


Abstract
The present study was aimed at characterizing the in vitro cellular uptake mechanism and kinetics of the bile salt analog cholylglycylamido-fluorescein (CGamF) in sandwich-cultured rat hepatocytes (SCRHs). Concentration-dependent inhibition of active CGamF accumulation by seven human immunodeficiency virus (HIV) protease inhibitors (PIs) was also determined and compared with inhibition data obtained with taurocholate (TC) as a substrate. A Km value of 9.3 ± 2.6 μM was obtained for saturable CGamF accumulation in SCRHs. The organic anion-transporting polypeptide (Oatp) inhibitor rifampicin (100 μM) inhibited CGamF (1 μM) accumulation in SCRHs by 72%; sodium depletion did not further reduce CGamF accumulation. In contrast, TC accumulation was reduced by only 25% in the presence of rifampicin, whereas additional sodium depletion resulted in a complete loss of TC accumulation. These data imply that Oatp(s) and sodium taurocholate-cotransporting polypeptide preferentially mediate hepatic uptake of CGamF and TC, respectively. Coincubation of CGamF with HIV PIs (amprenavir, atazanavir, darunavir, indinavir, nelfinavir, ritonavir, saquinavir) revealed that five of them had a concentration-dependent inhibitory effect on CGamF accumulation in SCRHs, with IC50 values between 0.25 ± 0.07 and 43 ± 12 μM. The rank order for inhibition of CGamF accumulation in SCRHs was: ritonavir >> saquinavir > atazanavir > darunavir > amprenavir. Indinavir (up to 100 μM) did not alter CGamF accumulation, whereas nelfinavir solubility was limited to 10 μM. Taken together, these findings illustrate the utility of CGamF as a suitable probe (complementary to TC) for rapid in vitro determination of interaction potential with sodium-independent uptake mechanisms (likely Oatps) in rat liver.
In the past decade, the key transport proteins mediating hepatic uptake and biliary excretion of endogenous compounds as well as xenobiotics have been identified, and their major roles have been recognized (Annaert et al., 2007). In the liver, the sodium taurocholate-cotransporting polypeptide [NTCP (human), Ntcp (rat), SLC10A1/Slc10a1] mediates sodium-dependent transport of conjugated and unconjugated bile salts across the basolateral (sinusoidal) membrane from blood to hepatocyte. Members of the organic aniontransporting polypeptide family [OATP (human), Oapt (rat); SLCO/Slco gene family] also support hepatic uptake of bile salts (in addition to a variety of organic anions, neutral compounds, and cations) but in a sodium-independent manner. At the apical (canalicular) membrane of the hepatocyte, the bile salt export pump [BSEP (human), Bsep (rat); ABCB11/Abcb11] and the multidrug resistance-associated protein-2 [MRP2 (human), Mrp2 (rat); ABCC2/Abcc2] are responsible for bile salt excretion from hepatocytes to bile. As also stated in the European Federation for Pharmaceutical Sciences report on the 2005 Copenhagen Drug Transporters conference (Meijer and Lennernas, 2005), knowledge in this field has increased rapidly, yet quantitative insights into the exact impact of various transport proteins on absorption, distribution, metabolism, and excretion processes and toxicity in vivo remain limited. Nevertheless, interference with hepatobiliary transport of endogenous compounds (i.e., bile salts) is now clearly established as a possible mechanism of hepatotoxicity (Funk et al., 2001; Kostrubsky et al., 2003; Mita et al., 2006; Marion et al., 2007). Compounds that inhibit one or more of the hepatic transporters may cause toxicity following alterations in the levels of various endogenous compounds, such as bile salts, but also thyroid hormones and bilirubin. The detergent effects exerted by elevated intracellular concentrations of bile salts on hepatocyte membranes have been put forward as a mechanism to explain hepatotoxicity (Spivey et al., 1993; Funk et al., 2001).
This emphasizes the need for a better mechanistic understanding of transport processes through the use of new and existing in vitro models. Therefore, thorough characterization of in vitro models, not only with respect to transporter/enzyme expression at the protein and mRNA level, but also at the level of transporter function and activity, is crucial. In addition, a sufficiently diverse panel of model substrates (and inhibitors) needs to be identified and profiled with respect to transporter affinity and interaction. This will ultimately lead to well characterized in vitro models enabling rapid and reliable screening for drug interaction and cellular toxicity potential.
In this context, the present study was aimed at characterizing the hepatic uptake mechanism and in vitro cellular kinetics of the conjugated bile salt cholyl-glycylamido-fluorescein (CGamF) in sandwich-cultured rat hepatocytes (SCRHs). The utility of CGamF as a convenient fluorescent probe simulating bile salt transport in various in vitro and in vivo models of different species has been demonstrated previously (Maglova et al., 1995; Bravo et al., 1998; Holzinger et al., 1998; Mita et al., 2006). Among the amido-fluorescein-tagged compounds, the cholylglycyl derivative (CGamF) was transported most efficiently. Its transport rate exceeded that of cholylamido-fluorescein and ursodeoxycholylamido-fluorescein, both of which were transported at the same rate (Holzinger et al., 1997). CGamF offers the advantages that no radiolabeled compound is needed and that it is amenable to fluorescence microscopy studies. However, the affinity profile of CGamF for human and rodent bile salt transporters appears to differ from the widely used model compound taurocholate (TC) (Mita et al., 2006). Hepatic TC transport is well characterized: its hepatic uptake is mediated by NTCP and OATPs for about 80 and 20% of total uptake, respectively (Meier et al., 1997; Kouzuki et al., 2000). Therefore, inhibition of OATPs alone would not be expected to affect TC accumulation kinetics to a large extent. The relative role of these transporters has not been reported in the case of the bile salt analog CGamF, potentially confounding interpretation of the actual impact of interference with its transport in vitro.
In addition to characterization of cellular CGamF kinetics in rat hepatocytes, the results of the present study illustrate the utility of this probe substrate for in vitro screening of interactions with hepatic uptake of endogenous and/or exogenous compounds by coincubations with seven HIV protease inhibitors (PIs). A study by McRae et al. (2006) already demonstrated that the antiretroviral drugs ritonavir, saquinavir, and efavirenz inhibit TC transport in sandwich-cultured human and rat hepatocytes. The results obtained in the present study with CGamF in sandwich-cultured rat hepatocytes support significant interaction of various HIV PIs with sodium-independent uptake transporters (most likely Oatps) in rat liver.
Materials and Methods
Chemicals. Amprenavir was kindly provided by GlaxoSmithKline (Uxbridge, Middlesex, UK). Ritonavir, indinavir sulfate, saquinavir mesylate, and nelfinavir mesylate were donated by Hetero Drugs Limited (Hyderabad, India). Atazanavir was obtained from Bristol-Myers Squibb Co. (Stamford, CT), darunavir was obtained from Cilag AG (Switzerland). 3H-Taurocholic acid (5 Ci/mmol) and 3H-estrone-3-sulfate (57.3 Ci/mmol) were obtained from PerkinElmer Life and Analytical Sciences (Waltham, MA). Williams' medium E, l-glutamine, penicillin-streptomycin mixture (contains 10,000 units of potassium penicillin and 10,000 μg streptomycin sulfate/ml in 0.85% saline), fetal bovine serum (FBS), and Hanks' balanced salt solution were purchased from Lonza Verviers SPRL (Verviers, Belgium). HEPES was purchased from MP Biomedicals (Irvine, CA). Insulin, collagenase (type IV), taurocholic acid, dexamethasone, rifampicin, choline chloride, and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). Cholyl-glycylamido-fluorescein was kindly provided by Prof. Alan Hofmann (University of California, San Diego). All other chemicals and reagents were of analytical grade and were readily available from commercial sources. Standard buffer consisted of Hanks' balanced salt solution, containing 10 mM HEPES and adjusted to pH 7.4. For experiments in which the effects on substrate accumulation were investigated, sodium-containing buffer and sodium-free buffer were prepared as described by Su et al. (2004). Sterile collagen was prepared in-house according to established procedures.
Animals. Male Wistar rats (240–300 g) were used for hepatocyte isolation from whole liver. The rats were housed according to the Belgian and European laws, guidelines, and policies for animal experiments, housing, and care in the Central Animal Facilities of the university. These facilities have the obligatory accreditation of the authorized Belgian Ministry and are registered under license number LA1210261. Approval for this project was granted by the Institutional Ethical Committee for Animal Experimentation.
Isolation of Rat Hepatocytes. Rat hepatocytes were isolated from male Wistar rats using a collagenase perfusion as described previously (Annaert et al., 2001), with slight modification; the recirculation system volume was 50 ml, and no trypsin inhibitor was added. The typical perfusion times were 10 min with Ca2+-free buffer, followed by about 6 min with buffer containing collagenase. Viability was determined using the trypan blue exclusion method. Viability was always >90% with a yield of 4 to 6 × 108 cells. Cells were resuspended in Williams' medium E and diluted to a final concentration of 1.33 × 106 cells/ml.
Culture of Sandwich-Cultured Rat Hepatocytes. Six-well plates were coated with ice-cold collagen solution (∼1.5 mg/ml final concentration; 0.1 ml/well) prepared by neutralizing a mixture of 4 ml of rat-tail collagen, 4 ml of deionized water, and 1 ml of 10× Dulbecco's modified Eagle's medium with 1 ml of 0.2 N NaOH solution (final pH, ∼7.4). Each well was hydrated with 1.5 ml of phosphate-buffered saline and placed overnight at 37°C in a humidified incubator. Two million cells were added to each well and cultured for 1 to 2 h, and then unattached cells were removed by aspirating the medium. The cells were overlaid with 100 μl of rat tail collagen solution (1.5 mg/ml, pH 7.4) to obtain a “sandwich” configuration (“day 0”). One hour later, Williams' medium E (1.5 ml/well, containing 5% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 4 mg/l insulin, and 0.1 μM dexamethasone) was added onto the cultures. The medium was changed every day with FBS-containing medium.
Transport Studies with CGamF, 3H-TC, and 3H-Estrone-3-Sulfate in Sandwich-Cultured Hepatocytes. SCRHs were rinsed twice with 1.5 ml/well standard buffer and preincubated for 10 min at 37°C. For experiments in which the effect of inhibitors was investigated, cells were preincubated for 10 min with the inhibitor at desired concentration. Subsequently, cells were coincubated in 1.5 ml of 1 μM substrate with or without inhibitors at desired concentration in standard buffer for 10 min (at 37°C unless mentioned otherwise) and subsequently rinsed four times with 1.5 ml of ice-cold standard buffer. Hepatocytes were lysed with 1 ml of 0.5% Triton X-100 solution (in phosphate-buffered saline) by placing plates on a shaker for 20 min at room temperature. Cell lysates were analyzed by fluorescence spectroscopy (excitation, 490 nm; emission, 524 nm) in a Tecan Infinite 200 plate reader (Tecan, Grödig, Austria) for determination of CGamF concentrations and by liquid scintillation spectroscopy (Wallac 1410; PerkinElmer Life and Analytical Sciences-Wallac Oy, Turku, Finland) for 3H-TC or 3H-estrone-3-sulfate. Accumulation was normalized to the protein content of the hepatocytes in each well and was measured by using the BCA Protein Assay kit (Pierce Chemical, Rockford, IL). All accumulation data were corrected for nonspecific binding to collagen-coated, hepatocyte-free culture plates.
Data Analysis. For the characterization of CGamF accumulation kinetics into SCRHs, the following equation was used:  with Kd representing the rate constant for the nonsaturable accumulation and Km and Vmax representing the kinetic parameters for the saturable (Michaelis-Menten) accumulation. The best fit of the equation to the individual accumulation data sets was obtained by Pharsight WinNonlin software version 5.2 (Pharsight, Mountain View, CA).
with Kd representing the rate constant for the nonsaturable accumulation and Km and Vmax representing the kinetic parameters for the saturable (Michaelis-Menten) accumulation. The best fit of the equation to the individual accumulation data sets was obtained by Pharsight WinNonlin software version 5.2 (Pharsight, Mountain View, CA).
In addition, the sigmoid inhibitory effect model was used to describe concentration-dependent inhibition of CGamF accumulation by taurocholate, rifampicin, and the various HIV PIs:  with E the accumulation of substrate in SCRHs, Emax the accumulation of substrate without inhibitor, E0 the accumulation of substrate at the maximum inhibitory effect of inhibitor, Emax - E0 the maximum inhibitory effect, and γ the shape parameter (Hill coefficient). The best fits of the above equations to the individual accumulation data sets were obtained in Pharsight WinNonlin software version 5.2 (Pharsight).
with E the accumulation of substrate in SCRHs, Emax the accumulation of substrate without inhibitor, E0 the accumulation of substrate at the maximum inhibitory effect of inhibitor, Emax - E0 the maximum inhibitory effect, and γ the shape parameter (Hill coefficient). The best fits of the above equations to the individual accumulation data sets were obtained in Pharsight WinNonlin software version 5.2 (Pharsight).

Concentration-dependent accumulation of CGamF in SCRHs. Day 1 SCRHs were incubated with 1 to 50 μM of CGamF in standard buffer at 37°C for 1.5 min, and cellular accumulation was measured as described under Materials and Methods. Points represent average (± S.D., n = 3) measured accumulation rates in a representative batch of SCRHs. The experiment was repeated in two additional batches; see text for average kinetic parameters.
Statistics. Two-tailed Student's t test and ANOVA were used to evaluate statistical differences (SPSS version 11.0 for Windows; SPSS Inc., Chicago, IL) at an α level of 0.05.
Results
Determination of CGamF Accumulation Kinetics in SCRHs. Time-dependent cellular accumulation profiles of CGamF (1–50 μM) in SCRHs were linear for incubation times up to 90 s. Initial accumulation rates were concentration-dependent and could be described by a model incorporating both a Michaelis-Menten (saturable) and a first order (linear) component. Average estimates for Km and Vmax were 9.3 ± 2.6 μM and 401 ± 73 pmol/mg protein/min, whereas an average Kd value of 12.5 ± 1.0 μl/mg protein/min was obtained for the linear accumulation component. A representative profile is shown in Fig. 1.
CGamF accumulation was compared between day 0 and day 1 cultures. In SCRHs, the accumulation rate remained essentially unchanged between days 0 (162 ± 9 pmol/mg protein/min) and 1 (149 ± 11 pmol/mg protein/min), whereas in hepatocytes cultured on a single layer of collagen (“conventional” configuration), a ∼ 35% reduction (from 160 ± 12 to 104 ± 8 pmol/mg protein/min) in CGamF accumulation was observed within this 24-h culture period. It follows that in day 0 hepatocytes, CGamF accumulation was independent of the culture configuration, thus excluding the possibility that the overlay collagen layer was a rate-limiting factor for CGamF uptake transport.
To further study the effect of culture time on CGamF accumulation in SCRHs, kinetic parameters for cultures between days 0 and 4 were determined (Table 1). Estimates of Km and Kd were comparable for day 0 to day 4 SCRHs, whereas the Vmax was decreased in day 4 SCRHs compared with the days 0 to 1 cultures.
Kinetic parameters describing CGamF accumulation in SCRH Values represent average ± S.D. obtained following triplicate measurements from the same batch of hepatocytes (days 0–4).
Based on these results, subsequent incubations with diagnostic inhibitors and HIV protease inhibitors were conducted in day 1 SCRHs at a CGamF concentration of 1 μM. At this incubation concentration, active uptake was calculated to represent 73% of total CGamF uptake.
Effect of Sodium Depletion and Diagnostic Inhibitors on Accumulation of CGamF and TC. To obtain further insight into the primary transport mechanisms mediating CGamF accumulation, the effect of extracellular sodium depletion on CGamF accumulation was investigated and compared with TC accumulation characteristics (Fig. 2). CGamF accumulation was reduced by 20% when extracellular sodium was replaced by choline. On the other hand, when using TC as a substrate, accumulation in the absence of sodium was clearly affected to a much larger extent, with a reduction of total TC accumulation to 40% of its control value. Coincubation of TC or CGamF with the known Oatp inhibitor rifampicin generated a different inhibition profile. Irrespective of the presence of sodium, rifampicin (100 μM) substantially reduced CGamF accumulation to the same level (∼28% of total accumulation in control conditions). On the other hand, the effect of rifampicin on TC accumulation in the presence of sodium remained limited to a 20% reduction in accumulation, whereas in the absence of sodium, TC accumulation was fully abolished by rifampicin coincubation. Coincubation with the nonspecific transporter inhibitor cyclosporin A (10 μM) affected TC and CGamF to a comparable extent.
The inhibitory effects of rifampicin and TC on CGamF accumulation kinetics were further studied at various concentrations of these inhibitors (Fig. 3). These results show that the extent of inhibition of CGamF accumulation was clearly greater with rifampicin compared with TC, whereas the IC50 values (as determined in three independent experiments, i.e., three batches of hepatocytes) corresponding to inhibition of CGamF accumulation by rifampicin (3.2 ± 1.0 μM) and TC (9 ± 4 μM) were rather comparable.
Effect of Temperature on Accumulation of CGamF and Estrone-3-Sulfate in Day 1 SCRHs. To confirm the prominent role of (sodium-independent) active transport mechanisms (likely Oatps) in CGamF accumulation in day 1 SCRHs, the effect of decreased temperature was investigated and compared with the effect on accumulation of the established Oatp substrate estrone-3-sulfate (Fig. 4). These data confirm that active transport mechanisms contribute significantly to the accumulation of CGamF and estrone-3-sulfate in days 0 and 1 SCRHs. Values for the extent of inhibition of CGamF accumulation on day 1, i.e., 50 ± 6% at 4°C and 67 ± 4% in the presence of rifampicin, are in line with a major contribution of an active component to overall CGamF accumulation as supported by the kinetic analysis (see Fig. 1). Rifampicin-inhibitable accumulation of estrone-3-sulfte in days 0 and 1 SCRHs is consistent with expression and activity of Oatps under these culture conditions.
Inhibition of CGamF Accumulation in SCRHs by HIV PIs. The effect of preincubation and coincubation of different HIV PIs [100 μM, except for nelfinavir (10 μM), ritonavir (50 μM), and saquinavir (50 μM), due to the solubility limitations] on CGamF and TC accumulation in SCRHs is shown in Fig. 5. The initial accumulation rate of CGamF was inhibited by amprenavir (100 μM), atazanavir (100 μM), saquinavir (50 μM), ritonavir (50 μM), and darunavir (100 μM) both in sodium-containing or sodium-free buffer. The accumulation rates were decreased to 144 ± 13, 130 ± 7, 109 ± 5, 144 ± 9, and 116 ± 10 pmol/mg protein/10 min, respectively, compared with control values of 323 ± 25 pmol/mg protein/10 min in sodium-containing buffer. The inhibition profiles obtained for these five PIs in sodium-free buffer only were only slightly lower than those obtained in the presence of sodium. In the case of darunavir, sodium depletion did not have any additive effect on CGamF accumulation. For nelfinavir (10 μM), there was no influence on CGamF accumulation in SCRHs and indinavir at 100 μM caused only slight inhibition of the CGamF accumulation. Except for the ∼50% reduction of TC accumulation by saquinavir, the effects of the HIV PIs on TC accumulation remained rather limited in comparison with the effects on CGamF. In addition, although the effect of sodium depletion on CGamF accumulation was relatively small, TC accumulation fell to ∼15% of control values when a combination of sodium-free buffer and HIV PIs was applied.
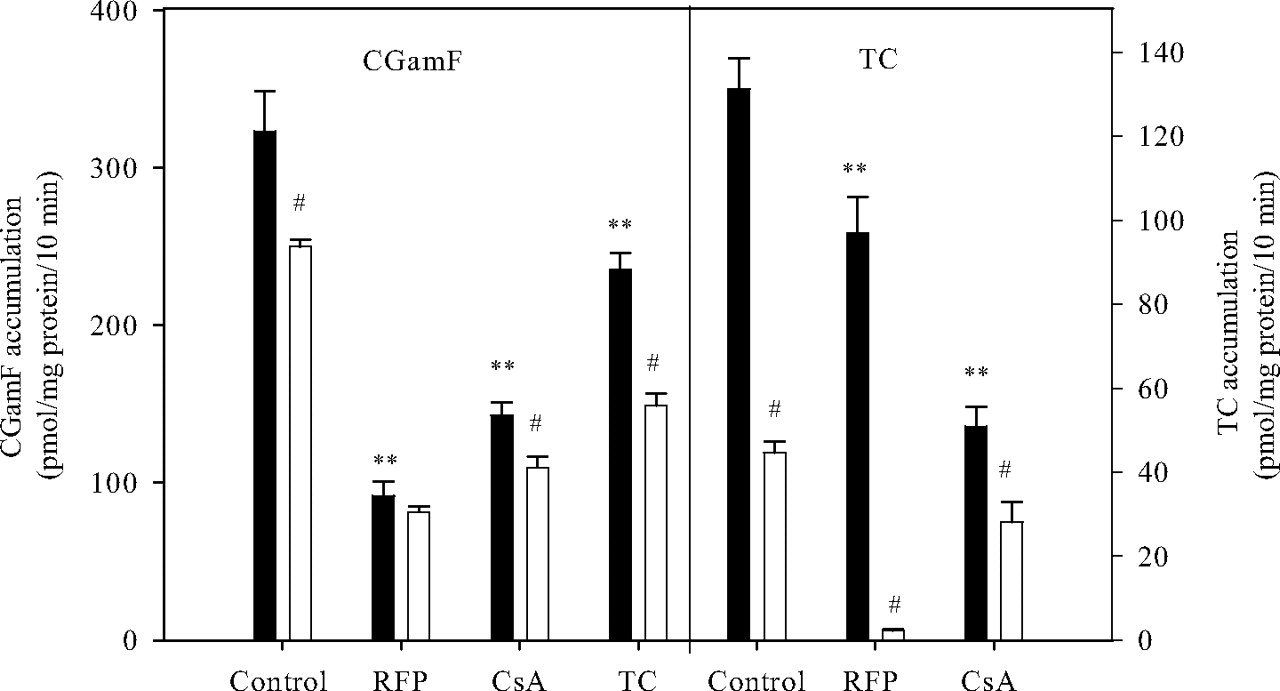
Effect of rifampicin (RFP; 100 μM), cyclosporin A (CsA; 10 μM), and TC (100 μM) on the accumulation of CGamF or TC in SCRHs. Day 1 SCRHs were preincubated with these inhibitors for 10 min, then coincubated with 1 μM CGamF or TC and inhibitors at same concentration for 10 min in sodium-containing (black bars) or sodium-free (white bars) buffer and accumulation measured as described under Materials and Methods. Bars, average (± S.D., n = 3) CGamF or TC accumulation. **, p < 0.05 (ANOVA, Dunnett), compared with the accumulation of CGamF or TC in sodium-containing control conditions. #, p < 0.05 (Student's t test) for pair-wise comparison between condition with sodium (black bars) and condition without sodium (white bars).
Figure 6 shows the concentration-dependent inhibition of CGamF accumulation in SCRHs by five HIV PIs. In this model system and under the present conditions, ritonavir, with an IC50 value of 0.25 ± 0.07 μM, was by far the most potent inhibitor. For the other four HIV PIs, more comparable inhibitory profiles were obtained. The rank order of inhibition of the CGamF accumulation in SCRHs is ritonavir >> saquinavir > atazanavir > darunavir > amprenavir. The IC50 values along with other in vitro kinetic parameters are listed in Table 2. Interestingly, the maximum extent of inhibition of CGamF accumulation that was predicted for ritonavir was lower than the values obtained for the other HIV PIs. The in vitro data obtained in the present study were obtained in rat hepatocytes, and species differences in transporter-based interactions are likely to exist. Nevertheless, reported literature values for peak plasma concentration ranges of PIs observed in patients are also included in Table 2. Evaluation of these in vivo and in vitro data shows that the IC50 value for ritonavir is comparable with the plasma level of unbound ritonavir observed in patients. For the other protease inhibitors (amprenavir, darunavir, atazanavir, saquinavir), plasma levels are lower than IC50 values observed in vitro in rat hepatocytes.
Kinetic parameters (Emax – E0, IC50) derived for inhibition of CGamF (1 μM) accumulation in SCRH by the HIV PIs APV, ATV, SQV, RTV, and DRV Literature values for maximum unbound plasma concentrations (Cmax, unbound) as observed in patients dosed with these PIs are also given.

Concentration-dependent inhibitory effect of RFP and TC on the accumulation of 1 μM CGamF in SCRHs. Day 1 SCRHs were preincubated with inhibitors for 10 min then coincubated with 1 μM CGamF and inhibitors at desired concentration for 10 min in standard buffer and intracellular accumulation measured as described under Materials and Methods. Points represent average (± S.D., n = 3) CGamF accumulation. Lines represent best fit of Emax - IC50 model to the accumulation data.

Effect of coincubation with RFP or incubation at reduced temperature (4°C) on accumulation (10 min) of estrone-3-sulfate (1 μM; top) or CGamF (1 μM; lower panel) in days 0 (black bars) and 1 (white bars) SCRHs. Control incubations and incubations in the presence of rifampicin were conducted at 37°C. Bars, average (± S.D., n = 3) CGamF or estrone-3-sulfate accumulation. **, p < 0.05 (ANOVA, Dunnett), compared with the accumulation of estrone-3-sulfate or CGamF under the corresponding control conditions.

Effect of different HIV PIs on the accumulation of CGamF (top) and TC (bottom) in SCRHs. Day 1 SCRHs were preincubated with amprenavir (APV; 100 μM), atazanavir (ATV; 100 μM), SQV (50 μM), RTV (50 μM), darunavir (DRV; 100 μM), indinavir (IDV; 100 μM), or nelfinavir (NFV; 10 μM) for 10 min, then coincubated with 1 μM CGamF or TC and inhibitors at same concentration for 10 min in sodium-containing (black bars) or sodium-free (white bars) buffer and intracellular accumulation measured as described under Materials and Methods. Bars, average (± S.D., n = 3) CGamF or TC accumulation. **, p < 0.05 (ANOVA, Dunnett), compared with the accumulation of CGamF or TC in sodium-containing control conditions. #, p < 0.05 (Student's t test) for pair-wise comparison between condition with sodium (black bars) and condition without sodium (white bars).
Discussion
The aim of the present study was to characterize the bile salt analog CGamF as a probe substrate for in vitro hepatic uptake in rat hepatocytes and to demonstrate its utility for mechanistic investigation of drug interactions at the hepatic uptake level. A series of HIV protease inhibitors were selected as interacting model drugs. In addition, a comparison was made with the established probe substrate taurocholate.
As compared with freshly isolated suspended hepatocyte preparations, which have to be used within 4 to 6 h after isolation, day 1 SCRHs offer the advantage that incubations can be done at higher throughput and for a more extended period of time. Data supporting the maintained expression and activity of the hepatic uptake transporters Ntcp and Oatp in rat hepatocytes early after isolation as well as in day 1 SCRHs have been generated previously. Liu et al. (1998) indeed showed superior expression of Ntcp in days 1 to 5 SCRHs compared with conventionally cultured rat hepatocytes as well as decreased Ntcp activity with increasing culture time. The data generated for TC accumulation (see Fig. 2) in day 1 SCRHs in the present study are consistent with these previous findings. More recently, Bow et al. published immunofluorescence images showing that Oatp1a1 remained expressed in the sinusoidal membrane of isolated rat hepatocytes, which was in contrast to the cytosolic internalization of efflux transporters (Mdr1, Mrp2). In the present study, we also showed rifampicin- and temperature-inhibitable accumulation of the Oatp substrate estrone-3-sulfate, supporting expression of fully functional Oatps in the plasma membrane of days 1 and 0 SCRHs. Furthermore, as shown by the data presented in Table 1, the use of day 1 versus day 4 SCRHs appears to offer the advantage of higher CGamF accumulation. A similar phenomenon was observed by Hoffmaster et al. (2004), who reported culture time-dependent decrease in accumulation of the Oatp substrate DPDPE in SCRHs.

Concentration-dependent inhibitory effect of different HIV PIs (APV, ATV, SQV, RTV, DRV) on the accumulation of 1 μM CGamF in SCRHs. Day 1 SCRHs were preincubated with inhibitors for 10 min, then coincubated with 1 μM CGamF and inhibitors at desired concentration for 10 min in standard buffer and intracellular accumulation measured as described under Materials and Methods. Points represent average (± S.D., n = 3) CGamF accumulation. Lines represent best fit of Emax - IC50 model to the accumulation data.

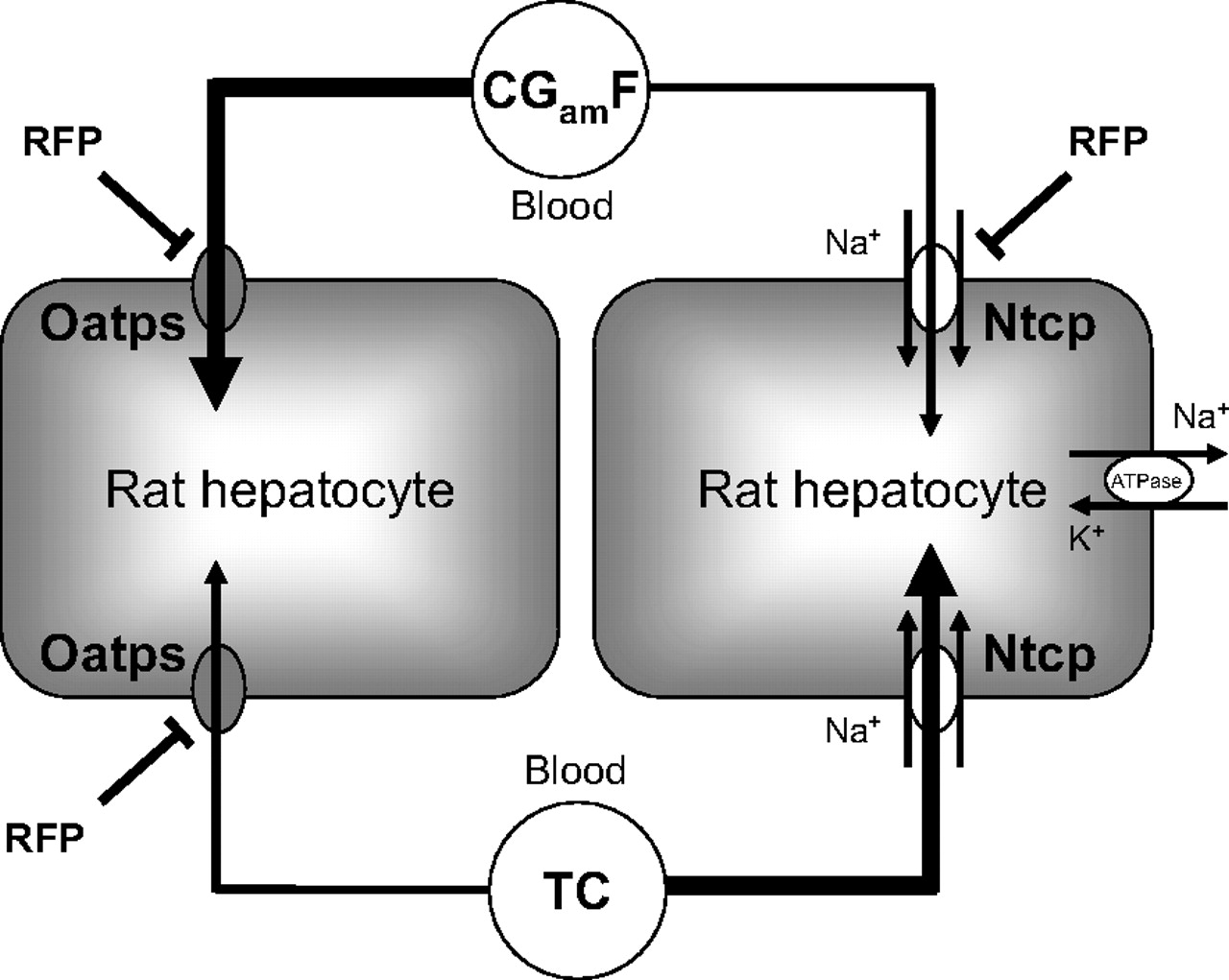
Scheme illustrating the different transporters primarily mediating the hepatic uptake of CGamF and TC in day 1 rat hepatocytes. As evidenced by the sodium dependence, Ntcp is supporting the majority of TC uptake with a minor role for one or more Oatps. In contrast, CGamF hepatic uptake occurs primarily by sodium-independent mechanisms (most likely Oatps), with limited contribution by Ntcp. Interestingly, the previously established Oatp inhibitor RFP altered both the Oatp- and Ntcp-mediated CGamF uptake, whereas the effect on TC accumulation was limited to the Oatp-mediated component.
The results obtained in the present study demonstrate a remarkable difference between CGamF and TC with respect to the transport mechanisms that are primarily mediating hepatic uptake of these substrates (for a schematic summary, see also Fig. 7). First of all, sodium depletion resulted in only a slight reduction in CGamF accumulation (Fig. 2), suggesting a limited role of Ntcp in active CGamF accumulation. Furthermore, in the presence of the known Oatp inhibitor rifampicin, CGamF accumulation was substantially reduced to ∼30% of control levels. This represents a complete loss of active CGamF uptake since about 70% of CGamF (1 μM) uptake was calculated to result from an active process. Rifampicin has indeed been shown to be a potent inhibitor of Oatp2 (Oatp1a4) in LLC-PK1 cells (Shitara et al., 2002), rat hepatocytes (Fattinger et al., 2000), and of various OATP/SLCO isoforms in human hepatocytes (Vavricka et al., 2002). At a concentration of 100 μM, rifampicin was also shown to inhibit Oatp1a4-mediated transport of digoxin by 60% (Shitara et al., 2002). To date, no reports have been published on the possible inhibition of Ntcp activity by rifampicin. In the present study, however, CGamF accumulation in SCRHs in the presence of rifampicin did not differ between sodium-containing buffer and sodium-free buffer. Together with the slight (but statistically significant) effect of sodium depletion alone on CGamF accumulation, these results suggest inhibition of Ntcp-mediated CGamF accumulation by rifampicin. Completely different effects of sodium depletion and rifampicin co-incubation were noted when taurocholate was used as a substrate; the effect of 100 μM rifampicin was limited to a reduction in total accumulation of only 20%, whereas additional sodium depletion completely abolished TC accumulation, demonstrating that rifampicin did not alter Ntcp-mediated TC uptake. These results suggest that affinity of rifampicin for Ntcp is lower than that of TC, but higher than that of CGamF. Incubation of CGamF in the presence of various concentrations of either rifampicin or TC further confirmed the above findings (Fig. 3). Indeed, the maximum extent of inhibition of CGamF accumulation was higher for rifampicin (mainly affecting Oatps) compared with TC (mainly affecting Ntcp). Comparison of the differential effects of sodium depletion and rifampicin in GCamF versus TC accumulation also strongly suggest that at least one Oatp isoform is a major player in CGamF accumulation in SCRHs. The rather limited role for Ntcp in CGamF accumulation does not conflict with previous findings. CGamF has been shown to display affinity for Ntcp, as based on the study of Boyer et al. (1994) in which the rat Ntcp was transfected into COS-7 cells. However, compared with TC transport in this transfected model system, CGamF accumulation rate was substantially lower. Taken together, these findings illustrate the utility of CGamF as a probe complementary to TC for accurate and rapid in vitro determination of drug interactions at the hepatic uptake level.
To further establish the usefulness of CGamF as a probe substrate for detecting Oatp-mediated interactions with active hepatic uptake of endogenous and exogenous substrates, a comparative in vitro study was conducted with HIV PIs as interacting drugs. Several HIV PIs are widely used in the clinic, mostly in combinations with other drugs. In addition, a few in vitro studies have provided evidence that HIV PIs display affinity for members of the Oatp family. Su et al. (2004) demonstrated for the first time that OATP-A (OATP1A2) was capable of transporting saquinavir in vitro. Concentration-dependent inhibition of [3H]estradiol-17β-d-glucuronide uptake by indinavir, nelfinavir, saquinavir, and ritonavir was observed in OATP-C (OATP1B1)-transfected HeLa cells (Tirona et al., 2003). In the present study, concentration-dependent inhibition of (Oatp-mediated) CGamF accumulation in SCRHs by several HIV PIs (ritonavir, saquinavir, atazanavir, darunavir, amprenavir) was observed. For indinavir, only a slight effect was observed at 100 μM, whereas incubations with nelfinavir were only possible up to 10 μM due to solubility limitations. The most remarkable result with respect to inhibition of active CGamF accumulation in SCRHs is the substantially higher potency of ritonavir (IC50 = 0.25 μM), compared with the other HIV PIs. Indeed, the IC50 value for saquinavir, which appeared to be the second most potent HIV PI tested in this study, was almost 25 times higher.
The preferential interference of HIV PIs with sodium-independent (likely Oatps) transport mechanisms is also reflected in the inhibition data for TC accumulation (Fig. 5). The extent of inhibition of TC accumulation in sodium-containing buffer (and reflecting mainly Ntcp-mediated processes) remains rather limited, except for saquinavir (50% inhibition). On the other hand, the relative inhibition (up to 80%) of TC accumulation by sodium-independent processes (likely Oatps) is much more pronounced. It is of interest to compare our data regarding inhibition of TC accumulation with those obtained previously for ritonavir (RTV) and saquinavir (SQV) in day 4 SCRHs (McRae et al., 2006). First of all, overall TC uptake in day 1 SCRHs observed in the present study (about 130 pmol/mg protein/min) was substantially higher than the TC uptake observed in day 4 SCRHs in the earlier study (35 pmol/mg protein/min). This is consistent with declining expression and activity of uptake transporters (including Ntcp) with increasing culture times in SCRHs (Liu et al., 1998). Interestingly, the maximal extent of inhibition (about 80%) observed in the study by McRae et al. at the highest SQV and RTV concentrations corresponds to the relative inhibition percentages of sodium-independent TC accumulation by several HIV PIs in our study. This may reflect relatively higher contribution of sodium-independent processes to overall TC accumulation in day 4 SCRHs.
The results of this study have provided a direct comparison of the inhibitory potential of seven clinically used HIV protease inhibitors toward hepatic uptake transport of CGamF in rat liver. These data may aid in understanding relative differences between these compounds with respect to hepatotoxicity in the rat as the main toxicological species. Clearly, comparable in vitro data in human hepatocytes are needed to determine clinical relevance of the present data as well as potential species differences in drug interactions. Nevertheless, it is interesting to note that free plasma concentrations observed clinically for ritonavir in patients (Table 2) range around the IC50 value obtained for this PI in the present study.
Inhibition of one or more Oatp isoforms by HIV protease inhibitors can potentially result in drug interactions with Oatp substrates. In addition, because certain endogenous substrates also rely on Oatp transporters (e.g., thyroid hormones), (hepato)toxicity may result. Hepatotoxicity has been reported as an adverse effect in the prescribing information for the U.S. Food and Drug Administration-approved HIV PIs, nelfinavir, indinavir, atazanavir, ritonavir, lopinavir/ritonavir, saquinavir, amprenavir, and fosamprenavir. The incidence values ranged from one to two cases per 100 patients for nelfinavir, which was the lowest among this group, and from 2.2 to 9.5 for lopinavir/ritonavir (Bruno et al., 2006). Sulkowski et al. (2000) also observed that the relative risk of severe hepatotoxicity in patients receiving a PI-containing regimen was more than twice that seen in patients who were prescribed regimens with dual nucleoside analogs. The possible role of interference with Ntcp/NTCP-mediated bile salt transport as a mechanism of hepatotoxicity associated with antiviral drugs has already been investigated in the aforementioned in vitro study by McRae et al. However, a simple relationship between inhibition of NTCP-mediated bile salt uptake in human hepatocytes and the risk for hepatotoxicity in the clinic could not be established (McRae et al., 2006). In light of the results obtained in the present study, inhibition of Oatp-mediated substrate transport (in addition to Ntcp-mediated transport) by HIV PIs may be considered as another potential mechanism in PI-associated hepatotoxicity.
Although the present data were obtained in rat hepatocytes, and species differences are expected to exist, it is interesting to note that ritonavir exhibits the highest risk for hepatotoxicity in clinical therapy. This finding may be especially important given the widespread combination of ritonavir as a pharmacokinetic booster with other HIV PIs to improve pharmacokinetics of these PIs.
In conclusion, our data have illustrated that thorough characterization and careful use of complementary probe substrates are important to get better insights in relative contributions of various transport mechanisms in drug interaction potential and risk for hepatotoxicity. Our data confirm that compared with hepatocytes cultured in a conventional configuration, SCRHs already after 1 day in culture represent an improved model system for in vitro evaluation of inhibition of hepatic uptake transporters by drugs. Quantitative in vitro data on inhibition of hepatic uptake transporters may help to understand mechanisms underlying side effects of certain HIV PIs.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.107.019398.
-
ABBREVIATIONS: Ntcp, sodium taurocholate-cotransporting polypeptide; Oatp, organic anion-transporting polypeptide; MRP, multidrug resistance-associated protein; CGamF, cholyl-glycylamido-fluorescein; SCRH, sandwich-cultured rat hepatocyte; TC, taurocholate; HIV, human immunodeficiency virus; PI, protease inhibitor; FBS, fetal bovine serum; ANOVA, analysis of variance; RFP, rifampicin.
- Received October 24, 2007.
- Accepted April 16, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}