


Abstract
The prediction of human pharmacokinetics from preclinical species is an integral component of drug discovery. Recent studies with a 103-compound dataset suggested that scaling from monkey pharmacokinetic data tended to be the most accurate method for predicting human clearance. Additionally, interrogation of the two-dimensional molecular properties of these molecules produced a set of associations which predict the likely extrapolative outcome (success or failure) of preclinical data to project human pharmacokinetics. However, a limitation of the previous analyses was the relative paucity of data for typical “discovery-like” molecules (molecular weight >300 and/or clogP >3). The objective of this investigation was to generate preclinical data required for extension of this dataset for additional discovery-like molecules and determine whether the aforementioned findings continue to apply for these molecules. In vivo nonrodent intravenous pharmacokinetic data were generated for 13 molecules, and data for 8 additional molecules were obtained from the literature. Additionally, the various scaling methodologies and molecular features analysis were applied to this new dataset to predict human pharmacokinetics. Whereas the predictive accuracies demonstrated across all of the various methodologies were lower for this higher clearance compound dataset, scaling from monkey liver blood flow continued to be an accurate methodology, and human volume of distribution was similarly well predicted regardless of scaling methodology. Lastly, application of the molecular feature associations, particularly data-dependent associations, afforded an improved predictivity compared with the liver blood flow scaling approaches, and provides insight into the extrapolation of high clearance compounds in the preclinical species to human.
The use of in vivo preclinical pharmacokinetic data generated during lead optimization to predict human pharmacokinetic properties is commonplace in the pharmaceutical industry, with a variety of methodologies available for data predictivity (Ings, 1990; Pogessi, 2004). Recent investigations using a diverse set of 103 nonpeptide xenobiotics with rat, dog, monkey, and human intravenous pharmacokinetic data have yielded a number of interesting and useful observations, including the value of monkey pharmacokinetic data in successful prediction of human pharmacokinetics, and the relative lack of added value provided by dog pharmacokinetic data (Ward and Smith, 2004a,b). Additionally, when several quantitative methodologies, including liver blood flow-based scaling and body weight-based allometry, were applied to this dataset for predicting human clearance (CL), the greatest accuracy was achieved using the monkey liver blood flow approach, rather than allometry, even with the inclusion of mechanistic or empirical allometric correction factors (Nagilla and Ward, 2004). Finally, the availability of this comprehensive dataset has allowed the identification of a set of associations between preclinical pharmacokinetic properties, two-dimensional molecular features, and extrapolative success or failure of human pharmacokinetics (Jolivette and Ward, 2005). These calculated physiochemical properties can then be used to predict the extrapolative success or failure of rat, dog, and monkey data to project human pharmacokinetic parameters.
Despite the clear utility of this extrapolation dataset, one of the concerns about the various relationships expounded to date has been the applicability of these observations beyond the chemical space originally circumscribed. Largely, the lack of additional molecules for which a complete set of clinical and preclinical data are available in the literature has hindered further exploration in this area; nonhuman primate literature data are particularly sparse. In the present investigation, the dataset is extended by an additional 21 compounds: 8 compounds for which rat, dog, monkey, and human data have already been published, and 13 compounds for which nonrodent preclinical data were not previously available and for which either monkey or dog data were generated de novo for this study. This dataset was of particular interest since it was enriched with molecules likely to be encountered in a modern drug discovery setting (Lipinski, 2000; Wenlock et al., 2003; Lajiness et al., 2004); 19 of the 21 compounds possessed molecular weights >300 g/mol or clogP >3, and 16 of the 21 compounds possessed both molecular features, a criterion largely unmet in the previous dataset. Interestingly, as a whole, compounds included in this dataset were biased toward higher CL compounds across all species than those found in the previous dataset. Using this dataset, human CL, distributional volume at steady state (Vd), and mean resident time (MRT) were predicted from each preclinical species alone using the liver blood flow technique, as well as allometric scaling either with or without application of empirical correction factors (Fig. 1). Additionally, the applicability of the previously developed molecular feature associations with rat, dog, and monkey predictivity of human pharmacokinetics was examined.

Schematic depicting the in vivo PK data generated for completion of this dataset, and the various methodologies used to predict human PK.
Materials and Methods
Literature Data Collection. Details underlying the collection of this dataset were identical to those detailed elsewhere (Ward and Smith, 2004a). Eight molecules, [3-{2-oxo-3-[3-(5,6,7,8-tetrahydro-[1,8]naphthyridin-2-yl)propylimidazolidin-1-yl}-3(S)-(6-methoxy-pyridin-3-yl)propionic acid (“αVβ3 antagonist”), epiroprim, flindokalner, garenoxacin, midazolam, ofloxacin, PNU-96391, and semaxanib] were identified with complete intravenous pharmacokinetic data in rat, dog, monkey, and human. Literature data were found for 11 additional xenobiotics (amiodarone, biperiden, chlorpromazine, diltiazem, felodipine, methadone, nifedipine, propafenone, remoxipride, verapamil, and vinorelbine) in rat, dog, and human, but no comparable data were reported for monkey. Likewise, two additional molecules (haloperidol and mifepristone) possessed published intravenous pharmacokinetic data in rat, monkey, and human, but no comparable data were reported for dog.
Materials. Haloperidol, mifepristone, amiodarone (HCl salt), chlorpromazine (HCl salt), felodipine, methadone (HCl salt), nifedipine, and propafenone (HCl salt) were purchased from Sigma-Aldrich (St. Louis, MO). Biperiden was obtained from Toronto Research Chemicals (North York, ON, Canada), diltiazem (HCl salt) from ICN Biomedicals (Costa Mesa, CA), remoxipride (HCl salt) from Tocris (Ellisville, MO), and verapamil (HCl salt) from BIOMOL Research Laboratories, Inc. (Plymouth Meeting, PA); in all instances purity was >98.6%. Vinorelbine was synthesized in-house and supplied by the GlaxoSmithKline Department of Medicinal Chemistry. Hydroxypropyl-β-cyclodextrin (Cavitron) was purchased from Cerestar USA, Inc. (Hammond, IN). All other reagents and materials were purchased from standard vendors and were of the highest available purity. All dosages are expressed as milligrams of parent molecule per kilogram, and final concentrations of each test compound in the administered dose were verified (and appropriate adjustments to calculations made) by procedures detailed below.
Animals. Male purebred beagle dogs (Marshall Research, North Rose, NY) weighing 9 to 12 kg and male cynomolgus monkeys (Macaca fascicularis; Charles River Primate Labs, Houston, TX) weighing 5 to 7 kg were used for the in vivo portions of this investigation. All animals were housed according to the Institute of Laboratory Animal Resources, National Research Council's Guide for the Care and Use of Laboratory Animals. All procedures on animals were reviewed and approved by the GSK Institutional Animal Care and Use Committee. A complete blood chemistry panel was performed before each study day to obtain baseline values and to ensure hematological recovery. Filtered tap water was available ad libitum, and the animals were fed a standard animal diet as appropriate for each species except on certain overnight periods before dosing as described below. Whenever overnight fasting was used, food was returned after the 240-min sample was obtained. Actual body weight-based doses and actual sampling times were recorded and used in all calculations.
In Vivo Dog and Monkey Pharmacokinetic Studies. On the study day, the dogs or monkeys (n = 2 per compound) received a temporary catheter in a saphenous vein for intravenous infusion and cephalic (dog) or contralateral saphenous (monkey) vein for blood sampling. The dogs (fasted overnight) or monkeys (not fasted) received test compounds as a 60-min intravenous infusion (0.03–1.0 mg/kg target dose; 4 ml/kg total volume). Dosages were selected based on the literature data available for other species and were designed to minimize the likelihood of overt pharmacology while maximizing the pharmacokinetic data obtained. Dosages were administered as solutions in 20% aqueous Cavitron (pH <5.0) with ≤1% dimethyl sulfoxide. Dogs were restrained in slings and monkeys in restraining chairs to facilitate drug administration and blood sampling for up to 4 h before being returned to their cages. Blood samples (0.25 ml) were obtained from a cephalic (dog) or contralateral saphenous (monkey) vein before dosing and at various timed intervals up to 24 h postdose. Plasma (50 μl) was isolated by centrifugation and stored at –80°C until analysis.
Analytical Procedures. Quantitative analysis was performed on plasma samples and dose solutions for each test compound using a liquid chromatography/tandem mass spectrometric detection method optimized for each individual analyte in the appropriate biological matrix. Plasma samples were thawed and each analyte was isolated from 50 μl of plasma using a method based upon protein precipitation with 95:5 acetonitrile/buffer (10 mM ammonium formate; pH = 3.0) containing an appropriate mass spectral internal standard, and the resulting mixture was vortex-mixed for 2 min followed by centrifugation for 30 min at >2000g. The resulting supernatant was injected by an HTS PAL autosampler (CTC Analytics, Zwingen, Switzerland) onto an appropriate analytical liquid chromatography column eluting to either a Sciex API 365 or API 4000 triple quadrupole mass spectrometer (Applied Biosystems, Foster City, CA). The eluent was subjected to Turbo IonSpray ionization multiple-reaction monitoring (positive ion mode for all compounds except for felodipine). Each analyte was characterized by an appropriate mass spectral transition of the parent [(M + H)+;(M–H)– for felodipine] precursor ion to an appropriate product ion, generated at an optimized collision energy. Data were reported as quantitative drug concentrations as determined by standard calibration curve analysis, using linear fitting of a 1/x-weighted plot of the analyte/internal standard peak area ratio versus analyte concentration. Using these optimized conditions, the lower limit of quantitation achieved for all compounds was 1.00 ng/ml except amiodarone (0.5 ng/ml).
Pharmacokinetic Parameter Generation. Plasma concentration versus time profiles were generated for each animal. Standard noncompartmental pharmacokinetic analysis was performed using WinNonlin Professional version 4.1 (Pharsight Corporation, Mountain View, CA) to determine CL, volume of distribution at steady state (Vd), and mean residence time (MRT). The terminal elimination phase was estimated by the selection of ≥3 points for all compounds, and the percentage extrapolated area under the curve was <30% for both compounds in the dog and <11% for all compounds in the monkey.
Clearance Data Analysis. Prediction of human CL from the preclinical data collected in this study was conducted using the liver blood flow (LBF) scaling technique from each individual species as described previously (Ward and Smith, 2004a). The pharmacokinetic parameters, CL and Vd, for all 21 compounds used in the present investigation, whether derived from the literature or experimentally determined by this laboratory, can be found in Appendix I. This technique involves a correction for LBF differences and the scaling of clearance from each preclinical species to humans as a percentage of liver blood flow. This technique involves the projection from rat, dog, and monkey as follows:  Liver blood flow in each species was defined as 85, 30, 45, and 21 ml/min/kg for rat, dog, monkey, and human, respectively (Davies and Morris, 1993; Uhing et al., 2004). In addition, both simple allometry (eq. 2) and exponent rule-corrected allometry using various correction factors including MLP and BrW (eqs. 3 and 4) were conducted for each molecule to predict human CL; allometric predictions were limited to all three preclinical species (rather than combinations of any two species) as described previously (Boxenbaum and DiLea, 1995; Mahmood and Balian, 1996; Nagilla and Ward, 2004):
Liver blood flow in each species was defined as 85, 30, 45, and 21 ml/min/kg for rat, dog, monkey, and human, respectively (Davies and Morris, 1993; Uhing et al., 2004). In addition, both simple allometry (eq. 2) and exponent rule-corrected allometry using various correction factors including MLP and BrW (eqs. 3 and 4) were conducted for each molecule to predict human CL; allometric predictions were limited to all three preclinical species (rather than combinations of any two species) as described previously (Boxenbaum and DiLea, 1995; Mahmood and Balian, 1996; Nagilla and Ward, 2004): 

 MLP in each species was 4.7, 20.0, 22.0, and 93.0 years for rat, dog, monkey, and human, respectively, and the BrW in each species was defined as 1.8, 80, 90, and 1400 g for rat, dog, monkey, and human, respectively (Davies and Morris, 1993). According to “rule of exponents,” when 0.55 ≤ b < 0.71, no correction factor was used; when 0.71 ≤ b < 1.00, MLP was incorporated as a correction factor into the scaling method; and when b ≥1, BrW was incorporated into the scaling method. The extrapolative success or failure of each human CL prediction was determined both qualitatively and quantitatively. For qualitative assessment, compounds were categorized as low (<30% LBF), moderate (30–70% LBF), or high (>70% LBF) CL. A qualitative extrapolative inlier (correct prediction) was declared when the extrapolation resulted in a CL prediction of the correct human CL category from a given preclinical species, whereas an extrapolative outlier (incorrect prediction) was declared when the predicted human CL was of a different CL category than the measured human CL. For quantitative assessment, when the predicted human CL from a given preclinical species was within 2-fold of the experimentally determined value, the prediction was considered an extrapolative inlier, and predicted values outside of a 2-fold window of the experimentally determined value were extrapolative outliers. Additionally, the mean absolute error (MAE) and %MAE relative to human CL were determined between the predicted and observed CL values, and were calculated as:
MLP in each species was 4.7, 20.0, 22.0, and 93.0 years for rat, dog, monkey, and human, respectively, and the BrW in each species was defined as 1.8, 80, 90, and 1400 g for rat, dog, monkey, and human, respectively (Davies and Morris, 1993). According to “rule of exponents,” when 0.55 ≤ b < 0.71, no correction factor was used; when 0.71 ≤ b < 1.00, MLP was incorporated as a correction factor into the scaling method; and when b ≥1, BrW was incorporated into the scaling method. The extrapolative success or failure of each human CL prediction was determined both qualitatively and quantitatively. For qualitative assessment, compounds were categorized as low (<30% LBF), moderate (30–70% LBF), or high (>70% LBF) CL. A qualitative extrapolative inlier (correct prediction) was declared when the extrapolation resulted in a CL prediction of the correct human CL category from a given preclinical species, whereas an extrapolative outlier (incorrect prediction) was declared when the predicted human CL was of a different CL category than the measured human CL. For quantitative assessment, when the predicted human CL from a given preclinical species was within 2-fold of the experimentally determined value, the prediction was considered an extrapolative inlier, and predicted values outside of a 2-fold window of the experimentally determined value were extrapolative outliers. Additionally, the mean absolute error (MAE) and %MAE relative to human CL were determined between the predicted and observed CL values, and were calculated as: 
 where N = the total number of observations in the set and i = individual drug molecule.
where N = the total number of observations in the set and i = individual drug molecule.
Finally, the role of two-dimensional molecular properties in predicting extrapolative success or failure of each preclinical species was also determined both qualitatively and quantitatively through the application of the molecular property associations described previously (Jolivette and Ward, 2005). The calculated two-dimensional molecular properties for all 21 compounds used in the present investigation can be found in Appendix II. Essentially, two different types of associations were investigated for the assessment of human CL. First, “data-independent” associations were applied, wherein extrapolative outcome for projecting human pharmacokinetics were predicted solely on each molecule's two-dimensional physicochemical features [calculated logarithm of the octanol-water partition coefficient (clogP), calculated molar refractivity (CMR), number of hydrogen bond acceptors (HBA), number of hydrogen bond donors (HBD), molecular weight (mol. wt.), number of rotatable bonds (nrot), and polar surface area (PSA)]. This exercise involved the calculation of a series of two-dimensional molecular properties for each compound, calculation of mean values for properties that provided statistically significant differentiation between extrapolative success and failure, and assignment of molecular property associations that provided this discrimination. For instance, the associations state that for molecules with a clogP >0, extrapolation based on rat data would result in a human CL prediction that would be a quantitative outlier (corrected for LBF differences between the species, the predicted human CL from rat would not be within 2-fold of the observed human CL value). Next, “data-dependent” associations were applied, in which preclinical predictivity was interrogated as a function of both molecular features and the experimentally determined pharmacokinetic parameters in a given preclinical species. This second approach utilized an additional descriptor, based on CL categories, which was also considered with the calculated molecular properties to determine parameters that provided differentiation between extrapolative inliers and outliers. For example, the molecular associations predict that for compounds with low CL in the dog and PSA >160 Å2, the resulting human CL prediction would be a quantitative outlier with respect to the dog CL (corrected for LBF differences between the species, the predicted human CL from dog would not be within 2-fold of the observed human CL value).
Volume of Distribution Data Analysis. The prediction of human Vd from the preclinical data was conducted both qualitatively and quantitatively as described previously (Ward and Smith, 2004b). For qualitative assessment, the compounds were categorized as possessing low (<0.7 l/kg), moderate (0.7–3.5 l/kg), or large (>3.5 l/kg) Vd in each species. An extrapolative inlier was declared when human Vd was in the same category as the preclinical species. For quantitative assessment, when predicted human Vd was within 2-fold of the experimentally determined value, the prediction was considered an extrapolative inlier, and outside 2-fold, the prediction was considered an extrapolative outlier; MAE and %MAE were also determined as described earlier. Human Vd was also predicted from three-species allometric scaling, with the same qualitative and quantitative inlier criteria applied. Finally, the role of two-dimensional molecular properties in the extrapolative success or failure was also determined both qualitatively and quantitatively through the application of molecular property associations as described previously (Jolivette and Ward, 2005). For example, the molecular associations revealed that for compounds with large categorical Vd in the rat and with HBA ≥5, the resulting human Vd prediction would be a qualitative outlier with respect to rat (human Vd would not be large, <3.5 l/kg).
Mean Residence Time Data Analysis. Human MRT was calculated from the experimentally determined and reported human Vd and human CL for each compound (where MRT = Vd/CL). The prediction of human MRT from the preclinical data was conducted both qualitatively and quantitatively as described previously (Ward and Smith, 2004b). Briefly, human MRT was predicted by assuming that human Vd would be identical to that of the preclinical species and that CL would simply be the same fraction of LBF in that preclinical species (no allometric scaling). Using the CL and Vd values derived from the three-species empirical allometric correction factor calculations (i.e., rule of exponents), human MRT was predicted. For qualitative assessment, the compounds were grouped into three MRT categories [<60 min (low), 60 to 480 min (moderate), and >480 min (high]] in each species. A correct prediction was declared when the predicted human MRT was in the same MRT category as the preclinical species. For quantitative assessment, a correct prediction was declared when the predicted human MRT was within 2-fold of the experimentally determined value. Lastly, MAE and %MAE were also determined for each predictive method as described previously.
Results
Intravenous Preclinical Pharmacokinetic Data in Dog and Monkey. The compounds studied in the dog for this investigation (haloperidol and mifepristone) exhibited high and moderate CL, respectively, with large Vd (Table 1), although haloperidol exhibited a much larger Vd than mifepristone. The n = 2 study design used to estimate pharmacokinetic parameters in the dog appeared to be adequate, with the same CL and Vd categories calculated for each animal for both compounds. A fairly even distribution of CL values was obtained in the monkey with low, moderate, and high CL values for three, five, and three compounds, respectively (Table 1). As was the case for the dog, the Vd values for these compounds in the monkey were weighted heavily toward large Vd; only one compound (nifedipine) exhibited a small Vd in the monkey, two compounds (felodipine, remoxipride) exhibited a moderate Vd, and the remaining eight compounds exhibited a large Vd. Overall, somewhat greater interindividual variability was observed in the monkey than in the dog, with more variability observed for Vd than for CL (data not shown). Different Vd categories would have been derived from the two study animals for three compounds (biperiden, diltiazem, and remoxipride), whereas different CL categories would have been determined for two molecules (diltiazem and remoxipride). Average data were used for all compounds for subsequent data analyses, although the borderline qualitative categorization of these molecules should be considered a caveat to this analysis.
Average pharmacokinetic parameters determined in the present investigation following intravenous administration in the male cynomolgus monkey or beagle dog (n = 2 animals)
Human Clearance Predictions. The combination of the intravenous pharmacokinetic data generated above in dog and monkey with published literature values provided intravenous pharmacokinetic parameters for 21 compounds in rat, dog, monkey, and human (Fig. 2; Appendix I). Plotting CL as %LBF in the three preclinical species versus human CL as %LBF, all three preclinical species were clearly biased toward overprediction of human CL, with 12, 13, and 12 of the 21 compounds falling above the +15% LBF line, and 3, 2, and 4 compounds falling below the +15% LBF line for rat, dog, and monkey, respectively (Fig. 2). The human CL predictions based on LBF scaling from each preclinical species and for allometric scaling were compared qualitatively (correct CL category) and quantitatively (within 2-fold), along with their associated MAE (Table 2). Overall, the qualitative predictivity of all five scaling methods was similar but poor, with only 7 to 9 of the 21 test compounds correctly classified in all instances. From a quantitative perspective, comparing the different predictive methods, rat LBF scaling, monkey LBF scaling, and empirically corrected three-species allometry all demonstrated similar predictivity with 11, 10, and 10 correct quantitative predictions, respectively. Of the approaches investigated, scaling of human CL based on both monkey LBF and empirically corrected three-species allometry yielded identical predictivity, and the lowest MAE from any method. Generally, the worst predictivity was demonstrated by dog LBF scaling and simple three-species allometry, both in terms of correct number of predictions and MAE. Under the rule of exponent assumptions, a BrW correction was required for 2 compounds (nifedipine and verapamil), and a MLP correction for 9 compounds (amiodarone, chlorpromazine, diltiazem, methadone, midazolam, PNU-96391, propafenone, vinorelbine, and semaxanib), with no correction required for the remaining compounds. The application of the rule of exponents improved the predictions compared with simple allometry in 10 of the 11 instances in this dataset where a correction was performed; vinorelbine was the only compound where the application of an empirical correction factor worsened the prediction of human CL. Rule of exponent correction of the vinorelbine human CL prediction resulted in nearly a 2-fold worsening of the prediction compared with simple allometry. Before application of the MLP correction, the three-species allometry prediction was a qualitative and quantitative inlier with respect to the measured human CL; however, application of the MLP correction worsened the prediction where it became both a qualitative and quantitative outlier.
Accuracy of human clearance predictions from preclinical species using various methodologies

Comparison of human CL with CL in rat (A), dog (B), and monkey (C). CL is expressed as a percentage of liver blood flow in each species; the solid line represents the line of unity and the dashed lines represent ± 15% LBF. Where necessary, CL values greater than 100% LBF were truncated to 100% LBF for graphical depiction.
For each molecule, the methods that quantitatively afforded the most and least accurate predictions in terms of absolute error in each prediction were tallied (Fig. 3). In terms of the number of occurrences, rat and dog yielded the most accurate predictions the least often, where human CL was most accurately predicted for only 4 of the 21 compounds by each of these species. The dog most often yielded the worst prediction of human CL, where the error was the greatest from this species for 10 of the 21 compounds. Monkey LBF and exponent rule-corrected allometry were the most accurate prediction methods the most often; these approaches yielded the most accurate predictions for 6 and 7 of the 21 compounds, respectively. Predictions from exponent rule-corrected allometry were slightly better than monkey LBF scaling in this regard, with one more correct prediction; however, in terms of predictivity for all predictive techniques, both methodologies yielded equivalent MAE, at 6.08 ml/min/kg.
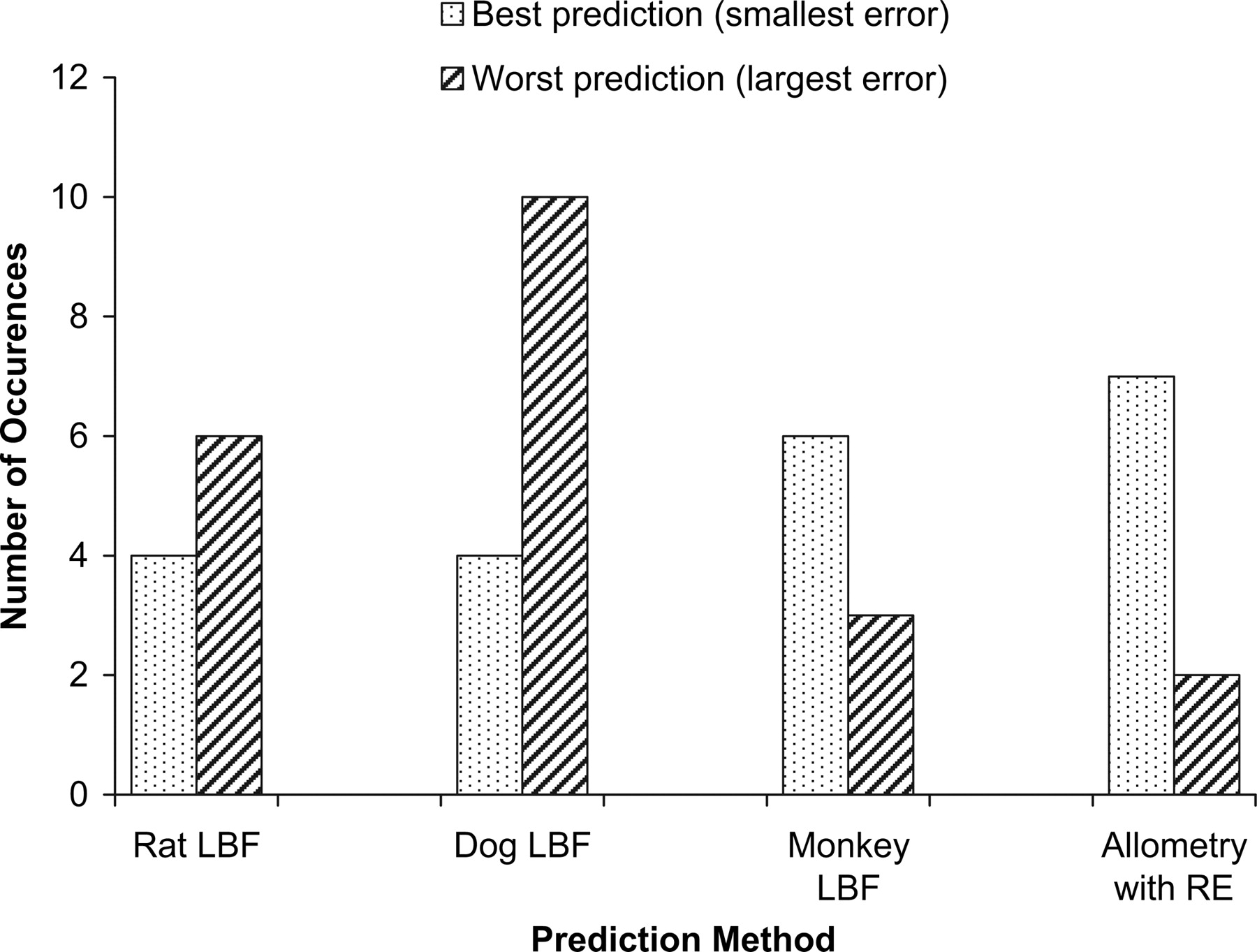
Comparison of the total number of molecules for which rat LBF, dog LBF, monkey LBF, and exponent-based rule-corrected allometry provided either the most or least quantitatively accurate human clearance prediction.
Molecular property associations (Jolivette and Ward, 2005) were next applied to the CL dataset (Table 3). Overall, application of these molecular feature criteria resulted in improved predictivity for each preclinical species, as well as adding the ability to discern when a prediction would likely be incorrect. As an example, using the results for data-independent quantitative predictions of human CL from dog as an example (Table 3, row 5) based on the associations, 21 predictions were made; 2 compounds were predicted to be extrapolative inliers (same CL category between dog and human), whereas 19 compounds were predicted to be extrapolative outliers (different CL category between dog and human). Of these 21 predictions, 15 were correctly predicted (71.4%); 1 of the 2 compounds predicted to be an extrapolative inlier was correctly predicted (50%) with an MAE of 1.59 ml/min/kg, whereas 14 of the 19 compounds predicted to be an extrapolative outlier were correctly predicted (73.7%) with an MAE of 12.9 ml/min/kg. Regarding the data-independent associations, assessment of molecules by qualitative class was not generally useful, since a prediction of extrapolative outcome could not be made based on the molecular features of most of the compounds in the dataset. However, in the few instances where an inlier was predicted, generally these molecules demonstrated decreased MAE compared with the entire dataset, where the MAE from qualitative predictions from monkey demonstrated a slight increase in overall error. Application of the data-independent associations in a quantitative manner was more useful, where the molecular property associations were used to make predictions for each compound; based on the molecular property associations, there was never an instance where a “no prediction” result was generated based on these associations. Again, when an inlier was predicted, MAE was lower for molecules than for the entire dataset; furthermore, MAE for predicted outliers was indeed higher than for the entire dataset. These trends were brought out even further when the data-dependent associations were applied (Table 4). In all instances, application of these associations resulted in lower MAE values for predicted inliers and higher MAE values for predicted outliers. For instance, predictions for data-dependent qualitative predictions of human CL from rat (Table 4, row 1) based on the associations of 15 extrapolative predictions were made and 6 compounds were not predicted; 3 compounds were predicted to be extrapolative inliers (same CL category between rat and human), whereas 12 compounds were predicted to be extrapolative outliers (different CL category between rat and human). Of these 15 predictions, 12 were correctly predicted (80.0%); 2 of the 3 compounds predicted to be an extrapolative inlier were correctly predicted (66.7%) with an MAE of 3.14 ml/min/kg, whereas 10 of the 12 compounds predicted to be an extrapolative outlier were correctly predicted (83.3%) with an MAE of 10.91 ml/min/kg. Furthermore, the overall predictivity of rat and dog increased substantially with the application of these molecular feature associations compared with LBF scaling alone. Rat LBF scaling to predict human CL was correct 33% and 52% for qualitative and quantitative predictions, respectively, whereas both qualitative accuracies increased to ∼80% after application of the molecular feature associations, and no improvement was noted in the quantitative predictions. However, only slight increases in predictive accuracy were noted for monkey after application of the associations.
Evaluation of the utility of data-independent molecular property associations applied to enhance preclinical predictivity of human CL
Evaluation of the utility of data-dependent molecular property associations applied to enhance preclinical predictivity of human CL
Human Distributional Volume Predictions. Examination of the intrinsic relationship between human Vd and the preclinical species revealed that both rat and dog had a tendency to overestimate human Vd with 8 and 9 of the 21 compounds falling above the +0.7 l/kg line, respectively; monkey was less biased with only 5 of the compounds above this line (Fig. 4). The preclinical Vd values for all 21 compounds were compared both qualitatively and quantitatively to human Vd and predicted using allometry (Table 5). On the basis of the number of compounds with correct qualitative predictions, dog appeared to be the most predictive, followed by the other predictive methodologies. When compared quantitatively, there were no substantial differences between rat, dog, and monkey. The ability of allometric scaling to predict human Vd was quantitatively similar to that of the preclinical species, whereas allometry exhibited a slightly higher MAE than any of the preclinical species alone. Although the rat had the lowest incidence of successful qualitative predictions and comparable quantitative predictions compared with the other preclinical species, the overall MAE associated with predictions from the rat were the lowest, followed by dog, monkey, and allometry. Even in the worst case, MAE from any single species was only approximately 100% (i.e., 2-fold error).
Accuracy of human Vd predictions from preclinical species using various methodologies
Finally, the data-dependent Vd molecular associations (Jolivette and Ward, 2005) were applied to the Vd predictions (Table 6). After application of the associations to the dog data to estimate human Vd, the number of “no predictions” between the qualitative and quantitative predictions remained the same; however, all of the qualitative inlier predictions became quantitative outlier predictions. Overall, the successful qualitative outcomes for predicting human Vd ranged from 57 to 78% of the time, whereas successful quantitative predictions were calculated to be accurate only 33 to 89% of the time. The performance of the molecular associations was somewhat disappointing when assembled by Vd class qualitatively, with no substantial improvement in predictivity for dog and monkey, and a decrease in predictivity for rat.
Evaluation of the utility of data-dependent molecular property associations applied to enhance preclinical predictivity of human Vd
Additionally, the MAE between predicted inliers and outliers is not discriminated appropriately from the overall dataset, regardless of species or prediction methodology. Similarly, the number of correct quantitative predictions from rat and dog also decreased compared with the other prediction methodologies described above, although the MAE for the predicted outliers was increased compared with the entire dataset. However, a substantial improvement in percentage correct was noted for quantitative predictions from monkey, where nearly 90% of the predictions were correct and the MAE for the predicted inliers was decreased compared with the entire dataset.
Human Mean Residence Time Predictions. Prediction of human MRT was qualitatively most accurate from monkey predictive approaches, whereas quantitatively, predictions were similar across all three preclinical species, with dog slightly more predictive (Table 7). Qualitatively and quantitatively, the least accurate results in human MRT predictions were obtained from three-species allometry-derived Vd and CL values. Human MRT predictions from rat yielded the smallest error, whereas error increased from dog, monkey, to allometry. Interestingly, amiodarone was responsible for a large amount of the MAE across all predictive methods, and removal of amiodarone from the calculations resulted in 60 to 80% decreases in the MAE for the various predictive methods. Overall, with amiodarone included, the predictions from allometry yielded the largest error. Interestingly, allometry-based prediction of human MRT trended to overpredict; specifically, this methodology overpredicted human MRT for 13 of the 21 compounds (61.9%).
Accuracy of human MRT predictions from preclinical species using various methodologies

Comparison of human Vd with Vd in rat (A), dog (B), and monkey (C). Vd is expressed in each species in l/kg; the solid line represents the line of unity and the dashed lines represent ±total body water (0.7 l/kg). Where necessary, Vd greater than 10 times total body water (>7 l/kg) was truncated to 7 l/kg for graphical depiction.
Discussion
Recently, this laboratory has performed extensive analyses around the in vivo pharmacokinetics of a 103-compound dataset in the rat, dog, monkey, and human (Nagilla and Ward, 2004; Ward and Smith, 2004a,b; Jolivette and Ward, 2005). From these analyses have arisen some intriguing conclusions, including the general utility of nonhuman primate data in lead optimization, the superiority of CL scaling from monkey data alone compared with allometric scaling, and the ability to apply “rules” based on two-dimensional molecular features to assess human predictivity from preclinical data. However, although the aforementioned dataset is the largest such species-comprehensive dataset compiled to date, the size of the dataset was particularly limited by data availability (particularly monkey data). Furthermore, although quite diverse, the original dataset was weighted somewhat toward lower molecular weight, less lipophilic molecules; average molecular weight and clogP values were 368.6 g/mol and 0.951, respectively. As noted in several surveys (Lipinski, 2000; Wenlock et al., 2003), this is somewhat smaller and more hydrophilic than many modern combinatorial libraries and successful drugs, and inclusion of more molecules with higher molecular weights and lipophilicity would be beneficial. The present investigation has fulfilled both of these criteria, and has substantially improved the diversity of the overall dataset. Nineteen of these 21 added compounds demonstrated molecular weights >300 g/mol or clogP >3, and 16 met both criteria; average molecular weight and clogP values for this dataset were 393.7 g/mol and 3.87, respectively. Although this increase in molecular weight between the two datasets is not particularly noteworthy, a 4-fold increase in clogP between the two datasets was demonstrated. Interestingly, introduction of this physiochemical diversity also inadvertently resulted in improved pharmacokinetic diversity. Whereas the previous dataset was more heavily weighted toward low CL compounds (53% of all compounds in the preclinical species and 65% of the compounds in human), the present dataset contained far fewer low CL compounds (14–33% in the preclinical species and 43% in human). Such diversity allowed a detailed interrogation of whether the observations from the original dataset would apply to molecules with these particular features.
In short, virtually all of the major findings from the previous dataset were confirmed and validated in this extended, more diverse test set. Among the major preclinical species, monkey afforded the most accurate prediction of human CL, and generating both dog and monkey added only minimal value to monkey data alone. Regarding scaling methodologies, the rule of exponent correction to allometric scaling improved CL predictivity compared with simple allometry, offering the same predictivity as monkey LBF scaling. Predictions from monkey LBF alone are as good as the rule of exponents but have the virtue of being from one species rather than three. Interestingly, application of the rule of exponents correction for vinorelbine worsened the human CL prediction, increasing the error from human nearly 2-fold compared with simple three-species allometry. This observation supports recent questions raised around allometry and the intrinsic defect associated with this correction method (Tang and Mayersohn, 2005).
Likewise, human Vd was similarly well predicted from any of the preclinical species, and allometry also provided comparable Vd predictivity. Finally and most importantly, the value of both the data-independent and data-dependent molecular associations for assigning preclinical predictivity was confirmed, especially for CL. Given the dataset concerns described above, the successful application of these findings to the present data are particularly encouraging. These observations lend further support to the continued use of nonhuman primates in pharmacokinetic lead optimization, and call further into question the need to generate both dog and monkey data in a lead optimization paradigm.

Discrimination of MAE for qualitative and quantitative human CL predictions after application of the molecular property associations to preclinical data from rat, dog, and monkey. Bars extending above the line represent MAE greater than that of the entire dataset; bars extending below the line have MAE values less than that of the entire dataset.
Despite the clear successes of this test set exercise, not all of the observations from the previous dataset applied here. First, whereas LBF scaling from rat and dog to human CL resulted in similar predictivity as in the original dataset, predictivity from monkey LBF scaling was somewhat lower in this dataset (∼48%) compared with the original dataset (68%). However, similar to the findings in the previous exercise, the error associated with the predictivity, in terms of MAE, was smallest from monkey LBF scaling when compared with that of the other preclinical species and equal to exponent rule-corrected allometry predictions. Although this decreased predictivity in the monkey was somewhat offset in the overall MAE as well as by the application of the molecular features analysis, it is interesting that these putatively more discovery-like molecules demonstrated more difficulty in extrapolating to human pharmacokinetics, and may suggest increased caution in future human predictions with these types of molecules. In addition, not all of the previously identified molecular associations applied as well to the present dataset. In particular, the monkey qualitative CL predictions and all of the quantitative volume predictions were not as useful as hoped; this could be due in part to some of the “borderline” molecule categorizations, specifically Vd classifications of methadone and remoxipride, which were borderline in multiple species. The importance in monkey data for prediction of human MRT was confirmed from the previous analysis.
Regardless of these limitations, the impact of the successful application of the molecular property “rules” (Jolivette and Ward, 2005) bears further emphasis. In nearly every case applied, the combination of two-dimensional molecular features with or without preclinical pharmacokinetic data provided a clear MAE discrimination between predicted human inliers and outliers (Fig. 5). This discrimination can be seen graphically in the fold difference in MAE between the MAE of correctly predicted extrapolative inliers and outliers compared with the MAE from the LBF scaling approach, where the predicted inliers possess ratios <1 (decreased MAE), and predicted outliers have ratios >1 (increased MAE). Furthermore, application of the data-dependent CL rules substantially improved the value of the rat and dog data, rendering these species even more predictive than the monkey (albeit the inlier prediction applied to fewer molecules). Numerous examples of the potential utility of such validated rules can be envisioned, in a drug discovery setting, for instance, as an aid for scientists to make more informed compound progression decisions early in the drug discovery process. A common practice in the pharmaceutical industry is the use of in vivo pharmacokinetic screening in the rat to make “go/no go” progression decisions during lead optimization (Korfmacher et al., 2001; Caldwell et al., 2004); and, as such, the progression of eight compounds in this dataset that demonstrate high CL in the rat would most likely have been terminated or de-prioritized. However, application of the data-dependent associations would correctly indicate that ∼40% of these compounds, i.e., chlorpromazine, methadone, and remoxipride, do not demonstrate high clearance in humans. Furthermore, in cases where molecular features indicate that the observed rat CL for a compound is likely to accurately extrapolate human CL, the need for additional animal studies would not be required to project human CL, resulting in a net reduction in animals and animal studies utilized in the drug discovery process. Additionally, the approach of combining in vivo and in silico methodologies is practically unprecedented; although previous studies have explored combining some aspects of in vivo and in silico data (Wajima et al., 2003), none have used the data in as direct a manner as exemplified here. This combination approach clearly adds substantial context and value to predictions made from preclinical data, and merits further consideration.
In summary, the findings in this investigation support the application of previous studies to molecules with more discovery-like molecular features, particularly with respect to the use of nonhuman primate data in pharmacokinetic screening and the application of two-dimensional molecular features analysis to improve preclinical predictivity of human pharmacokinetics, particularly the data-dependent associations. The relationships derived for this 21-compound dataset should be considered preliminary since differences between the two compound datasets were already noted; further refinement to these approaches may be necessary as more compounds can be included in the analysis. These observations also support the value of alternative approaches to allometric scaling in predicting human CL, Vd, and MRT, where comparable predictive power was noted from monkey alone, and in some cases, superior predictivity of human PK was observed. The ability to accurately estimate human PK in fewer preclinical species certainly represents an attractive resource savings; compounds can be selected to progress into selected species based on their molecular features and predicted PK, e.g., conducting PK studies in the rat and not in the monkey for compounds with molecular properties associated with successful extrapolation of human CL from rat data and unsuccessful extrapolation from monkey data, thereby resulting in fewer in vivo PK studies. Finally, the predictive power of combining in silico and in vivo approaches is exemplified. Future investigations in this laboratory will continue to focus on the integration of diverse data to form a comprehensive human pharmacokinetic prediction paradigm, particularly the use of in vitro plasma protein binding and metabolic stability data.
Appendix I: Pharmacokinetic parameters of compounds used in the present investigation
Units for CL and Vd are ml/min/kg and l/kg, respectively.
Appendix II:Calculated two-dimensional molecular properties of compounds used in the present investigation
Acknowledgments
We gratefully acknowledge Len Azzarano, Kelly Frank, Lauren Hardy, Melanie Nord, Katrina Rivera, and Rebecca Trejo for conducting the in vivo portions of this study; and Stephen Eisennagel, Harvey Fries, and Laura Suhadolnik for completing the bioanalytical portions of this study.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.105.006619.
-
ABBREVIATIONS: CL, clearance (ml/min/kg); BrW, brain weight (g); clogP, calculated logarithm of the octanol-water partition coefficient; CMR, calculated molar refractivity (ml/mol); HBA, number of hydrogen bond acceptors; HBD, number of hydrogen bond donors; LBF, liver blood flow; MAE, mean absolute error; MLP, maximum life span potential (years); MRT, mean residence time (minutes); nrot, number of rotatable bonds; PSA, polar surface area (Å2); Vd, volume of distribution (l/kg); PK, pharmacokinetic(s).
- Received July 20, 2005.
- Accepted April 12, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}