


Abstract
Raltegravir is a potent human immunodeficiency virus 1 (HIV-1) integrase strand transfer inhibitor that is being developed as a novel anti-AIDS drug. The absorption, metabolism, and excretion of raltegravir were studied in healthy volunteers after a single oral dose of 200 mg (200 μCi) of [14C]raltegravir. Plasma, urine, and fecal samples were collected at specified intervals up to 240 h postdose, and the samples were analyzed for total radioactivity, parent compound, and metabolites. Radioactivity was eliminated in substantial amounts in both urine (32%) and feces (51%). The elimination of radioactivity was rapid, since the majority of the recovered dose was attributable to samples collected through 24 h. In extracts of urine, two components were detected and were identified as raltegravir and the glucuronide of raltegravir (M2), and each accounted for 9% and 23% of the dose recovered in urine, respectively. Only a single radioactive peak, which was identified as raltegravir, was detected in fecal extracts; raltegravir in feces is believed to be derived, at least in part, from the hydrolysis of M2 secreted in bile, as demonstrated in rats. The major entity in plasma was raltegravir, which represented 70% of the total radioactivity, with the remaining radioactivity accounted for by M2. Studies using cDNA-expressed UDP-glucuronosyltransferases (UGTs), form-selective chemical inhibitors, and correlation analysis indicated that UGT1A1 was the main UGT isoform responsible for the formation of M2. Collectively, the data indicate that the major mechanism of clearance of raltegravir in humans is UGT1A1-mediated glucuronidation.
HIV-1 is the etiologic agent of AIDS. HIV infection continues to be a major problem with more than 40 million individuals currently infected with the virus worldwide ([UNAIDS] Joint United Nations Programme on HIV/AIDS 2006 Report on the global AIDS epidemic. http://www.unaids.org). The current standard of care for treating HIV infection, called HAART, is a regimen typically consisting of three or more drugs from two or more available classes. Current HAART medications (of which there are >20) include members from four classes of drugs: nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, protease inhibitors, and fusion inhibitors. Although the advent of HAART has significantly reduced AIDS-related morbidity and mortality, it has been estimated that 78% of treatment-naive patients harbor viruses that are resistant to one or more of the three classes (nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, and protease inhibitors) (Richman, 2001; Little et al., 2002). Because of this factor and issues of tolerability, toxicity, and patient noncompliance due to the rigorous drug administration schedules, there is a critical need for new HIV therapies capable of addressing the deficiencies inherent with currently used drugs.
Integrase is one of the three HIV-1 enzymes required for viral replication (Esposito and Craigie, 1999; Asante-Appiah and Skalka, 1999) and catalyzes the stepwise process, which results in the integration of the HIV-1 DNA into the genome of the host cell. This ordered series of reactions includes the assembly of integrase in a stable complex with the viral DNA, the endonucleolytic processing of the viral DNA ends, and the subsequent strand transfer or joining of the viral and cellular DNAs. Integration is required for stable maintenance of the viral genome as well as for efficient viral gene expression. To date there are no approved drugs targeting this enzyme. Raltegravir is an HIV-1 integrase DNA strand transfer inhibitor that has potent in vitro activity against HIV-1 (IC95 = 33 nM in 50% human serum). Raltegravir has been shown to be efficacious and well tolerated when administered to treatment-naive patients over a 10-day period (Markowitz et al., 2006). Due to the unique and specific mechanism of action (no integrase homolog in humans), it is anticipated that raltegravir will be a safe addition or alternative to other antiretroviral therapies. Understanding the metabolism and disposition of a new drug is an integral part of the safety evaluation of a new chemical entity and is crucial to the safe use of drugs such as raltegravir, which would be used in a poly-therapy setting. Therefore, the objectives of this study were to investigate the pharmacokinetics, metabolism, and routes of excretion of this novel drug.

The structures of [14C]raltegravir and its glucuronide metabolite (M2).
Materials and Methods
Chemicals. [14C]Raltegravir and the glucuronide metabolite of raltegravir (M2) were synthesized at Merck Research Laboratories (Rahway, NJ). The C-14 in [14C]raltegravir was incorporated at the C-2 carbon on the substituted pyrimidinone ring (Fig. 1) and was supplied as the potassium salt. All other materials were of HPLC or analytical grade.
Liver Microsomes and Enzymes. A pool of human liver microsomes was obtained from XenoTech, LLC (Lenexa, KS), and a reaction phenotyping kit containing liver microsomes of individual organ donors (n = 10 subjects) fully characterized with respect to their UGT1A1, UGT1A4, and UGT1A9 activities was obtained from BD Gentest (Woburn, MA). Microsomes prepared from baculovirus-infected insect cells containing cDNA-expressed UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17 were also obtained from BD Gentest.
In Vivo Human Study. The clinical study was conducted at SFBC International (Miami, FL). The study participants were healthy male subjects 18 to 45 years of age. Before initiation of the study, each subject (or legal representative) signed and received a dated copy of an informed consent form. After an overnight fast, the subjects (n = 8) received a single oral dose of 200 mg (200 μCi) of [14C]raltegravir administered as four 50-mg capsules. Subjects continued fasting for 4 h and were given water ad libitum. Blood, urine, and fecal samples were obtained at selected time points for up to 240 h postdose. Blood was collected in EDTA-containing tubes, and the plasma was separated via centrifugation and stored at -20°C. Urine and fecal samples were stored at -20°C and -70°C, respectively until analysis.
Animal Study. Adult male Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA). Rats were housed in metabolic cages and allowed food and water ad libitum. Cannulas were surgically placed in the jugular vein under aseptic conditions. A cannula port system was also surgically implanted into the bile duct of some of the animals (n = 3). The rats were administered an i.v. dose of [14C]raltegravir (3 mg/kg; ∼40 μCi/animal), which was prepared as a solution in DMSO/saline (50:50). Bile samples were collected at various time intervals from the bile duct-cannulated rats, whereas fecal samples were collected from the intact animals. The protocol was reviewed and approved by the Merck Institutional Animal Care and Use Committee before study commencement.
In Vitro Studies. Stock solutions for in vitro studies (0.5 and 5 mM) were prepared by dissolving [14C]raltegravir or unlabeled raltegravir in acetonitrile.
Oxidative metabolism. In a total incubation volume of 0.5 ml, microsomal protein (1.0 mg/ml) was mixed with potassium phosphate buffer (100 mM, pH 7.4), MgCl2 (5 mM), and raltegravir (5 and 50 μM). The mixture was preincubated for 3 min at 37°C before the addition of NADPH (1 mM). Samples were incubated at 37°C for 60 min; then, the reaction was terminated with the addition of acetonitrile (1 ml).
Glucuronidation. Incubations (total volume of 0.5 ml) were carried out at 37°C for 20 or 60 min and contained the following: phosphate buffer (100 mM, pH 7.4), MgCl2 (5 mM), d-saccharic acid-1,4-lactone (10 mM), alamethicin (0.02 mg/mg protein), microsomal protein (native liver microsomes pool or from individual donors, 1 mg/ml; cDNA-expressed UGTs, 0.25 mg/ml), and [14C]raltegravir (specific activity 120 μCi/mg; 5-200 μM). The mixture was preincubated for 3 min before initiation of reaction with the addition of UDPGA (4 mM). The reaction was terminated using acetonitrile (1 ml). The rate of formation of the glucuronide metabolite of raltegravir (M2) by microsomes from individual donors was correlated with the activities (provided by BD Gentest) of estradiol 3-glucuronidation (UGT1A1), trifluoperazine glucuronidation (UGT1A4), and propofol glucuronidation (UGT1A9).
Incubations were also performed in the presence of typical substrates for UGT1A1 and UGT1A3/1A4, namely, bilirubin and β-estradiol, and imipramine, respectively. The inhibitors were dissolved in DMSO and the final concentration of organic solvent in the reaction mixture was ∼1.0% (v/v). Each inhibitor was incubated with pooled microsomes as described above using a concentration of 200 μM[14C]raltegravir. Control incubations contained 1.0% (v/v) DMSO but no inhibitor. The IC50 values of the inhibitors were obtained using nonlinear regression (SigmaPlot; Systat Software, Inc., Point Richmond, CA) by fitting data with a four-parameter symmetric logistic equation (with r indicating the goodness of fit).
For the determination of the kinetic parameters of raltegravir glucuronidation, cDNA-expressed UGT1A1 and UGT1A9 (0.25 mg/ml), and pooled human liver microsomes (0.4 mg/ml) were incubated, as described above, with various concentrations of [14C]raltegravir (8-1000 μM). Linear conditions for the assay were determined over a range of protein concentrations (0.125-0.5 mg/ml) and times (5-40 min). Kinetic parameters were estimated from the fitted curves using nonlinear regression analysis (KaleidaGraph; Abelbeck/Synergy, Reading, PA). The simple hyperbolic Michaelis-Menten equation, V = Vmax · S/(Km + S), was applied with r indicating the goodness of fit.
Preparation of Samples for LC-MS/Radiochromatography.Urine. Samples from 0- to 4- and 4- to 8-h collection intervals were thawed at room temperature and mixed vigorously. Aliquots from each time interval were combined in proportion to their respective volumes to produce a representative 0- to 8-h sample for each subject. The pooled samples were then centrifuged for 10 min and transferred to autosampler vials for LC-MS and radiochromatographic analysis. The volume injected was 100 μl.
Feces. Homogenized fecal samples (0- to 24-, 24- to 48-, and 48- to 72-h) from each subject were thawed at room temperature and mixed thoroughly. Aliquots from each time interval were weighed and combined in proportion to their respective weights to produce a representative 0- to 48-h or 0- to 72-h sample for each subject. To each sample acetonitrile containing 0.2% formic acid was added (volume/weight ratio of 5:1); the sample was mixed and then centrifuged (1811g). The supernatant was then transferred to a glass tube and evaporated to dryness in a SpeedVac concentrator (Thermo Fisher Scientific, Waltham, MA). The residue was reconstituted in DMSO (300 μl), centrifuged for 10 min, and transferred to an autosampler vial for LC-MS and radiochromatographic analysis (aliquot injected was 50 μl).
Plasma. Samples from each subject collected at 0, 0.5, 1, 2, 3, 4, 5, and 6 h were thawed at room temperature. Appropriate aliquots from each sample were pooled such that the pooled sample had a concentration proportional to the pharmacokinetic area under the curve (AUC0-6 h) for a total volume of 1 to 3 ml of plasma (Hop et al., 1998). The pooled plasma samples were extracted using solid-phase extraction cartridges (Oasis HLB, 3 ml; Waters, Milford, MA). The cartridges were conditioned with methanol (1 ml) followed with water (1 ml). The plasma samples were then added to the cartridge and washed with water (1 ml). The samples were finally eluted with methanol (2 ml) and dried under nitrogen gas (Zymark TurboVap) at 37°C. The residue was reconstituted in 30% acetonitrile in water (250 μl) and centrifuged, and the supernatant was transferred to an autosampler vial for LC-MS and radiochromatographic analysis (volume injected was 100 μl).
Rat bile. Bile samples were generated by pooling samples in proportion to the volumes excreted in each collection period over 0 to 8 h. The samples were diluted with 1 volume of water, vortex-mixed, and centrifuged (16,000g). The supernatant was then analyzed directly using LC-MS/radiochromatography.
Rat feces. Fecal samples (collected 0-24 h) were homogenized in water using a Polytron homogenizer (Kinematica Polytron; Brinkmann Instruments, Westbury, NY), and the homogenate was subsequently extracted with acetonitrile containing 0.2% formic acid. The extract was centrifuged and the resulting supernatant was dried (SpeedVac concentrator; Thermo Fisher Scientific) and reconstituted in DMSO for LC-MS/radiochromatographic analysis.
In vitro samples. After the termination of microsomal incubates with acetonitrile, the mixture was vortex-mixed and then centrifuged. The supernatant was evaporated to dryness under nitrogen (Zymark TurboVap). The residue was reconstituted into 30% acetonitrile in water (300 μl). Aliquots of 20 to 50 μl of the resolubilized material were injected for LC-MS/radiochromatographic analysis.
Radioactivity Measurement. To determine total radioactivity in human feces, the subject's samples from each 24-h collection period were pooled to form one sample. An aliquot of 10:90 (v/v) denatured alcohol/water was added to each sample to form approximately a 20% (w/w) homogenate. Feces with diluent were homogenized by using a probe-type homogenizer (Kinematica Polytron; Brinkmann Instruments). Triplicate weighed aliquots (0.2-0.5 g) of homogenate were placed into cones and pads, dried for at least 4 h at ambient temperature, and combusted using a model 307 PerkinElmer sample oxidizer (PerkinElmer, Boston, MA). The 14CO2 was trapped in 9 ml of Carbo-Sorb E (PerkinElmer) and mixed with 9 ml of Permafluor E+ (PerkinElmer) scintillation fluid and then analyzed by liquid scintillation counting. Triplicate aliquots (0.3-0.4 g) of urine were mixed with scintillation fluid (15 ml; Emulsifier-Safe, PerkinElmer) and analyzed directly by liquid scintillation counting (5 min using a Beckman Coulter model LS 6500 liquid scintillation counter; Beckman Coulter, Fullerton, CA). Total radioactivity in plasma was determined in 500-μl aliquots after addition of 15 ml of liquid scintillation cocktail (Ultima Gold; PerkinElmer). The samples were counted for a period of 10 min (PerkinElmer Tri-Carb 2900TR). The limit of quantitation of total radioactivity in plasma was ∼15 ng Eq/ml.
LC-MS/MS Radiochromatographic Analysis. HPLC analysis was carried out on a Hewlett Packard (Palo Alto, CA) HP1100 gradient system. Separation was achieved on a Luna C18-2 column (4.6 mm × 25 cm, 5 μm) (Phenomenex, Torrance, CA) using a mobile phase consisting of 0.1% formic acid in water (solvent A) and 0.1% formic acid in acetonitrile (solvent B) at a constant flow rate of 1.0 ml/min. The gradient started with 10% B and was increased linearly to 50% B in 40 min. The system was equilibrated for 6 min at 10% B before the next injection. The HPLC apparatus was interfaced to a Finnigan TSQ Quantum tandem mass spectrometer (Thermo Fisher Scientific) and mass spectral analyses were carried out using electrospray ionization in the positive ion mode. The capillary temperature was 320°C and the electrospray ionization ionizing voltage was maintained at ∼4 kV for all analyses. Tandem mass spectrometry (MS/MS) was based on collision-induced dissociation of ions entering the rf-only octapole region where argon was used as the collision gas at a pressure of 1.0 mtorr. A collision offset of -25 eV was used for MS/MS analyses. The presence of [14C]raltegravir and 14C-labeled metabolites was monitored using a β-RAM (IN/US, Tampa, FL) radiochemical detector for HPLC. The HPLC effluent was split between the β-RAM detector and mass spectrometer at a ratio of 8 to 2. The β-RAM was operated in homogeneous liquid scintillation counting mode with a 600-μl flow cell and a scintillant (Tru-Count) flow rate of 3 ml/min. The area of the radioactive peaks was used to determine the relative contributions of M2 and raltegravir to the radioactivity in each analyzed sample, which in turn was related to the initial concentration of raltegravir in the incubation.
Quantitation of Raltegravir. The concentration of raltegravir in human plasma was determined by LC-MS/MS in the positive ion mode using the heated nebulizer interface on a Sciex 4000 mass spectrometer (Applied Biosystems, Foster City, CA). Aliquots of plasma samples (0.2 ml) to which internal standard (13C6-raltegravir) was added were extracted using a liquid-liquid extraction method (hexane/methylene chloride). Chromatography consisted of an Ace C18 (50 × 3.0 mm, 3 μm, titanium frits) column with 42.5:57.5% (v/v) 0.1 mM EDTA in 0.1% formic acid/methanol mobile phase at a flow rate of 0.5 ml/min. Quantitation was based on multiple reaction monitoring of the following precursor/product ion pairs: m/z 445→109 (raltegravir); m/z 451→367 (13C6-raltegravir). The lower limit of quantitation was 2 ng/ml (4.5 nM) and the linear calibration range was 2 to 1000 ng/ml.
Pharmacokinetic Calculations. Concentrations below the assay limit of quantification for raltegravir (2 ng/ml) and radioactivity (∼15 ng Eq/ml) were replaced with “0.” An accurate determination of the terminal half-life of raltegravir or radioactivity was not possible due to the fact that the concentrations of raltegravir and radioactivity were below the assay limit of quantitation over much of the terminal phase. AUC0-last was therefore reported for both analytes instead of AUC0-∞. The AUC was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. Cmax and Tmax values were obtained by inspection of the plasma concentration data. The sample pooled according to AUC and analyzed by HPLC with radiochemical detection was used to calculate the relative AUC of M2.
Results
Excretion of Radioactivity. After the oral administration of [14C]raltegravir to humans, radioactivity was eliminated in substantial amounts in both urine and feces (Table 1). The mean (n = 7) recovery of radioactivity in urine and feces was 31.8% and 51.2%, respectively, resulting in a mean total recovery of 83.0% (range 64-102%). The elimination of radioactivity was rapid, since >95% of the recovered dose in urine was attributable to samples collected through 8 h postdose. Examination of individual data (Table 1) indicates that the recovery of radioactivity in urine was comparable for each subject. For feces, however, the recoveries of radioactivity were similar in seven of the eight subjects, but the recovery for subject 1 (12.9%) was significantly lower than that for the other subjects (36.2-64.9%). Consequently, the data for this subject were not included in the calculation of the mean values.
Recovery of radioactivity in urine and feces after administration of a single oral dose of [14C]raltegravir to healthy volunteers

Mean concentration-time profiles of raltegravir and radioactivity in plasma after the administration of a single oral dose of [14C]raltegravir. Raltegravir was quantitated using a validated LC-MS/MS assay, whereas levels of radioactivity were determined by liquid scintillation counting.
Pharmacokinetics. The mean concentration versus time profiles of raltegravir and radioactivity are shown in Fig. 2 and the resulting pharmacokinetic parameters are summarized in Table 2. The compound was absorbed rapidly such that peak plasma concentrations (Cmax) for both raltegravir (4.94 μM) and radioactivity (6.18 μM Eq) were achieved within 1 h of dosing. Plasma levels of MK-0518 and radioactivity declined rapidly and were below the limit of quantitation after 24 h. Raltegravir accounted for approximately 70% of the radioactivity in plasma, as determined by the AUC ratio of raltegravir to radioactivity.
Individual pharmacokinetic parameters of raltegravir and radioactivity after administration of a single oral dose of [14C]raltegravir to healthy volunteers
Metabolite Profiles in Human Urine, Feces, and Plasma.Figure 3 shows a radiochromatogram obtained from pooled urine (0-8 h) from one of the subjects and is representative of the urinary profiles from all subjects studied. Only two peaks were detected and were identified as the glucuronide derivative (M2; Fig. 1) and unchanged parent compound by comparison of retention times and MS/MS spectra with those obtained from authentic standards. As shown in Table 3, M2 and raltegravir accounted for an average of 22.9 and 8.8% of the dose, respectively.
Percentage of dose accounted for by raltegravir and its glucuronide metabolite (M2) in urine
In radiochromatograms obtained from fecal extracts, only one peak was observed and was identified as the parent compound (data not shown). Thus, raltegravir accounts for the entire radioactivity (51.2% of the dose; Table 1) recovered in the feces.
Radiochromatographic analysis of extracts of plasma (from individual subjects and individual time points) revealed the presence of two radioactive peaks which were identified as the parent compound and M2 (data not shown). The AUC (0-6 h) ratio of M2 to radioactivity averaged ∼33% (range 23-41%). Although glucuronidation plays an important role in the metabolism of raltegravir, no evidence of enterohepatic circulation was observed (Fig. 2).
Metabolism Profiles in Rat Bile and Feces. The metabolite profile in rat bile after an i.v. dose of raltegravir is shown in Fig. 4A. The two peaks shown in Fig. 4A were identified as M2 and the parent compound and were determined to represent 85 and 15% of the biliary radioactivity, respectively. In a parallel experiment in which fecal samples were collected, but the bile duct was not exteriorized, all fecal radioactivity was attributable to the parent compound (Fig. 4B). These results indicate that the major source of the parent compound in feces is the glucuronide metabolite M2.
In Vitro Studies. In human liver microsomal incubations fortified with NADPH, no metabolism of raltegravir was observed (data not shown), whereas M2 was readily formed in the presence of UDPGA (Fig. 5). This result suggests that raltegravir is not a substrate of cytochrome P450, consistent with the in vivo data.
In studies using cDNA-expressed UGTs, raltegravir (5 and 50 μM) was converted to M2 by UGT1A1, 1A3, and 1A9 but not by UGT1A4, 1A6, 1A7, 1A8, 1A10, 2B4, 2B7, 2B15, or 2B17 (Fig. 6). The apparent Km values for the glucuronidation of raltegravir by UGT1A1 and UGT1A9 were 99 ± 16 and 296 ± 55 μM, respectively. By comparison, the Km for M2 formation in pooled human liver microsomes was 206 ± 23 μM. The corresponding Vmax values (nmol/min/mg) were 0.89 ± 0.05 (UGT1A1), 0.53 ± 0.06 (UGT1A9), and 0.86 ± 0.04 (human liver microsomes).
The formation of M2 in pooled human liver microsomes was inhibited by typical UGT1A1 substrates, bilirubin (IC50 = 7.1 μM) and β-estradiol (IC50 = 53 μM). However, no inhibitory effect (IC50 > 500 μM) was observed using imipramine, a substrate for UGT1A3 and 1A4 (Fig. 7). In a correlation study with a bank of human liver microsomes, the formation of M2 correlated highly (r = 0.88 and 0.91 at 5 μM and 50 μM raltegravir, respectively) with β-estradiol 3-glucuronidation (marker for UGT1A1 activity), whereas the correlation with two other UGT marker activities was weak [r = 0.02-0.15 for trifluoperazine glucuronidation (UGT1A4) and r = 0.15-0.21 for propofol glucuronidation (UGT1A9)]. Together, these in vitro results demonstrate that UGT1A1 is the main enzyme responsible for raltegravir glucuronidation. Based on the magnitude of maximum inhibition (∼80%) of raletgravir glucuronidation by bilirubin and β-estradiol and the fact that ∼9% of the dose is excreted in human urine as unchanged parent compound, the fraction of the dose metabolized by UGT1A1 can be estimated to be ∼0.7.

A representative radiochromatogram of a urine extract from a subject administered [14C]raltegravir. Urine samples from each time interval were combined in proportion to their respective volumes to produce a representative 0- to 8-h sample for each subject. The pooled samples were centrifuged and an aliquot of the supernatant was analyzed by LC-MS/MS in conjunction with radiometric detection.

Representative radiochromatograms of bile (A) and fecal extracts (B) from rats administered an i.v. dose of [14C]raltegravir. Bile was diluted with 1 volume of acetonitrile, vortex-mixed, and centrifuged. The supernatant was then analyzed directly using LC-MS/radiochromatography. Fecal samples were homogenized using water and the homogenate was extracted with acetonitrile containing 0.2% formic acid. The extract was dried and reconstituted in DMSO for LC-MS/radiochromatographic analysis.
Discussion
The objective of this study was to determine the absorption, metabolism, and mass balance of [14C]raltegravir. In studies in healthy subjects, the pharmacokinetics of raltegravir were found to be generally dose-proportional across a wide range of oral doses (10-1600 mg) (Petry et al., 2006), indicating that the results of this study using a 200-mg dose are applicable to the anticipated clinical dose (400 mg b.i.d.). It is generally required that the absorption, distribution, metabolism, and excretion properties of a new chemical entity intended for human administration are established; this is especially important for an HIV drug which, by definition, will be used in a poly-therapy setting. In the current study, an average of 83% (range 64-102%) of the [14C]raltegravir dose was excreted in urine (32%) and feces (51%), indicating that a reasonable mass balance was achieved. Radioactivity was eliminated rapidly, consistent with the relatively short half-life reported for raltegravir (Petry et al., 2006), and demonstrating that no long-lived metabolites of raltegravir were formed. The oral absorption of raltegravir was at least 32% (range 20-43%) based on the fraction of the dose recovered in urine in this oral study.
Analysis by LC-MS/MS in conjunction with radiochromatography revealed the presence of only two raltegravir-derived components in urine, which were identified as the parent compound and its phenolic hydroxyl glucuronide, M2. Raltegravir and M2 were shown to account for 28% and 72% of the dose recovered in urine, respectively. The parent compound was the only drug-related species observed in extracts of human feces. However, in bile duct-cannulated rats given an i.v. dose of raltegravir, the majority of radioactivity was recovered as M2 in bile (Fig. 4A), whereas in a parallel experiment in intact animals, all the radioactivity in feces was in the form raltegravir (Fig. 4B). These data indicate that in rats, raltegravir is eliminated largely by glucuronidation (>50% of an i.v. or p.o. dose of [14C]raltegravir is recovered in rat feces; unpublished data) and the majority of raltegravir in feces results from the secretion of the metabolite M2 in bile and subsequent hydrolysis in the gut to the parent compound.
Studies in rats and dogs indicate a high level (>70%; unpublished data) of absorption of raltegravir, and since human is not expected to be different from other species, the parent compound observed in human feces is unlikely to represent large unabsorbed drug. In addition, although raltegravir is a P-glycoprotein substrate in both rats and humans (M. Yamazaki, unpublished data), the rat data described here indicate that P-glycoprotein does not contribute significantly to the clearance of raltegravir (mass balance data in rats also indicate little or no P-glycoprotein-mediated intestinal secretion of raltegravir). Consequently, the majority of the raltegravir in human feces is likely derived from the hydrolysis of M2 secreted in human bile as observed in rats (Fig. 4, A and B) and dogs (unpublished observation). Therefore, based on the likely scenario that M2 gave rise to a significant fraction of the radioactivity in human feces, and given the fact that the majority of the dose recovered in urine was attributable to M2, it is reasonable to conclude that metabolism via glucuronidation is the principal mode of clearance of raltegravir in humans (as was the case in rats and dogs).
The metabolite profile obtained from plasma extract indicated the presence of only raltegravir and M2, consistent with what was observed in urine. The major circulating entity was raltegravir and represented 70% of the plasma AUC; the remaining radioactivity was accounted for by M2. In vitro studies show that M2 is devoid of anti-HIV-1 activity. Thus, it can be concluded that the anti-HIV activity observed after administration of raltegravir is entirely due to the parent compound.

A representative radiochromatogram of an extract after incubation of [14C]raltegravir (5 μM) with human liver microsomes in the presence of UDPGA. Incubations were carried out for 60 min using pooled human liver microsomes in the presence of alamethicin.

Metabolism of [14C]raltegravir (5 and 50 μM) to its glucuronide by cDNA-expressed human UDP-glucuronosyltransferases. Incubations were carried out for 20 min with cDNA-expressed UGTs (0.25 mg/ml) in the presence of alamethicin. M2 was quantitated by radiochromatography.
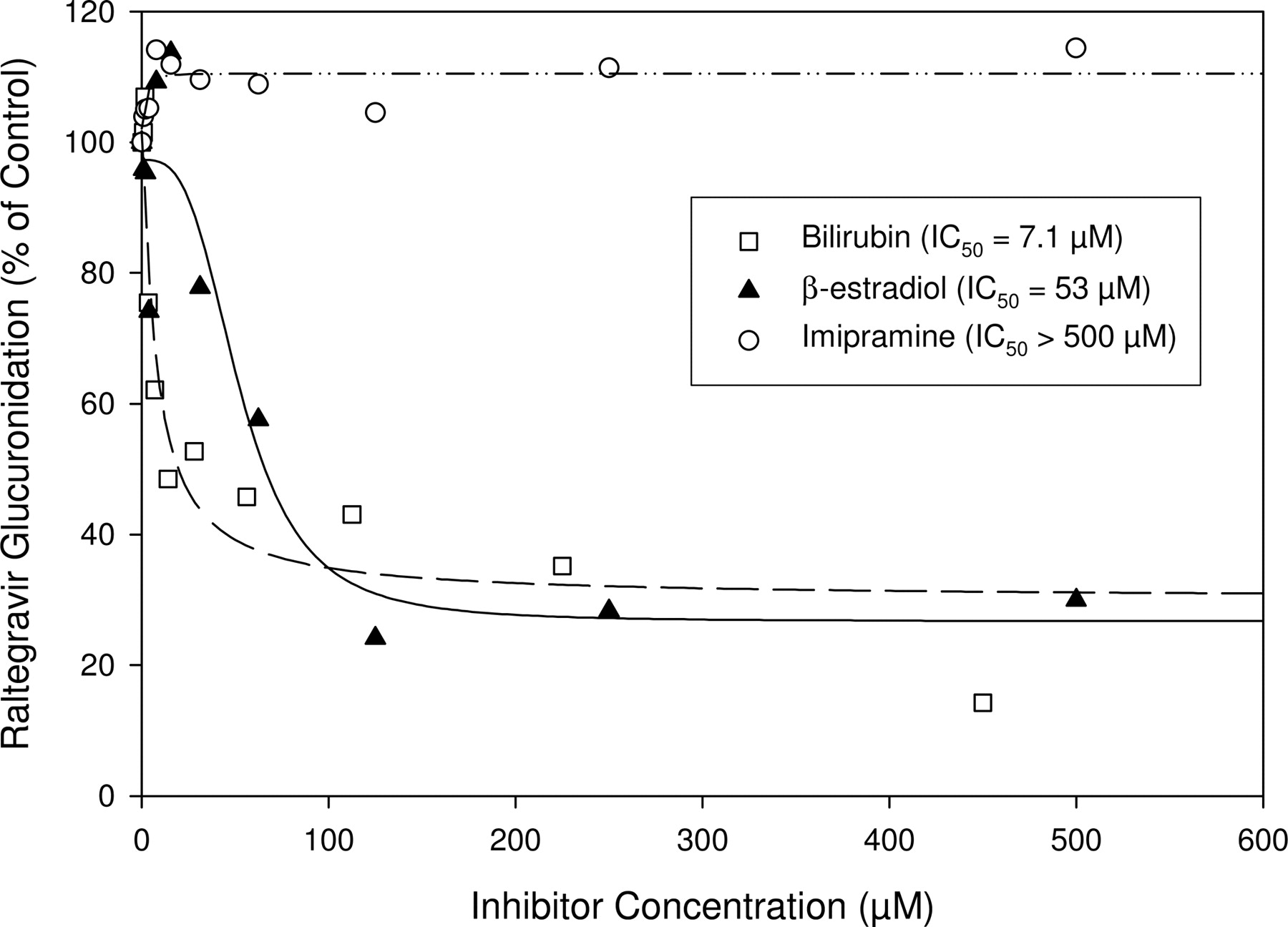
Inhibition of the formation of raltegravir glucuronide (M2) by bilirubin, β-estradiol, and imipramine in incubations of raltegravir (200 μM) with UDPGA-fortified human liver microsomes. The IC50 values of the inhibitors were obtained using nonlinear regression (SigmaPlot) by fitting data with a four-parameter symmetric logistic equation with r indicating the goodness of fit (r = 0.95, 0.95, and 0.77 for bilirubin, β-estradiol, and imipramine, respectively).
A number of in vitro studies were conducted to identify the human UGTs involved in the glucuronidation of raltegravir and the overall data indicate that raltegravir is metabolized mainly by UGT1A1 with minor contributions from UGT1A3 and UGT1A9. It is important to note that although the tools for phenotyping UGT reactions are not as well established as those for cytochrome P450 enzymes (Miners et al., 2006), the conclusion regarding the identity of the major UGT involved in the metabolism of raltegravir is reasonable based on the following: 1) of the 12 tested cDNA-expressed UGTs, only UGT1A1, UGT1A3, and UGT1A9 metabolized raltegravir and UGT1A1 exhibited the highest activity (albeit expression levels in these preparations are not known); 2) the Km for the glucuronidation of raltegravir by cDNA-expressed UGT1A1 and native human liver microsomes was similar; 3) bilirubin, a selective UGT1A1 substrate, inhibited raltegravir glucuronidation with an IC50 of 7.1 μM [although bilirubin may also inhibit UGT1A4 (Bosma et al., 1994), raltegravir was not a substrate of cDNA-expressed UGT1A4]; 4) the formation of M2 was highly correlated with UGT1A1 activity, but not UGT1A4 or UGT1A9, in a bank of human liver microsomes characterized with respect to their activities of these three UGTs.
Collectively, the in vivo and in vitro data presented in this article indicate that the major mechanism for the clearance of raltegravir in humans is UGT1A1-mediated glucuronidation. Since UGT1A1 is polymorphically expressed, a study looking at the effect of UGT1A1 polymorphism (specifically the UGT1A1*28 allele) on the pharmacokinetics of raltegravir has been initiated (but not yet completed); however, UGT polymorphism is not expected to have a major impact on the pharmacokinetics of raltegravir based on the overall clinical data gathered to date (which is bound to include data from individuals with polymorphisms).
Raltegravir may be subject to drug-drug interactions when co-administered with drugs that are known to be UGT1A1 inhibitors such as atazanavir (an HIV-protease inhibitor) or inducers such as rifampin. The AUCi/AUC ratio in the presence of atazanavir, a potent UGT1A1 inhibitor with Ki = 1.9 μM (Reyataz Product Information; Bristol-Myers Squibb, Stamford, CT), was 1.7 (Mistry et al., 2007), and induction by rifampin resulted in 40% reduction in raltegravir AUC (Iwamoto et al., 2006a). These effects on plasma levels of raltegravir by the potent UGT1A1 inhibitor atazanavir and the potent inducer of drug-metabolizing enzymes rifampin might represent the worst case scenario and thus should be considered the upper bounds (i.e., ∼2-fold increase and 2-fold decrease in the presence of potent inhibitors and potent inducers, respectively) with respect to any metabolically mediated pharmacokinetic drug-drug interaction. It should be noted these values were obtained despite the fact that the fraction of the dose metabolized by UGT1A1 is estimated to be high (∼0.7). The magnitude of change in the AUC of raltegravir as a result of UGT1A1 inhibition is consistent with the fact that raltegravir is a high Km UGT1A1 substrate and also is unlikely to undergo high first-pass liver extraction; both of these are factors that reduce the risk for a large AUCi/AUC ratio (Williams et al., 2004). The results from the induction study are in keeping with the concept that the magnitude of drug interaction via induction of glucuronidation is generally less than the induction of cytochrome P450-mediated pathways (Lin and Wong, 2002). Similarly, since raltegravir is neither a substrate nor an inhibitor of cytochrome P450 enzymes and is not an inducer of CYP3A4 (A. Rhoton, unpublished data), raltegravir is not expected to be a perpetrator of metabolic drug interactions with substrates of cytochromes P450. Thus, raltegravir is anticipated to be an anti-AIDS drug with a favorable drug-drug interaction profile (low propensity to be a victim or perpetrator of drug interaction), and the clinical data generated to date (Iwamoto et al., 2006b; Wenning et al., 2006a,b) attest to this property of the compound.
Acknowledgments
We thank the subjects who participated in this study. We also thank Matt Braun, Dennis Dean, Allen Jones, David Schenk, and Joe Simeone (Merck Research Laboratories) for the synthesis of M2 and synthesis and characterization of the [14C]raltegravir used in the study; Ralph Laufer, Odalys Gonzalez, Edith Monteagudo, and Enzo Summa (Merck Research Laboratories) for their contribution to the preclinical studies; and Paul Zavorskas (Charles River Laboratories) for the human urinary and fecal excretion data.
Footnotes
-
doi:10.1124/dmd.107.016196.
-
ABBREVIATIONS: HIV-1, human immunodeficiency virus type 1; HAART, highly active antiretroviral therapy; raltegravir (MK-0518), N-(4-fluorobenzyl)-5-hydroxy-1-methyl-2-(1-methyl-1-{[(5-methyl-1,3,4-oxadiazol-2-yl)carbonyl]amino}ethyl)-6-oxo-1,6-dihydropyrimidine-4-carboxamide; HPLC, high-performance liquid chromatography; UGT, UDP-glucuronosyltransferase; UDPGA, UDP-glucuronic acid; LC-MS, liquid chromatography-mass spectrometry; MS/MS, tandem mass spectrometry; AUCi/AUC, ratio of the area under the plasma concentration vs. time curve in the presence of an inhibitor compared with control curve.
- Received April 11, 2007.
- Accepted June 21, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}