


Abstract
Glucuronidation of morphine in humans is predominantly catalyzed by UDP-glucuronosyltransferase 2B7 (UGT2B7). Since our recent research suggested that cytochrome P450s (P450s) interact with UGT2B7 to affect its function [Takeda S et al. (2005) Mol Pharmacol 67:665–672], P450 inhibitors are expected to modulate UGT2B7-catalyzed activity. To address this issue, we investigated the effects of P450 inhibitors (cimetidine, sulfaphenazole, erythromycin, nifedipine, and ketoconazole) on the UGT2B7-catalyzed formation of morphine-3-glucuronide (M-3-G) and morphine-6-glucuronide (M-6-G). Among the inhibitors tested, ketoconazole was the most potent inhibitor of both M-3-G and M-6-G formation by human liver microsomes. The others were less effective except that nifedipine exhibited an inhibitory effect on M-6-G formation comparable to that by ketoconazole. Neither addition of NADPH nor solubilization of liver microsomes affected the ability of ketoconazole to inhibit morphine glucuronidation. In addition, ketoconazole had an ability to inhibit morphine UGT activity of recombinant UGT2B7 freed from P450. Kinetic analysis suggested that the ketoconazole-produced inhibition of morphine glucuronidation involves a mixed-type mechanism. Codeine potentiated inhibition of morphine glucuronidation by ketoconazole. In contrast, addition of another substrate, testosterone, showed no or a minor effect on ketoconazole-produced inhibition of morphine UGT. These results suggest that 1) metabolism of ketoconazole by P450 is not required for inhibition of UGT2B7-catalyzed morphine glucuronidation; and 2) this drug exerts its inhibitory effect on morphine UGT by novel mechanisms involving competitive and noncompetitive inhibition.
A number of endogenous and exogenous compounds are biotransformed by phase I and phase II drug-metabolizing enzymes (Oguri et al., 1994; Radominska-Pandya et al., 1999; Ritter, 2000). The cytochrome P450 (P450) and UDP-glucuronosyltransferase (UGT) families are two major enzyme groups responsible for phase I and II reactions. UGT-catalyzed glucuronidation is assumed to play a role in up to 35% of phase II reactions. UGT is anchored to the endoplasmic reticulum membrane by the transmembrane domain, and the main body of the enzyme, including the catalytic site, is believed to be present on the luminal side of the endoplasmic reticulum. This model of type I topology causes “latency,” a characteristic feature of UGT, whereby UGT activity is activated by disruption of the microsomal membranes with detergent. Among the UGTs identified in humans, UGT2B7 is of particular interest because it is a major isoform involved in morphine glucuronidation (Coffman et al., 1997). To date, UGT2B7 is the sole enzyme reported capable of forming morphine-6-glucuronide (M-6-G) (Coffman et al., 1997), a metabolite having more potent analgesic activity than morphine. Although increasing evidence suggests that there are a number of factors that affect morphine UGT activity (Narayan et al., 1991; Ishii et al., 2001, 2004, 2005; Antonilli et al., 2003; Takeda et al., 2005a,b), little is known about inhibitors of UGT2B7.
Experimental evidence in humans indicates that the glucuronidation of acetaminophen, codeine, zidovudine, carbamazepine, lorazepam, and propafenone is influenced by concomitantly administered drugs (Kiang et al., 2005). However, the glucuronidation of morphine is not affected by any of the drugs examined so far (Kiang et al., 2005). Although diclofenac is a potent inhibitor of codeine glucuronidation in vitro (Ki = 7.9 μM; Ammon et al., 2000), it has only a low ability to inhibit the glucuronidation of morphine by human liver microsomes (IC50 = approximately 500 μM; King et al., 2001). In contrast, using human liver microsomes, it has been reported that ketoconazole, a P450 inhibitor (Wrighton and Ring; 1994), competitively inhibits the glucuronidation of the UGT2B7 substrate zidovudine with a Ki of 80 μM (Sampol et al., 1995). A recent study by Yong et al. (2005), also using human liver microsomes, showed that ketoconazole inhibited UGT1A-mediated glucuronidation of 7-ethyl-10-hydroxycamptothecin (SN-38) and cDNA-expressed UGT1A isoforms. Since glucuronidation of zidovudine is catalyzed by human liver microsomal UGT (Sampol et al., 1995), it is conceivable that UGT2B7 is inhibited by ketoconazole. However, there are no data that demonstrate the direct inhibition of UGT2B7 by ketoconazole.
Since P450s are suggested to interact with UGTs, including UGT2B7, to modulate its function (Taura et al., 2000; Fremont et al., 2005; Ishii et al., 2005; Takeda et al., 2005a,b), it is conceivable that P450 inhibitors modulate UGT2B7-catalyzed reactions. To examine this possibility, we carried out a comparative study to clarify whether P450 inhibitors are able to inhibit morphine UGT activity catalyzed by hepatic microsomes and recombinant UGT2B7. The results indicated that, among the inhibitors tested, ketoconazole is the strongest inhibitor for UGT2B7-mediated morphine glucuronidation.
Materials and Methods
Chemicals and Enzyme Sources. UDP-glucuronic acid (UDPGA, trisodium salt) was purchased from Wako Pure Chemical Ind., Ltd. (Osaka, Japan). Morphine hydrochloride and codeine phosphate were obtained from Takeda Chemical Ind. Co., Ltd. (Osaka, Japan). NADPH, ketoconazole, sulfaphenazole, egg yolk l-α-phosphatidylcholine, saccharolactone, and bovine serum albumin (fraction V) were purchased from Sigma Chemical Co. (St. Louis, MO). Morphine-3-glucuronide (M-3-G) and M-6-G were synthesized in our laboratory (Yoshimura et al., 1968). Cimetidine, erythromycin, and nifedipine were purchased from Nacalai Tesque Co. (Kyoto, Japan). Pooled human liver microsomal samples were obtained together with agreement for their use from donors from BD Gentest (Woburn, MA), and stored at –80°C until use. Recombinant UGT2B7 microsomes (Supersomes) were obtained from BD Gentest. Protein assay reagents were provided by Bio-Rad (Hercules, CA). All other reagents were of analytical grade and used without further purification.
Glucuronidation Assay. Morphine glucuronidation activity was determined by the method described elsewhere (Takeda et al., 2005a). Briefly, the assay mixture consisted of 50 mM Tris-HCl buffer (pH 7.8), 10 mM MgCl2, 0.45 mg protein/ml human liver microsomes or recombinant UGT2B7 microsomes, 2 mM UDPGA, 5 mM saccharolactone, and morphine in a final volume of 300 μl. Morphine was dissolved in H2O and added to the incubation mixture at the desired concentrations. Sonicated l-α-phosphatidylcholine, at a concentration of 0.5 mg/ml, was included in all assays. Solubilization of the microsomes with sodium cholate was performed by the methods described previously (Taura et al., 2000; Takeda et al., 2005a). In a separate experiment, 1 mM NADPH was added to the above reaction mixture. The reaction at 37°C was started by adding ice-cold microsomes and stopped by the addition of 100 μl of ice-cold 1 M trichloroacetic acid; then, the mixture was chilled on ice and centrifuged. An aliquot of the supernatant was analyzed by high-performance liquid chromatography (Ishii et al., 2001). Formation of M-3-G and M-6-G in relation to time and protein concentration was linear under the conditions used. No production of M-3-G and M-6-G was observed in blank experiments which were performed in the absence of UDPGA.
Inhibition Studies. Before the inhibition studies, the apparent Michaelis-Menten values (Km) of M-3-G and M-6-G formation were determined using human liver microsomes and cDNA-expressed UGT2B7 microsomes as the enzyme source. The activity was measured at eight morphine concentrations ranging from 0.1 to 5 mM. The apparent Km of morphine glucuronidation was determined by the Michaelis-Menten equation by the nonlinear regression method using SigmaPlot software (SPSS Inc., Chicago, IL). Inhibition of human liver microsomal UGT-catalyzed morphine glucuronidation by inhibitors was examined at a morphine concentration of 3.8 mM. The inhibitory effects on morphine glucuronidation of cimetidine, sulfaphenazole, erythromycin, nifedipine, and ketoconazole were investigated at concentrations of 6.25, 12.5, 25, 50, 100, and 250 μM. Cimetidine and codeine were dissolved in H2O. The other drugs were dissolved in dimethyl sulfoxide, the concentration of which did not exceed 0.5% (v/v) in the incubation mixture. The reference incubation contained the same volume of solvent. It was confirmed that the 0.5% dimethyl sulfoxide had little effect on morphine glucuronidation (data not shown). Control incubations were carried out by omitting substrate.
Data Analysis. The concentration of the inhibitor that is required to produce 50% inhibition of the enzymatic activity (IC50) was determined from the curves plotting enzymatic activity versus inhibitor concentrations. The inhibition mechanism was estimated from the figure plotting 1/V versus 1/S (Segel, 1993). The apparent inhibitory constant (Ki) was determined from the x-intercept of a replot of the mean slopes of the double reciprocal plot versus inhibitor concentration. The Ki and the mode of inhibition were determined by fitting the kinetic data to a mixed inhibition model using nonlinear regression analysis using the SigmaPlot enzyme kinetics module. The mode of inhibition that best described the data was determined by comparing r2 values for the fit of the various models. The Ki/Km ratio was used to compare the relative inhibition potential by ketoconazole for UGT1A1/9 or UGT2B7-mediated reactions (Murray and Butler, 1996). Differences were considered significant when p value was calculated to be less than 0.05. All statistical analyses were performed by Scheffe's F test, which is a type of posthoc test for analyzing results of ANOVA testing. These calculations were done using Satview5.0J software (SAS Institute Inc., (Cary, NC).

Morphine 3- and 6-glucuronide formation activity of human liver microsomes in the presence of nifedipine and ketoconazole. The activities of M-3-G and M-6-G formation in the absence of inhibitors were 312.3 ± 12.0 and 49.0 ± 5.9 pmol/min/mg protein, respectively. See Materials and Methods for details of the assay method. Each plot represents the mean ± S.D. of triplicate determinations.
Results
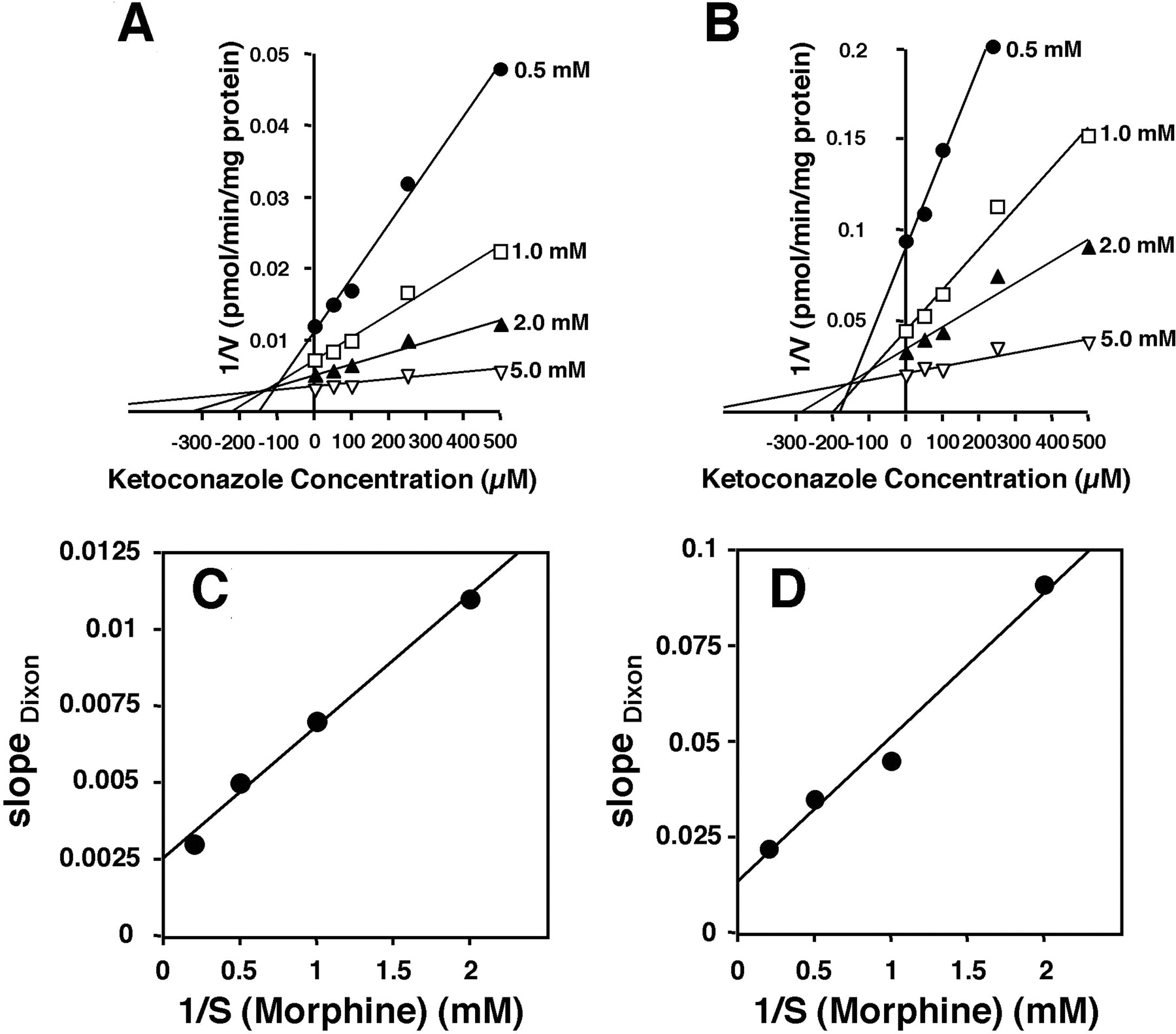
Effects of P450 Inhibitors on Morphine Glucuronidation. The inhibitory effect of five P450 inhibitors, cimetidine, sulfaphenazole, erythromycin, nifedipine, and ketoconazole, on the activity of morphine UGT was examined using human liver microsomes as the enzyme source. From kinetic analyses in the absence of inhibitor, the Km values for M-3-G and M-6-G formation were determined to be 2.66 ± 0.32 and 2.62 ± 0.46 mM, respectively. The apparent Km values for morphine glucuronidation were similar between the human liver microsomes and cDNA-expressed UGT2B7 microsomes (approximately 3 mM). Although M-3-G formation can be mediated by UGT isoforms other than UGT2B7 at high morphine concentrations ( mM) (Court et al., 2003), the pooled human liver microsomes used in this study exhibited apparent monophasic Michaelis-Menten kinetics. The monophasic nature of M-3-G formation is reasonable because morphine is a highly specific substrate of UGT2B7 and is poorly glucuronidated by other UGTs (Coffman et al., 1997; Court et al., 2003; Stone et al., 2003). These results were consistent with the reports by other workers (Fisher et al., 2000; Court et al., 2003). The effects of the five drugs on the activity of morphine glucuronidation by human liver microsomes were determined at a morphine concentration around the Km. Ketoconazole inhibited both M-3-G and M-6-G formation, with apparent IC50 values of 196 and 168 μM, respectively (Fig. 1). However, cimetidine, sulfaphenazole, and erythromycin did not exhibit any inhibitory effect on morphine glucuronidation (data not shown). Although nifedipine produced a reduction in M-6-G formation (IC50 186 μM), the IC50 value for M-3-G formation could not be determined because of the weak inhibition (Fig. 1). It should be noted that there have been no reports describing the glucuronidation of ketoconazole and nifedipine by UGTs including UGT2B7. Figure 2A illustrates the Dixon plots for the inhibition of M-3-G formation by ketoconazole. The Dixon plot was characterized by straight lines at different fixed substrate concentrations intersecting in the second quadrant (Fig. 2A). The Dixon plot was not able to distinguish between the inhibition types. A replot of the slopes produced a straight line not going through the origin, suggesting a “linear mixed-type” mode of inhibition (Fig. 2C) (Segel, 1993). Double reciprocal plots of the substrate concentration data (1/V versus 1/S) in the presence of fixed concentrations of ketoconazole showed that both the slopes and the y-intercepts increased along with increasing inhibitor concentrations, consistent with a “mixed-type” mechanism, and this was also found for the M-6-G inhibition (data not shown) (Segel, 1993). The Ki of ketoconazole for M-3-G formation was calculated to be 118 ± 27 μM. In the case of the M-6-G formation catalyzed by human liver microsomal UGT, ketoconazole also exhibited a linear mixed-type inhibition (Fig. 2, B and D) with an apparent Ki of 108 ± 35 μM.
mM) (Court et al., 2003), the pooled human liver microsomes used in this study exhibited apparent monophasic Michaelis-Menten kinetics. The monophasic nature of M-3-G formation is reasonable because morphine is a highly specific substrate of UGT2B7 and is poorly glucuronidated by other UGTs (Coffman et al., 1997; Court et al., 2003; Stone et al., 2003). These results were consistent with the reports by other workers (Fisher et al., 2000; Court et al., 2003). The effects of the five drugs on the activity of morphine glucuronidation by human liver microsomes were determined at a morphine concentration around the Km. Ketoconazole inhibited both M-3-G and M-6-G formation, with apparent IC50 values of 196 and 168 μM, respectively (Fig. 1). However, cimetidine, sulfaphenazole, and erythromycin did not exhibit any inhibitory effect on morphine glucuronidation (data not shown). Although nifedipine produced a reduction in M-6-G formation (IC50 186 μM), the IC50 value for M-3-G formation could not be determined because of the weak inhibition (Fig. 1). It should be noted that there have been no reports describing the glucuronidation of ketoconazole and nifedipine by UGTs including UGT2B7. Figure 2A illustrates the Dixon plots for the inhibition of M-3-G formation by ketoconazole. The Dixon plot was characterized by straight lines at different fixed substrate concentrations intersecting in the second quadrant (Fig. 2A). The Dixon plot was not able to distinguish between the inhibition types. A replot of the slopes produced a straight line not going through the origin, suggesting a “linear mixed-type” mode of inhibition (Fig. 2C) (Segel, 1993). Double reciprocal plots of the substrate concentration data (1/V versus 1/S) in the presence of fixed concentrations of ketoconazole showed that both the slopes and the y-intercepts increased along with increasing inhibitor concentrations, consistent with a “mixed-type” mechanism, and this was also found for the M-6-G inhibition (data not shown) (Segel, 1993). The Ki of ketoconazole for M-3-G formation was calculated to be 118 ± 27 μM. In the case of the M-6-G formation catalyzed by human liver microsomal UGT, ketoconazole also exhibited a linear mixed-type inhibition (Fig. 2, B and D) with an apparent Ki of 108 ± 35 μM.

Dixon plots of the inhibition by ketoconazole of morphine glucuronidation catalyzed by human liver microsomes. Effect of ketoconazole (25 μMto500 μM) on the M-3-G (A) and M-6-G (B) formation was estimated using four concentrations of morphine (closed circles, 0.5 mM; open squares, 1.0 mM; closed triangles, 2.0 mM; and open triangles, 5.0 mM). Replots of the slopes of Dixon plot (slopes versus 1/[S]) are shown in C (M-3-G) and D (M-6-G). Details of the assay conditions are described under Materials and Methods. Each plot represents the mean of duplicate measurements. In experiment B, M-6-G formation in the presence of 0.5 mM morphine and 0.5 mM ketoconazole was not determined because of the low activity.
Effect of NADPH on the Inhibition of Morphine Glucuronidation by Ketoconazole. It has been suggested that the inhibition of CYP2D6 by cimetidine is likely due to a metabolite(s) rather than the parent compound (Madeira et al., 2004). Similarly, antifungal compounds are suggested to exert their inhibitory effects on P450s through oxidative metabolism by P450 (Isoherranen et al., 2004). To examine the hypothesis that the potency of morphine UGT inhibitors is also modified by oxidative metabolism, we examined the inhibitory effects of cimetidine and ketoconazole in the presence of NADPH, an obligate cofactor of P450. The results showed that cimetidine does not exhibit any inhibitory effect on the activity of hepatic microsomal morphine UGT, even in the presence of NADPH (Fig. 3A). Similarly, NADPH did not influence the inhibitory effect of ketoconazole (Fig. 3A). This result does not support the possibility that ketoconazole requires oxidative metabolism to inhibit morphine glucuronidation by human liver microsomes.
Inhibition by Ketoconazole of Morphine Glucuronidation Catalyzed by Solubilized Microsomes and Recombinant UGT. To examine the possibility that the condition of the microsomal membrane may affect the inhibition of morphine glucuronidation, an inhibition study was performed for cimetidine and ketoconazole using solubilized microsomes. The results (Fig. 3B) showed that ketoconazole inhibits the M-3-G and M-6-G formation catalyzed by solubilized microsomes in a concentration-dependent manner, with IC50 values comparable to those obtained after incubation with intact microsomes (Fig. 1). Cimetidine had hardly any effect on the activity obtained with both microsomal preparations (data not shown). When recombinant UGT2B7 expressed in insect cells was used as the enzyme, ketoconazole (100 μM) significantly reduced morphine glucuronidation at both the 3- and 6-positions (Fig. 3C). This observation, together with the data showing that NADPH failed to affect the inhibitory effect of ketoconazole (Fig. 3), suggested that ketoconazole directly inhibits UGT2B7-catalyzed morphine glucuronidation without requiring preactivation by P450.
Effect of Codeine and Testosterone on the Inhibition by Ketoconazole of Morphine Glucuronidation. As shown in Fig. 2, the inhibition mode of ketoconazole for M-3-G and M-6-G formation was suggested to be a mixed type consisting of competitive and noncompetitive inhibition. Since codeine is a substrate for UGT2B7 (Coffman et al., 1997), it is reasonable to expect that the competitive phase in ketoconazole-produced inhibition of morphine glucuronidation should be additively affected by codeine. To assess this possibility, codeine was added to the incubation mixture containing pooled human liver microsomes, ketoconazole, UDPGA, and morphine. When codeine was added to the incubation mixture not containing ketoconazole, higher concentrations (over 2.5 mM) of this opioid were needed for the inhibition of both M-3-G and M-6-G formation (Fig. 4, A and B). Codeine at lower concentrations (below 1.0 mM) did not exert any inhibitory effect (Fig. 4, A and B). However, in the presence of ketoconazole, codeine reduced the activities of M-3-G and M-6-G formation more markedly than did the opioid alone. Such an effect of codeine was observed even at the lowest concentration (0.25 mM). Similarly, the degree of the inhibition with ketoconazole plus codeine was significantly greater than that of ketoconazole alone (Fig. 4, A and B). The combination of ketoconazole and codeine seemed to produce an inhibitory effect that was synergic rather than additive. On the contrary, testosterone, another UGT2B7 substrate, exhibited a quite different effect from codeine on ketoconazole inhibition. Although testosterone itself inhibited morphine glucuronidation, this compound hardly exhibited any additive and/or synergic effect on ketoconazole-produced inhibition (Fig. 4, C and D). As far as M-6-G formation was concerned, testosterone significantly weakened the inhibitory effect of ketoconazole (Fig. 4, A and B). Taken together, these results suggested that ketoconazole affects not only the morphine binding site in UGT2B7 but also other possible effector site(s).

Inhibitory effect of ketoconazole on morphine glucuronidation by solubilized (A) and NADPH-supplemented human liver microsomes (B), and on the activity catalyzed by UGT2B7 expressed in insect cells (C). In A, the effect of NADPH on the ketoconazole-induced inhibition of morphine glucuronidation by human liver microsomes was assessed. The values are expressed as a percentage of the control which was determined in the absence of NADPH or ketoconazole (UDPGA alone). Each bar represents the mean ± S.D. of triplicate determinations. *, significantly different from UDPGA alone (p < 0.01). Control (UDPGA alone) activities of M-3-G and M-6-G formation were 352.0 ± 9.4 and 48.6 ± 3.5 pmol/min/mg protein, respectively. In B, morphine 3- and 6-glucuronidation was determined using solubilized human liver microsomes in the presence of ketoconazole. The values are expressed as percentages relative to the control, which was determined in the absence of ketoconazole. Each plot represents the mean ± S.D. of triplicate determinations. In C, incubation was performed with insect cell microsomes expressing recombinant UGT2B7, 3.8 mM morphine, and 2 mM UDPGA for 60 min. The values are expressed as percentages relative to the control, which was determined in the absence of ketoconazole. Details of the assay conditions are described under Materials and Methods. Each bar represents the mean ± S.D. of triplicate determinations. *, significantly different compared with the control group (p < 0.01). Control activities of M-3-G and M-6-G formation were 351.4 ± 7.0 and 53.2 ± 6.2 pmol/min/mg protein, respectively.
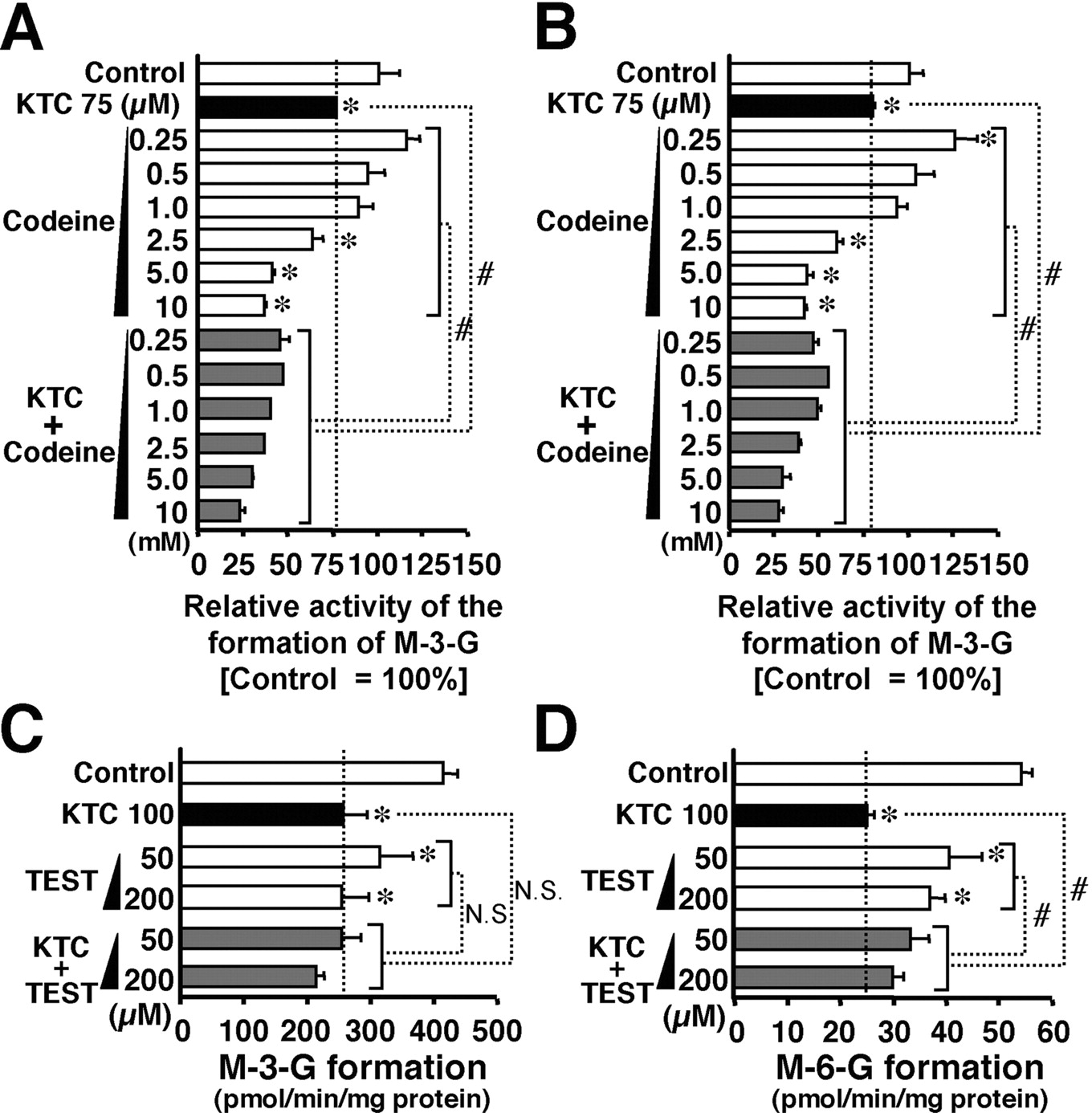
Effects of codeine and testosterone on the inhibition of human liver microsomal morphine glucuronidation by ketoconazole. In A, and B, morphine glucuronidation (A, M-3-G; B, M-6-G) by pooled human liver microsomes was examined in the presence of either ketoconazole (KTC), codeine, or their combination. Control activities of M-3-G and M-6-G formation were 293.2 ± 16.3 and 54.4 ± 12.4 pmol/min/mg protein, respectively. In C and D, morphine glucuronidation (C, M-3-G; D, M-6-G) by pooled human liver microsomes was examined in the presence of either ketoconazole (KTC), testosterone (TEST), or their combination. Inhibitor concentrations are shown in the figure. Details of the assay conditions are described under Materials and Methods. Each bar represents the mean ± S.D. (triplicate determinations) of the relative activity to the control. *, significantly different (p < 0.05) from control, which was determined in the absence of inhibitor. #, significantly different (p < 0.05) from the activity in the presence of KTC alone, codeine alone, or TEST alone. In A and B, the comparison of codeine alone versus codeine plus KTC was made for pairs with the same concentration of codeine. In C and D, the comparison of TEST alone versus TEST plus KTC was made for pairs with the same concentration of TEST. N.S., not significant.
Discussion
Several drugs have been reported thus far as inhibitors of UGT-mediated glucuronidation (Kiang et al., 2005). However, to our best knowledge, UGT-selective inhibitors have not been known. For example, diclofenac is a potent UGT inhibitor, but the effect is not selective to this enzyme (Kiang et al., 2005). Since CYP3A4 and other forms of P450 interact with UGT2B7 to modulate the function (Taura et al., 2000; Fremont et al., 2005; Ishii et al., 2005; Takeda et al., 2005a,b), it seemed worthwhile to investigate whether the inhibition of P450 function gives rise to a reduction in morphine glucuronidation. This was examined in this study by using P450 inhibitors, including ketoconazole, capable of inhibiting CYP3A4 and other forms of P450 (Ong et al., 2000). However, the above hypothesis was not supported by observations that ketoconazole inhibits morphine glucuronidation catalyzed by a UGT2B7 preparation not containing P450 (Fig. 3C), and NADPH does not cause any modification toward the ketoconazole inhibition of morphine glucuronidation by hepatic microsomes (Fig. 3A). However, NADPH itself produced a minor, but significant, increase in M-3-G formation by human liver microsomes. The mechanism whereby NADPH stimulates UGT function remains to be determined.
Biphasic kinetics of M-3-G formation by human liver microsomes and/or recombinant UGT2B7 has been reported (Miners et al., 1988; Stone et al., 2003), although neither our experiments nor those of other workers observed the same form of kinetics (Fisher et al., 2000; Court et al., 2003; Takeda et al., 2005a). The Km values of the low- and high-affinity phases in the kinetics of M-3-G formation have been reported to be 2.5 μM and 798 μM, respectively (Miners et al., 1988). If the kinetics of UGT2B7-mediated glucuronidation is biphasic, the inhibition by ketoconazole of UGT2B7 function observed in our study may only affect the low-affinity phase reaction.
Inhibitory effects of ketoconazole on UGT function that have been reported so far are summarized in Table 1. On the basis of data shown in this article, the mode of inhibition of M-3-G and M-6-G formation by ketoconazole is suggested to be a mixed type (Fig. 2; computer fitting program, Table 1). Since ketoconazole is a CYP3A4 inhibitor and morphine is a substrate of both CYP3A4 (Projean et al., 2003) and UGT2B7, it is conceivable that ketoconazole shares structural similarities with morphine and functions as a competitive inhibitor for morphine glucuronidation. If ketoconazole only affects substrate binding to the catalytic site, addition of codeine, which is a UGT2B7 substrate, would result in additive inhibitory effects. However, although codeine itself had no effect on morphine UGT activity at 0.25 to 1.0 mM concentrations, the same concentrations of codeine potentiated ketoconazole-produced inhibition. Thus, codeine is suggested to modify enzyme structure to facilitate the inhibitory effect by ketoconazole. Similarly, binding of testosterone may alter UGT structure so that the inhibitory effect of ketoconazole is decreased somehow. It has been suggested that UGT2B7 has an opioid binding site in its N-terminal domain (Coffman et al., 2001). In addition, a possible steroid binding site has been proposed in UGT2B7 (Coffman et al., 2001). Taken together, it is suggested that ketoconazole binds to a site(s) distinct from opioid- and testosterone-binding sites in the UGT2B7. If this is true, the following may be the reason codeine synergistically enhances the inhibitory effect of ketoconazole: that is, ketoconazole may bind to its specific site to modulate efficacy and/or specificity in the interaction of opioid and its association site. Ketoconazole is suggested to be a competitive inhibitor of zidovudine UGT in human liver microsomes (Sampol et al., 1995) (Table 1). Since zidovudine glucuronidation is catalyzed by UGT2B7 (Court et al., 2003), it is reasonable to consider that ketoconazole also directly inhibits UGT2B7-catalyzed zidovudine glucuronidation. Although ketoconazole is not a UGT substrate (Yong et al., 2005), this drug appears to bind to the substrate binding site of UGT2B7. This assumption is supported by our present data together with the above information showing that ketoconazole inhibits UGT2B7 in a competitive manner. Further studies are necessary to clarify the clinical impact and significance of the ketoconazole-produced inhibition of UGT.
Kinetic parameters for the in vitro inhibition of UGT-mediated glucuronidation by ketoconazole
The apparent Km and Ki values of morphine UGT-catalyzing M-3-G/M-6-G formation were determined using microsomal preparations from pooled human liver, as described under Materials and Methods.
The Ki values of ketoconazole for M-3-G and M-6-G formation were far greater than the maximal serum concentrations (7.9–11.7 μM, 1–2 h) of this antifungal agent given to patients at a dose of 200 mg/man/day (Huang et al., 1986). However, several lines of investigation described below seem to support the in vivo inhibitory effect of ketoconazole. First, the Ki values of ketoconazole for the inhibition of M-3-G and M-6-G formation were close to the Ki (80 μM) seen for the inhibition of zidovudine glucuronidation (Sampol et al., 1995). In addition, fluconazole, another azole drug, is known to affect the bioavailability of zidovudine in vivo (Kiang et al., 2005). Second, azole antifungal agents including ketoconazole are effectively taken up from plasma into liver cells, and this leads to an increase in the hepatic concentration of the inhibitor to a level that is much higher than the plasma concentrations (von Moltke et al., 1998). This observation supports the view that ketoconazole inhibits UGT activity in vivo, although its plasma concentration is lower than the Ki. In support of this view, it has been reported that ketoconazole significantly increases the in vivo level of active metabolite (SN-38) of irinotecan, an anticancer drug (Yong et al., 2005). When the Ki/Km ratio is used as an index (Murray and Butler, 1996), ketoconazole seems to be a more potent inhibitor for morphine glucuronidation than SN-38 glucuronidation (Table 1). Further studies are necessary to clarify the clinical impact (and significance) by ketoconazole.
Human hepatic UGT1A1 and 1A3 also catalyze morphine-3-glucuronidation, although their activities are lower than that of UGT2B7 (Stone et al., 2003). Therefore, it is reasonable to assume that the estimated Ki of ketoconazole with regard to morphine UGT activity by human liver microsomes is a combination of Ki values toward different UGTs. Therefore, the combined Ki should vary as a function of the molar ratio of the UGT1A1, 1A3, and 2B7 expressed in the liver. However, due to the lower expression of UGT1A3 mRNA in the liver (5- to 20-fold less than other UGT1As; Mojarrabi et al., 1996), this UGT would not substantially contribute to the glucuronidation of morphine. The contribution of UGT1A1 to M-3-G formation is also less than expected (Stone et al., 2003). It has been reported that whereas neither UGT1A1 nor UGT1A3 catalyzes M-6-G formation (Stone et al., 2003), UGT2B7 catalyzes this reaction (Coffman et al., 1997). Thus, it is suggested that the Ki of ketoconazole for M-6-G formation reflects the effect on the UGT2B7-catalyzed reaction.
It has been reported that estradiol-17-glucuronidation by human liver UGTs is modulated by polymethoxyflavonoids, nobiletin and tangeretin (Williams et al., 2002). In this report, enzyme activities were used as a marker for UGT2B7. However, nobiletin and tangeretin (100 μM) had no effect on the activity of human microsomal morphine-UGT (data not shown). Thus, it seems that the inhibitory effect of ketoconazole to UGT2B7 is relatively specific for morphine glucuronidation. In this study, although we focused on the inhibitory effect of P450 inhibitor toward morphine-UGT (UGT2B7), the activity of human liver microsomal morphine-UGT was a little but significantly stimulated by erythromycin and sulfaphenazole (data not shown). It has been suggested that estradiol-3-glucuronidation activity by human liver UGT1A1 is stimulated by the substrate of this UGT (Williams et al., 2002). Thus, it is likely that the interaction of P450 substrates and UGTs including UGT2B7 is complex.
Taking all these findings into consideration, we have shown that ketoconazole is a potent inhibitor of UGT2B7-catalyzed morphine glucuronidation in vitro as well as of P450-catalyzed oxidation. Our findings also suggest that further studies are needed to identify potential drug-drug interactions based on the inhibition and stimulation of UGT.
Footnotes
-
This work was supported in part by a Grant-in-Aid for Scientific Research (C) (Research No. 17590128, recipient Y.I.) from the Japan Society for Promotion of Science.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.009738.
-
ABBREVIATIONS: P450, cytochrome P450; UGT, UDP-glucuronosyltransferase; UDPGA, UDP-glucuronic acid; M-3-G, morphine-3-glucuronide; M-6-G, morphine-6-glucuronide; SN-38, 7-ethyl-10-hydroxycamptothecin.
- Received February 6, 2006.
- Accepted April 25, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}