2013b). The main advantage of this mechanistic tool is the ability to simulate the concentration-time course of the perpetrator at relevant site(s) of interaction (often intracellular), to account for differential membrane location of transporters, and to assess effects on multiple transporters/enzymes. Theoretically, PBPK models for the parent can be extended to investigate the contribution of the major metabolite(s) to DDI potential, as illustrated in this review for the cyclosporine AM1 metabolite. However, this approach requires rich data sets and an understanding of metabolite disposition and inhibitory potencies against transporters/enzymes of interest, which typically are limited in early stages of drug development.
Case studies discussed in this review highlight the need for careful interpretation of the >25% of parent exposure cut-off proposed by regulatory agencies. The net reduction in enzyme/transporter activity will depend on inhibition potency, metabolite concentration at the relevant site of interaction, and the mechanism of inhibition. The latter may differ from the parent, as illustrated with the effect of gemfibrozil glucuronide on CYP2C8. Alternatively, the interaction mechanism may not be completely understood, as illustrated with the cyclosporine/AM1 interaction with OATP1B1/1B3. In this case, PBPK modeling predicted relatively fast recovery of the activity of these uptake transporters under the assumption of reversible inhibition (Gertz et al., 2013; Zamek-Gliszczynski et al., 2013b). Further work is required to confirm whether this mechanism holds true for cyclosporine/AM1 inhibition of OATP1B1 and OATP1B3 in vivo.
Sensitivity analyses represent another highly useful application of mechanism-based modeling within the PBPK framework. Availability of metabolite in vitro inhibition data can enable initial simulation of the dynamic reduction in transporter activity over a range of assumed metabolite exposures (e.g., from the recommended 25% to >100% of the parent) in both plasma and relevant tissues (Zamek-Gliszczynski et al., 2013b).
PBPK models can be an extremely useful tool to investigate the impact of alteration in transporter activity on the intracellular concentrations (Pang et al., 2008; Fan et al., 2010; Gertz et al., 2013) and hence DDI potential of both parent and metabolite, particularly in cases of complex disposition and interplay of different cellular processes, as illustrated here with ezetimibe and mycophenolic acid. Without an adequate mechanistic model for the metabolite in place, initial analysis can use reported systemic metabolite concentration-time data as a forcing function to simulate the magnitude of DDI. This analysis can be conducted in combination with the PBPK model developed for the parent (Gertz et al., 2013; Varma et al., 2013). However, the reliance on plasma metabolite data as a surrogate for tissue exposure represents a limitation if the actual DDI target is intracellular.
Expert Opinion
The objective of this commentary is to foster further research and discussion around the transport properties of metabolites, in particular phase II conjugates, which may be overlooked during drug development as “inactive” (http://www.ema.europa.eu). The perspectives presented herein are not meant to be prescriptive, but aim to encourage drug metabolism scientists to critically review, on a case-by-case basis, the potential role of transporters in the disposition of metabolites. Relative to parent drug, metabolites generally exhibit greater polarity and decreased permeability, increasing their reliance on active transport for movement across biologic membranes, and thus increasing the likelihood of transporter-mediated interactions. In addition, current efforts to minimize P450 metabolism and to select compounds with enhanced solubility increase the potential for interactions with phase II metabolism and transporters. This is a trend that has been observed for many recently registered drugs and their metabolites (e.g., the first approved sodium/glucose cotransporter 2 inhibitor, canagliflozin; http://www.invokanahcp.com/prescribing-information.pdf).
A conjugated metabolite is one possible trigger to further investigate the importance of transporter-mediated interactions, as the disposition of these metabolites is often transporter-dependent and can contribute to efficacy or safety issues. Phase II conjugates, even when nonreactive and lacking pharmacologic activity, are the most likely to exhibit transporter-mediated disposition and DDIs, especially when they are involved in enterohepatic cycling. Thus, pharmacological activity assessment cannot be limited to testing only the metabolite in vitro against the target, but needs to consider whether a “nonactive metabolite” can contribute indirectly to the pharmacology by conversion back to the parent compound. Although a phase II metabolite may not exhibit direct activity, characterizing its transport properties may be prudent to anticipate alterations in pharmacokinetics/pharmacodynamics during drug interactions. Notably, characterization of phase II metabolism is itself challenging (Miners et al., 2010; Gill et al., 2012; Rowland et al., 2013), and the study of phase II conjugate transport adds an additional level of complexity.
Understanding transport pathways for major excretory metabolites is important, but this investigation must proceed thoughtfully. Excreta may not accurately reflect excretory metabolites, since intestinal and bacterial metabolism, as well as chemical degradation, may contribute to the observed metabolite profile. As outlined in this commentary, Entero-Test could become a practical clinical approach to identify and obtain qualitative estimates of biliary metabolites in humans. An ongoing review of metabolites in excreta should be completed and a strategy developed to address the current regulatory recommendations with respect to safety coverage, as well as to consider the absorption, distribution, metabolism, and excretion (ADME) and pharmacology studies required to elucidate a metabolite’s disposition and clinical importance. This exercise may require no action, but at other times may necessitate ADME, pharmacology, and/or toxicology characterization (e.g., LY2090314, faldaprevir).
Investigation of metabolite transport properties should also consider changes in intracellular concentrations in targeted tissues, such as the liver, to understand how metabolites may contribute to drug-drug interactions, pharmacology, and/or toxicity. Broader use of PBPK modeling/simulation and MSI imaging will aid in the understanding of tissue and intracellular concentrations of parent and metabolites and enable testing of the hypothesis that metabolites contribute to drug interactions and/or toxicities.
One area of drug transport research that is currently underappreciated and is in great need of more basic research is the importance of basolateral efflux, particularly in liver, intestine, and kidney. There is currently a limited understanding as to why metabolites are directed to either the apical or basolateral membrane for excretion, i.e., why certain metabolites circulate, whereas others do not, and instead are excreted immediately following their formation. These differences are presumably due to affinity for, or expression levels of, the individual transporters present in the relevant membrane domain (e.g., hepatic canalicular MRP2 versus sinusoidal MRP3 and MRP4); however, the exact reasons are unknown. Elucidation of the mechanistic basis for excretory directionality will improve the understanding of systemic metabolite exposure versus biliary/fecal or urinary recovery.
We believe a more proactive approach to explore the transport properties of major metabolites in both systemic circulation and in excreta will enhance the development of new medicines by providing a richer understanding of a drug’s pharmacology, disposition, drug interaction risk, and toxicity. Clinical efficacy, toxicity, or DDIs involving metabolites can be complex and require a thorough understanding of drug and metabolite disposition. As demonstrated in the case studies presented here (Table 1), characterization of metabolites with respect to transporter properties can provide greater insight into overall drug pharmacokinetics, dynamics, and safety. However, the decision to assess metabolites requires a significant commitment of resources and time (e.g., synthesis of metabolites, running assays, etc.), and thus should be triggered judiciously as scientifically justified on a case-by-case basis. Even if the final decision is not to progress any metabolite transport work, such a decision should be supported through integration of all available metabolite knowledge.
In summary, the transport properties of major circulating and excretory metabolites merit consideration during drug development, such that consequences of the modulation of these transport mechanisms can be anticipated instead of discovered empirically in clinical practice.
Authorship Contributions
Performed data analysis: Zamek-Gliszczynski, Chu, Polli, Paine, Galetin.
Wrote or contributed to the writing of the manuscript: Zamek-Gliszczynski, Chu, Polli, Paine, Galetin.
Footnotes
- Received October 25, 2013.
- Accepted December 17, 2013.
Abbreviations
- BCRP
- breast cancer resistance protein
- BSEP
- bile salt export pump
- CNS
- central nervous system
- DDI
- drug-drug interaction
- GSK1325756
- N-(4-chloro-2-hydroxy-3-(3-piperidinylsulfonyl)phenyl)-N′-(3-fluoro-2-methylphenyl)urea
- HEK
- human embryonic kidney
- MRP
- multidrug resistance-associated protein
- MSI
- mass spectrometry imaging
- LY2090314
- 3-[9-fluoro-2-(piperidin-1-ylcarbonyl)-1,2,3,4-tetrahydro[1,4]diazepino[6,7,1-hi]indol-7-yl]-4-imidazo[1,2-a]pyridin-3-yl-1H-pyrrole-2,5-dione
- OAT
- organic anion transporter
- OATP
- organic anion transporting polypeptide
- P450
- cytochrome P450
- PBPK
- physiologically based pharmacokinetic
- P-gp
- P-glycoprotein
- TR−
- Mrp2-deficient
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}