


Abstract
We generated the organic anion transporting polypeptide (Oatp) 1b2 knockout (KO) mouse model and assessed its utility to study hepatic uptake using model compounds: cerivastatin, lovastatin acid, pravastatin, simvastatin acid, rifampicin, and rifamycin SV. A selective panel of liver cytochromes P450 (P450s) (Cyp3a11, Cyp3a13, Cyp3a16, Cyp2c29, and Cyp2c39) and transporters [Oatp1b2, Oatp1a1, Oatp1a4, Oatp1a5; organic anion transporter (Oat) 1, Oat2, Oat3; multidrug resistance gene 1 (Mdr1) a, Mdr1b; bile salt export pump, multidrug resistance associated protein (Mrp) 2, Mrp3; breast cancer resistance protein] were measured by reverse transcription-polymerase chain reaction in both KO and wild-type (WT) male mice. Male KO and WT mice received each model compound s.c. at 3 mg/kg. Blood and liver samples were obtained at 0, 0.5, and 2 h postdose and analyzed using liquid chromatography/tandem mass spectrometry. Liver/plasma concentration ratio (Kp,liver) was calculated. Student's t test was used to compare the mRNA and Kp,liver between the KO and WT mice. A similar mRNA expression was observed between the KO and WT for the selected P450s and transporters except for Oatp1b2, for which the level was negligible in the KO but prominent in the WT mice with P < 0.0001. The in vivo results showed a differential effect of Oatp1b2 on hepatic uptake of the model compounds, indicating that Oatp1b2 plays a more significant role in the hepatobiliary disposition of rifampicin and lovastatin than the other compounds tested. This study suggests the Oatp1b2 mouse as a useful in vivo tool to understand drug targeting and disposition in the liver.
Organic anion transporting polypeptides (rodents: Oatps, human: OATPs) belong to the superfamily of solute carriers (Hagenbuch and Meier, 2004). Rodent Oatp1a1, Oatp1a4, Oatp1b2, and Oatp2b1 and human OATP1B1, OATP1B3, and OATP2B1 are expressed in liver (Hagenbuch et al., 2000; Cheng et al., 2005). Among them, Oatp1b2 is predominantly if not exclusively expressed in liver (Hsiang et al., 1999; Konig et al., 2000; Cheng et al., 2005) and is orthologous to OATP1B1 and OATP1B3 (Hagenbuch and Meier, 2003). OATP1B1 has been shown to actively uptake not only endogenous compounds but also xenobiotics including statins such as pravastatin (Hsiang et al., 1999), cerivastatin (Shitara et al., 2004), simvastatin (Hsiang et al., 1999), and antibiotics such as rifampicin (Tirona et al., 2003). Inhibition of OATP1B1-mediated uptake of statins to the liver is believed to cause clinically relevant drug-drug interactions between statins and concomitant medicines such as cyclosporin A and gemfibrozil that are strong inhibitors of OATP1B1 (Poirier et al., 2007). There is accumulated evidence pointing to the importance of OATP1B1 in the hepatobiliary disposition and interaction of many organic anion drugs (Poirier et al., 2007). However, experimental methods, specific substrates, and inhibitors are less well established for this family of transporters, and this presents challenges in the study of OATP uptake-related drug interactions using in vitro tools expressing OATPs or using wild-type (WT) animals.
In vitro approaches including OATP1B1 stably transfected transporter express systems and hepatocytes with a general organic anion inhibitor have been used to study the contribution of OATP1B1 in clinically observed drug interactions. The transport expressing systems allow a mechanistic insight into the particular transporter(s) involved in the transport of compounds of interest (Shitara et al., 2003, 2004). Likewise, hepatocyte assays with an intended transporter inhibitor can also be used (Hoffmaster et al., 2005). However, whether the in vitro transport of a test compound has any in vivo significance such that it changes the systemic exposure of the test compound if the particular transport process is altered via chemical inhibitors or inducers is challenging to predict. Animal models with deficient Oatps as in the case of multidrug resistance gene 1 (Mdr1) a- and/or Mdr1a/b-deficient mouse models (Schinkel et al., 1996; Chen et al., 2003) will be instrumental to our understanding of the in vitro-in vivo relationships, the species-related transport, and the potential significance of OATP1B1/Oatp1b2 in disposition and drug interactions before clinic studies.
Similar to OATP1B1, Oatp1b2, which shares a 65% homology in amino acid sequence with human OATP1B1, is exclusively expressed in liver (Abe et al., 1999; Konig et al., 2000; Cheng et al., 2005). Limited information is available regarding the substrate specificity between the mouse Oatp1b2 and human OATP1B1. Recently, an Oatp1b2 knockout (KO) mouse model was generated by Lu et al. (2008), and their studies showed that Oatp1b2 KO mice are useful in elucidating the role of Oatp1b2 and its human orthologs OATP1B1/1B3 in hepatic uptake and systemic disposition of toxins such as phalloidin and microcystin-LR. The present study describes the generation of the Oatp1b2 KO mouse model in our laboratory followed by the examination of the role of Oatp1b2 in the hepatic uptake of six model compounds: cerivastatin, lovastatin acid, pravastatin, simvastatin acid, rifampicin, and rifamycin SV. All of them have been shown to interact with human OATP1B1 (Hsiang et al., 1999; Shitara et al., 2003; Tirona et al., 2003; Chen et al., 2005).
Materials and Methods
Materials. Cerivastatin was purchased from Sequoia Research Products Ltd. (Pangbourne, UK). Lovastatin, simvastatin, pravastatin, rifampicin, and rifamycin SV were purchased from Sigma (St. Louis, MO). The acid form of lovastatin and simvastatin was prepared by hydrolysis of the corresponding lactone under alkaline condition. All the other chemicals and reagents were the highest grade available from commercial sources. Structures and physical-chemical parameters of all the compounds used in the present study were described previously (Chen et al., 2007).
Generation of Oatp1b2 KO Mice. The detailed information regarding the generation of Oatp1b2 KO mice was described recently (Zaher et al., 2008). Briefly, a targeting vector to disrupt the Oatp1b2 murine gene was constructed using 4.1 kilobase (kb) of 5′ homology and 5.0 kb of 3′ homology. Linearized targeting vector was electroporated into DBA/1lacJ embryonic stem (ES) cells using procedures previously described (Roach et al., 1995). Two Oatp1b2-targeted ES cell clones were identified and microinjected into C57bl6 (Charles River Laboratories, Wilmington, MA) blastocysts using standard procedures (Nagy et al., 2003). Resulting chimeric males were backcrossed to DBA/1lacJ (The Jackson Laboratory, Bar Harbor, ME) females to generate inbred germline heterozygous offspring. Offspring from these and all the subsequent matings were genotyped by polymerase chain reaction (PCR) analysis using two oligo sets, neomycin and Oatp1b2. The neomycin set (NeoF, 5′-gcaggatctcctgtcatctcacc-3′ and NeoR, 5′-gatgctcttcgtccagatcatcc-3′) amplified a 190-base pair (bp) product from an Oatp1b2-targeted allele, whereas the Oatp1b2 set (Oatp1b2F, 5′-tggacaatgtgcaatgggagc-3′ and Oatp1b2R, 5′-gaaagagctgattagagatacg-3′) amplified a 497-bp product from an Oatp1b2 WT allele.
Sample (Double-Stranded cDNA) Preparation. Total RNA from the livers of six mice (n = 3/strain) was isolated using Qiagen's RNeasy Mini Kit (Qiagen, Inc., Valencia, CA). The quality of the RNA was determined using the Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA). A mixture of 3 μg of high quality total RNA and 5 μM oligo dT24 was heated at 70°C for 10 min to promote primer annealing. First-strand cDNA reactions (20 μl), containing the annealed mixture, 5 mM DTT, 0.5 mM dNTP mix, along with Invitrogen (Carlsbad, CA) First Strand Buffer (1×) and SuperScript II ribonuclease H reverse transcriptase (200 U), were heated at 42°C for 1 h. To each first-strand reaction, the following was added (for a final volume of 150 μl): 91 μl of RNase-free H2O, 30 μl of 5× Second Strand Buffer (Invitrogen), 3 μl of 10 mM dNTP mix, 1 μl of 10 U/ml Escherichia coli DNA ligase, 4 μl of 10 U/ml E. coli DNA polymerase I, and 1 μl of 2 U/μl RNaseH. These second-strand synthesis reactions were incubated at 16°C for 2 h; then 10 U of T4 DNA polymerase was added and the incubation continued for an additional 5 min at 16°C. The reactions were stopped by the addition of EDTA, and the double-stranded cDNA (precleaned) was stored at –20°C.
Real-Time Quantitative PCR. A panel of genes was selected for real-time quantitative PCR analysis. Amplification of each target cDNA was carried out in triplicate in a 96-well format using Applied Biosystems' (Foster City, CA) GeneAmp 5700 Sequence Detection System and TaqMan Gene Expression Assays. Using the 5′ nuclease activity of Taq DNA polymerase to generate a real-time quantitative analysis (Heid et al., 1996), 50-μl amplification reactions contained 5 μl of 1:30 dilution of precleaned double-stranded cDNA (1 part cDNA, 29 parts molecular biology grade water), 1× TaqMan Universal PCR Master Mix (Applied Biosystems), and 1× Target Assay Mix (ABI's TaqMan Gene Expression Assay). Similar 50-μl reference reactions were prepared in triplicate for amplification of glyceraldehyde-3-phosphate dehydrogenase (GAPD), the reference/housekeeping gene. Thermal cycling conditions for all the reactions were as follows: 2 min at 50°C, 10 min at 95°C, then 40 cycles of 15 s at 95°C and 1 min at 60°C.
To use the cycle time (CT) method (Meijerink et al., 2001) for relative quantitation, separate reactions were carried out to ensure that the efficiency of the reference amplification was equal to the efficiency of the gene-of-interest amplification (Applied Biosystems User Bulletin 2). Template cDNA (precleaned) was diluted in molecular biology grade water and used in reactions as described above. Reaction efficiencies were found to be close to 1.0 as desired (data not shown).
The relative mRNA expression level of each gene of interest (target) was determined by first normalizing its average reaction CT to the GAPD average CT, where CT (or threshold cycle) is the cycle number at which emitted fluorescence exceeds 10 times the S.D. of baseline emission (measured from cycle 6 to 15). This ΔCT value, the difference in threshold cycles for target and reference, is calculated by subtracting the GAPD average CT from the CT for the gene-of-interest target average. Finally, the normalized amount of target for a knockout sample was divided by the normalized amount of target for a WT (calibrator) sample to obtain the ΔΔCT value. This ΔΔCT value was then substituted into the formula 2.0–[ΔΔCt] to provide the relative mRNA expression.
Disposition of Cerivastatin, Lovastatin Acid, Pravastatin, Simvastatin, Rifampicin, and Rifamycin SV in Oatp1b2 KO and WT Mice. Four- to 6-week-old male Oatp1b2 KO and WT littermates both with body weight approximately 20 g were housed at controlled temperature and humidity in an alternating 12-h light and dark cycle with free access to food and water before the study. Cerivastatin, pravastatin, lovastatin acid, and simvastatin acid were dissolved in 20% cyclodextrin, and rifampicin and rifamycin SV were dissolved in dimethyl sulfoxide initially and then diluted with water with final dimethyl sulfoxide content of less than 1%. The compound preparations were injected s.c. (10 ml/kg) at 3 mg/kg to mice (n = 3 mice/strain/time point). Blood and liver tissues were collected at 0, 0.5, and 2 h postdose. Plasma was obtained by centrifugation (3000 rpm for 10 min) of heparinized blood. The liver tissues were rinsed in saline, blot-dried, weighed, and homogenized with saline (w/v, 1:3). All the samples were stored at –20°C until analysis.
Sample Analysis. Separate standard curves were prepared for plasma and liver samples. Plasma (10 μl) and liver (50 μl) samples were transferred to a 96-well plate, followed by adding the appropriate internal standard. Atorvastatin was used as the internal standard for all the statins and rifampicin for rifamycin SV and rifamycin SV for rifampicin. After vortexing, the mixture was centrifuged at 3000 rpm for 10 min, and an aliquot (10 or 20 μl) of the supernatant was analyzed using a high-performance liquid chromatography (HPLC)-mass spectrometric (MS) method.
The HPLC/tandem MS system consisted of either a Shimadzu ternary pump (Shimadzu LC-10A; Kyoto, Japan) or an Agilent quaternary pump HPLC system (Hewlett Packard, Palo Alto, CA), an autosampler, and a PE Sciex API 4000 or 3000 (PerkinElmer Sciex Instruments, Foster City, CA) MS fitted with an electrospray ionization operated in the positive ion mode. Phenomenex Aqua C18 (4.6 × 50 mm, 5 μm; Phenomenex, Torrance, CA; for all the statins) and Phenomenex PrimeSphere C18 (2.0 × 30 mm, 5 μm; Phenomenex; for rifampicin and rifamycin SV) columns were used. The statins were eluted with a mobile phase consisting of acetonitrile/ammonium acetate (10 mM, solvent A) and water/ammonium acetate (10 mM, solvent B) at a flow rate of 0.5 ml/min with the following gradient: 20% A from 0 to 2 min, 20 to 90% A from 2 to 4 min, 90% A from 4 to 4.5 min, and 90 to 20% A from 4.5 to 4.7 min. Rifampicin and rifamycin SV were eluted with a mobile phase consisting of acetonitrile (95%)/ammonium acetate (20 mM, solvent A) and acetonitrile (5%)/ammonium acetate (20 mM, solvent B) at a flow rate of 0.5 ml/min with the following gradient: 5% A from 0 to 1 min, 5 to 95% A from 1 to 4 min, 95% A from 4 to 4.2 min, 95 to 5% from 4.2 to 4.5 min, and 5% A from 4.5 to 5.0 min. The ion transition for the analytes and internal standards, retention times, and dynamic ranges for the assay were described previously (Chen et al., 2005).

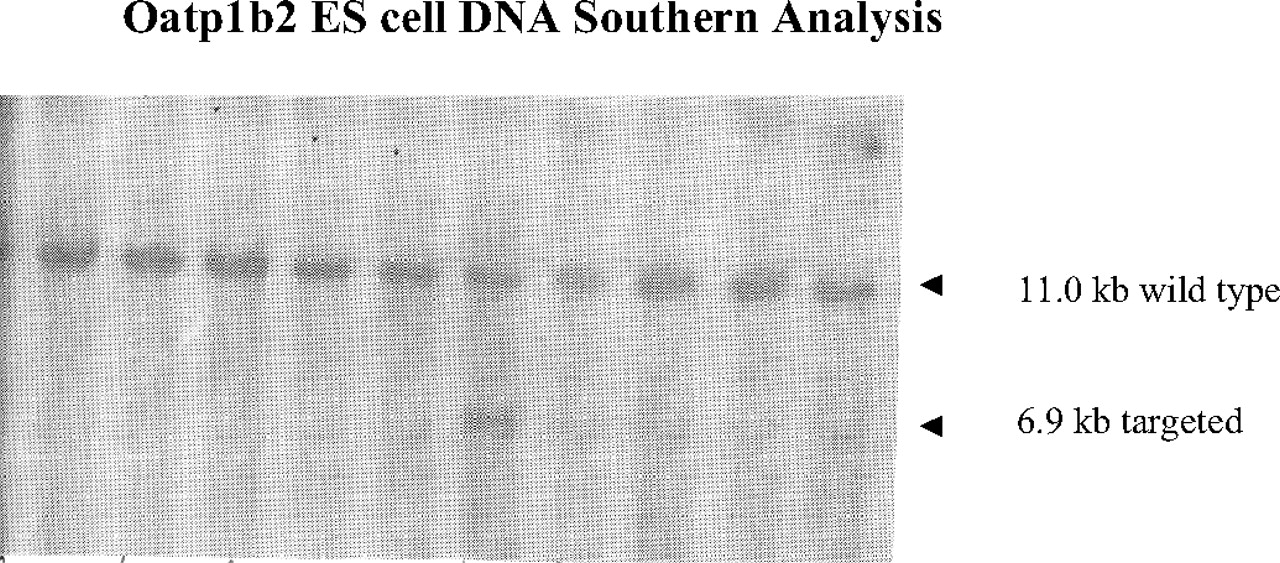
Oatp1b2 ES cell DNA Southern blot analysis from 10 ES clones. ES cell DNA was digested with BamHI and XhoI restricted enzymes, and Southern blot analysis was performed using the 3′ external probe. The probe hybridizes to an 11.0-kb endogenous (WT) Oatp1b2 fragment. The probe also hybridizes to a 6.9-kb targeted (KO) Oatp1b2 fragment as a result of the introduction of a new XhoI site in the neomycin cassette.
Data Analysis. The mean and S.D. of mRNA for the selective liver cytochromes P450 (P450s) and transporters, model compound concentrations, and the hepatic uptake index based on liver-to-plasma concentration ratios (Kp,liver) at 0.5 and 2 h postdose were calculated for both KO and WT mice. Student's t test (two-tailed) was used to compare the difference between the KO and WT mice for the reverse transcription-PCR, concentration, and Kp,liver data. P < 0.05 was considered statistically significant.
Results
Generation of Oatp1b2 KO Mice.Figure 1 shows the Southern blot analysis of Oatp1b2 ES cell DNA using a 3′ external probe. As expected, the probe not only hybridized to an 11.0-kb endogenous (WT) Oatp1b2 fragment but also to a 6.9-kb targeted (KO) Oatp1b2 fragment as a result of the introduction of a new XhoI site contained within the neomycin cassette. Genotyping by PCR using the neomycin set and the Oatp1b2 KO region specific set is shown in Fig. 2. The KO (–/–) animals are negative for the Oatp1b2 PCR and positive for the Neo PCR. A heterozygous (+/–) mouse was positive for both PCR sets, and a WT (+/+) mouse displayed positive for the Oatp1b2 PCR but negative for the Neo PCR.
Analysis of mRNA for a Selective Panel of P450s and Transporters in Liver. No product was yielded for Cyp2c39, Oatp1a5, organic anion transporter (Oat) 1, and Oat3 in either strain of mice. Oatp1b2 showed a significantly higher expression in the WT compared with the KO mice (P < 0.0001), but no statistically significant difference was detected in other P450s and transporters (Fig. 3).
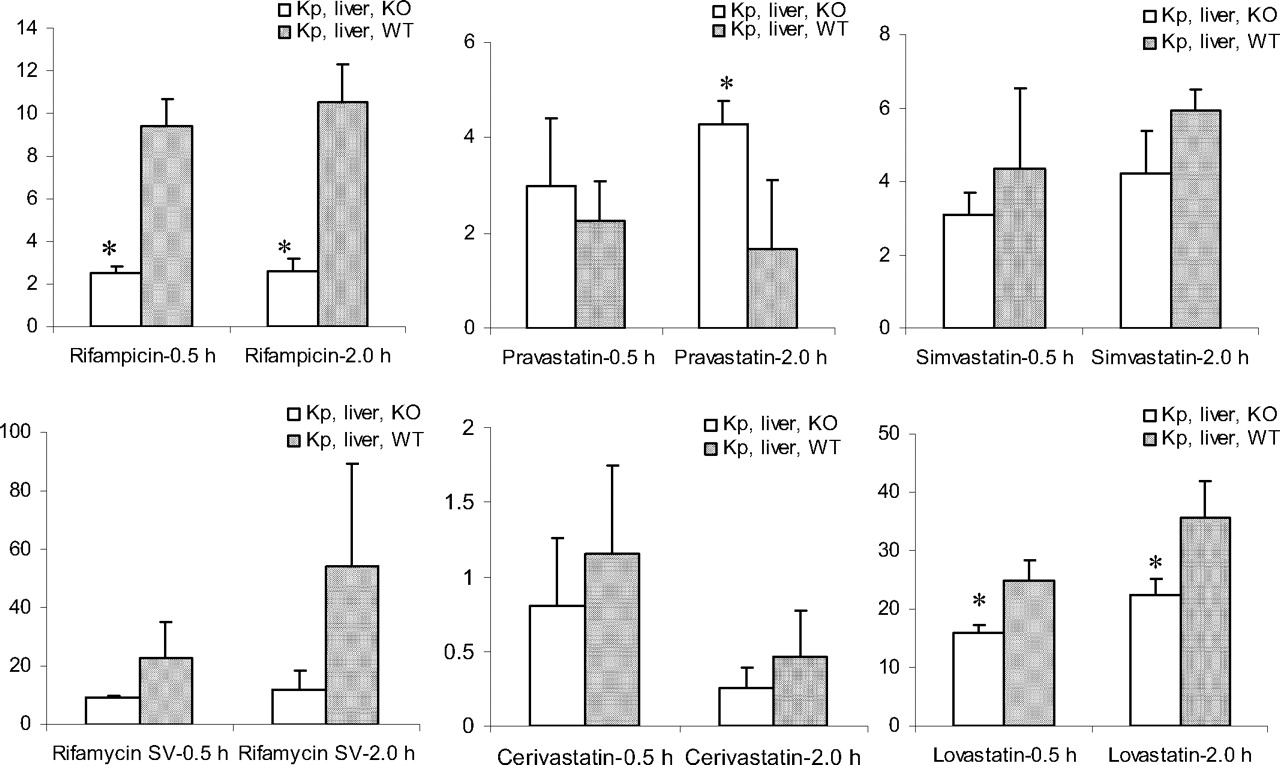
Hepatic Uptake of Six Model Compounds in Oatp1b2 KO and WT Mice. As shown in Table 1 and Fig. 4, rifampicin, rifamycin SV, and lovastatin acid displayed a Kp,liver of approximately 10 or higher, simvastatin acid and pravastatin between 1 to 5, and cerivastatin about 1 or lower in WT mice. This holds true in most cases in KO mice except for rifampicin, for which the Kp,liver value in KO is reduced 4-fold compared with the KO mice. Student's t test showed a statistically significant higher Kp,liver or liver uptake for rifampicin and lovastatin acid in WT compared with the KO mice.
Plasma and liver concentrations (mean ± S.D., n = 3 mice/strain/time point) and liver-to-plasma concentration ratio (Kp,liver) following an s.c. administration of the model compounds at 3 mg/kg to 4- to 6-week-old male Oatp1b2 KO and WT mice
Discussion
OATP1B1 has attracted much attention since it was cloned from human liver (Abe et al., 1999; Hsiang et al., 1999; Konig et al., 2000), especially in statin disposition and drug interaction (Charatan 2001; Launay-Vacher et al., 2005; Tiwari et al., 2006). As more statins enter the generic market, the possibility of coadministration of these statins with other drugs that are OATP1B1 substrates and/or inhibitors increases, and consequently the possibility of drug interaction and adverse effects also increases. Therefore, there is a need to have robust in vitro and in vivo tools to further understand the role of OATP1B1 in the hepatobiliary disposition of its substrates to predict prospectively the drug interactions, thus reducing and eradicating the incidence of adverse events.
P-glycoprotein (P-gp), the most well studied drug transporter, has been studied using both in vitro systems such as the MDR1-transfected cell lines and in vivo tools such as the Mdr1a- and Mdr1a/b-deficient mice (Chen et al., 2003, 2005, 2007). These studies have allowed us to relate data such as efflux ratio from in vitro P-gp–transfected systems to in vivo brain penetration (i.e., brain/plasma concentration ratio between the KO and WT mice). Similarly, the stably transfected OATP1B1 in various cell lines has allowed mechanistic studies to examine whether a compound is transported via OATP1B1 and whether the transport is inhibited by a concomitant drug. However, it is important to translate the in vitro transport property such as Km and IC50 to in vivo effects on plasma concentration of substrate and inhibitor of OATP1B1/Oatp1b2. Ideally, the Oatp1b2 KO mouse model would serve as an in vivo confirmation of the in vitro transport data.
We generated the Oatp1b2 KO mice and used them as an in vivo preclinical model to examine the effects of Oatp1b2 on hepatic uptake of model compounds. Our data show that Oatp1b2 KO mice lacked expression of Oatp1b2 but have similar expressions of the drug-metabolizing enzymes and other transporters examined compared with the WT mice.
Recently, Lu et al. (2008) published the generation of Oatp1b2 mouse model and use it to elucidate the role of Oatp1b2 in hepatic uptake and toxicity of phalloidin and microcystin-LR. Consistent with our findings, Lu et al. (2008) also showed no significant change in mRNA expression between the Oatp1b2 KO and WT mice in bile salt export pump (Bsep), Oatp1a1, breast cancer resistance protein (Bcrp), and multidrug resistance associated protein (Mrp) 2. Extrapolation of the difference observed in mRNA to protein level and activities requires further experiments.
The selection of the metabolizing enzymes and other transporters was based on the knowledge that they are potentially involved in the disposition of the model compounds. For example, CYP3A4 has been shown to metabolize both simvastatin and lovastatin, whereas CYP2C8/9 metabolizes cerivastatin (Shitara and Sugiyama, 2006). Rifampicin is known to be an inducer of CYP3A4 (Silva et al., 1999; LeCluyse et al., 2000), and no information is available for rifamycin SV, but it is likely to be metabolized by CYP3A. Therefore, both Cyp3a and Cyp2c families were examined. Similarly, studies have shown that multiple hepatic uptake and efflux transporters are involved in the hepatobiliary disposition of statins. For example, cerivastatin, pravastatin, simvastatin, and rosuvastatin are transported by OATP1B1 (Hsiang et al., 1999; Shitara et al., 2003, 2004; Niemi et al., 2004; Huang et al., 2006). Furthermore, lovastatin and simvastatin are weak to moderate substrates for P-gp (Chen et al., 2005), pravastatin is a substrate for MRP2/Mrp2 (Yamazaki et al., 1997) and BSEP (Hirano et al., 2005a,b), and BCRP is responsible for the biliary excretion of rosuvastatin (Huang et al., 2006). Therefore, the Mdr1, Mrp, Bsep, and Bcrp families were selected and examined. Other transporters in the Oatp1a and Oat families were also investigated to ensure no potential alteration as a result of knockout of Oatp1b2; these transporters have been also shown to be important in hepatobiliary handling of xenobiotics (Sekine et al., 1998; Kusuhara et al., 1999; Lickteig et al., 2007; Rizwan and Burckhardt, 2007). Our data showed that no alteration occurred in enzymes and transporters that are known to be involved in the disposition of the model compounds in the Oatp1b2 KO mice, suggesting the validity of using the model to examine the role of Oatp1b2 in the hepatobiliary disposition of these model compounds.

Oatp1b2 KO mouse genotyping of 11 blinded samples. PCR analysis using two oligo sets, neomycin and Oatp1b2, was performed. The neomycin set (NeoF, 5′-gcaggatctcctgtcatctcacc-3′ and NeoR, 5′-gatgctcttcgtccagatcatcc-3′) amplified a 190-bp product from an Oatp1b2-targeted allele, whereas the Oatp1b2 set (Oatp1b2F, 5′-tggacaatgtgcaatgggagc-3′ and Oatp1b2R, 5′-gaaagagctgattagagatacg-3′) amplified a 497-bp product from an Oatp1b2 WT allele.

Reverse transcription-PCR analysis of a selective panel of liver P450s and transporters. Data are expressed as mean ± S.D. (n = 3 livers from male mice that had similar age as those used for the liver uptake study in vivo).
Our observation in the expression of relevant drug-metabolizing enzymes and transporters was consistent and complement with Zaher et al. (2008). Two studies have overlap in eight genes examined: Oatp1b2, Oatp1a1, Oatp1a4, Mdr1b, Mrp2, Mrp3, Bcrp, and Bsep. However, our study examined 10 new genes (five P450s and five transporters: Mdr1a, Oat1, Oat2, Oat3, Oatp1a5, Cyp2c39, Cyp2c29, Cyp3a13, Cyp3a16, and Cyp3a11) and their six genes (Mrp4, Ostα, Ostβ, Ntcp, Oat7, and Oatp2b1). Our selection of genes was to fulfill two purposes: characterization of the mouse model and to ensure the potential responsible transporters and metabolizing enzymes were not altered for the six probe compounds. In the case of rifampicin, a known substrate/inhibitor of P-gp, it is important to examine Mdr1a gene expression; we did, but Zaher et al. (2008) did not. Moreover, overlap in substrate specificity and interplay between P450s and transporters make it critical to examine the potential alteration in P450 expressions in KO mice. We did, but Zaher et al. (2008) did not. Nevertheless, together data from both studies have strengthened the characterization of the model as a useful tool for the study of hepatobiliary disposition of many organic anion compounds/drugs. Comparison of Kp,liver, an index for hepatic uptake between the WT and KO mice, assesses the contribution of Oatp1b2 to the overall hepatobiliary disposition of a model compound. Similar to OATP1B1, Oatp1b2 is expressed at the sinusoidal membrane of liver (Cheng et al., 2005) and is responsible for the uptake of its substrates from the blood to the liver, resulting in a higher than unity of liver-to-plasma concentration ratio (Kp,liver). Assuming there is no difference in relevant metabolizing enzymes and other transporters that are relevant to the hepatobiliary disposition of a compound between the Oatp1b2 KO and WT mice, when Oatp1b2 uptake does not represent a rate-limiting step, Kp,liver would be near unity and similar between the two strains of mice. In contrast, when Oatp1b2 uptake plays a critical role in the hepatobiliary disposition of the compound, Kp,liver values in WT mice would be significantly higher than unity and that of KO mice. Our data showed that Oatp1b2 played a significant part in the uptake of rifampicin and lovastatin acid to the liver and in their overall hepatobiliary disposition processes.

Comparison of Kp,liver (mean ± S.D., n = 3 mice/strain/time point) between 4- to 6-week-old male Oatp1b2 KO and WT mice.
In the case of rifampicin, our data are consistent with Zaher et al. (2008). Furthermore, the in vivo mouse data are consistent with data from in vitro human OATP1B1-expressed cells (Vavricka et al., 2002), suggesting lack of substantial species-dependent substrate specificity. This observation is in good agreement with Lau et al. (2006), who have shown that rifampicin significantly reduced the uptake of atorvastatin, another known OATP1B1 substrate, to rat liver. Unlike rifampicin, rifamycin SV did not exhibit significant liver uptake by Oatp1b2. The difference between the two analogs is consistent with an in vitro interaction study where they showed that the two antibiotics interact differently with OATP1B1-mediated sulfobromophthalein uptake (Vavricka et al., 2002).
Among the statins tested, we showed for the first time that Oatp1b2 apparently played a significant role in the uptake of lovastatin acid to the liver, consistent with our previous in vitro results from OATP1B1 (Chen et al., 2005), suggesting negligible species difference for lovastatin acid. Interestingly, pravastatin Kp,liver at 2 h was significantly higher in KO than WT mice. The underlying mechanism is not known and requires further study. Pravastatin was also examined by Zaher et al. (2008) following an s.c. infusion for 24 h at two doses. Although the WT mice showed a higher liver-to-plasma concentration ratio at both doses compared with the KO mice and the difference diminished as dose increased, a similar magnitude in dose-related reduction in liver-to-plasma ratio in both strains suggests factors other than Oatp1b2 affecting the hepatobiliary disposition of pravastatin. In contrast, lack of differences in Kp,liver between the KO and WT mice for cerivastatin and simvastatin acid suggests possible species difference between rodent Oatp1b2 and human OATP1B1/1B3 and/or lack of critical role of Oatp1b2 in the overall hepatobiliary disposition of these compounds. Further studies using vitro tools such as OATP1B1/1B3 and Oatp1b2 transfected systems will provide additional evidence regarding species-related substrate specificity.
In summary, the present study describes the generation of the Oatp1b2 KO mouse model for the study of the potential impact of Oatp1b2 on hepatic uptake of four statins (cerivastatin, lovastatin acid, pravastatin, simvastatin acid) and two antibiotics (rifampicin and rifamycin SV) in Oatp1b2 KO and WT mice. Our data showed that Oatp1b2 plays a significant role in the access of the model compounds to the liver and that potential species differences in substrate specificity exist between rodent Oatp1b2 and human OATP1B1. Together with studies by Lu et al. (2008) and Zaher et al. (2008), we have shown that the Oatp1b2 KO mouse is a valuable tool to understand active drug/toxin uptake processes and enable clinical predictions.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.020594.
-
ABBREVIATIONS: Oatp/OATP, organic anion transporting polypeptide; WT, wild type; MDR1/Mdr1, multidrug resistance gene 1; KO, knockout; kb, kilobase; ES, embryonic stem; PCR, polymerase chain reaction; bp, base pair; HPLC/MS, high-performance liquid chromatography/mass spectrometry; P450, cytochrome P450; Oat/OAT, organic anion transporter; P-gp, P-glycoprotein; Bsep/BSEP, bile salt export pump; Bcrp/BCRP, breast cancer resistance protein; Mrp/MRP, multidrug resistance associated protein; GAPD, glyceraldehyde-3-phosphate dehydrogenase; CT, cycle time.
- Received January 25, 2008.
- Accepted June 10, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}