


Abstract
Although primary human hepatocytes are commonly used for induction studies, the evaluation method is associated with several problems. More recently, a reporter gene assay has been suggested to be an alternative, although the contribution of only transfected nuclear receptors can be evaluated. The aim of the present study was to establish a method by which the extent of in vivo CYP3A4 induction in humans can be quantitatively predicted based on in vitro results with a reporter gene assay. From previous reports, we calculated in vivo induction ratios (Rin vivo) caused by prototypical inducers based on the alterations in the hepatic intrinsic clearance of probe drugs. Next, we derived equations by which these Rin vivo values can be predicted from the results of a reporter gene assay. To use the data obtained from a reporter gene assay, rifampicin was used as a reference drug. The correction coefficient (CC), which is used to quantitatively correlate the activity of inducers between in vitro and in vivo situations, was calculated by comparing the predicted data with the observed Rin vivo values for rifampicin. With the calculated CC value, good correlations were found between the predicted and observed Rin vivo values for other inducers such as phenobarbital, phenytoin, and omeprazole. Taken together, with the equations derived in the present study, we have been able to predict the extent of in vivo induction of human CYP3A4 by inducers in a time-dependent and quantitative manner from in vitro data.
For proper drug therapy, coadministration of drugs should be closely monitored, because drug-drug interactions (DDIs) potentially entail an increase or a decrease in the pharmacological action of coadministered drugs, which consequently affects the risk of adverse reactions and the effectiveness of drug therapy (Periti et al., 1992; Backman et al., 1996). In some cases, DDIs have resulted in serious clinical problems including patient death due to drug adverse reactions (Okuda et al., 1997). Therefore, for successful drug development, it is important to investigate the potential of candidate drugs to cause DDIs at an early stage in their development (Hewitt et al., 2007). The most common mechanisms of DDIs are inhibition and induction of drug-metabolizing enzymes (Kato et al., 2005; Ohno et al., 2007). Many attempts have been made to predict the extent of in vivo enzyme inhibition from in vitro data (Nomeir et al., 2001; Zhao et al., 2005).
In contrast, there are difficulties in predicting the extent of in vivo enzyme induction in a quantitative manner from in vitro data. Human hepatocytes have often been used to examine enzyme induction, because they are considered to exhibit the properties necessary for enzyme induction (Rodriguez-Antona et al., 2001; Gómez-Lechón et al., 2004). This methodology may be reasonable if we consider the fact that human hepatocytes can be used as a good model to estimate the extent of drug metabolism and transport (Sandker et al., 1994; Salonen et al., 2003). However, quantitatively speaking, the evaluation method often used in the prediction of enzyme induction is associated with several problems. In most reports, the potency of an inducer is given as the ratio of the enzyme amount or activity after induction to that before induction (Table 1). The accuracy of this evaluation method depends entirely on the accuracy of evaluating the enzyme expression level before initiation of the induction experiments. We have summarized the data on in vitro induction experiments using human primary cultured hepatocytes from previous reports in Table 1, focusing in particular on CYP3A4 as the clinically most important metabolizing enzyme. Large variability in the reported values for the induction ratio is associated with the variability of the enzyme expression level before initiation of the induction experiments (LeCluyse et al., 2000a). The variability before induction is associated with the quite marked interindividual differences in the activity of CYP3A4 to some extent (Ponsoda et al., 2001). For example, by analyzing the enzyme activities in 64 human hepatocyte preparations, Parkinson et al. (2004) reported that the values deviate so much that the S.D. value is almost the same as the mean value (3910 ± 3370 pmol/mg of protein/min, n = 64) (Parkinson et al., 2004). The activities of metabolizing enzymes in human hepatocytes are also affected by differences in the preparation methods and culture conditions among laboratories (LeCluyse et al., 2000b; Hamilton et al., 2001). Moreover, down-regulation of CYP3A4 expression has also been observed during the continued culture of human hepatocytes (Hamilton et al., 2001; Rodriguez-Antona et al., 2001). Taken together, the evaluation method for the potency of inducers based on the results expressed as the induction ratio obtained from in vitro experiments with human hepatocytes may not be appropriate for quantitatively validating the potency of drugs to induce CYP3A4.
Variability of response to induction in primary human hepatocytes
The previously reported values for the extent of induction of CYP3A4 and/or its metabolizing activity are summarized to indicate its variability. The induction ratio was defined as the ratio of the expression level and/or metabolizing activity of CYP3A4 before and after inducer treatment.
More recently, it was reported that the reporter gene assay may be used as an alternative method for evaluating the induction responses of drugs (Goodwin et al., 1999; Moore et al., 2000). Because established cell lines can be used in the reporter gene assay, the assay stability and experimental reproducibility may be much more uniform among laboratories than those for the assay using human hepatocytes. With regard to the nuclear receptors used in the reporter gene assay, pregnane X receptor (PXR) and constitutive androstane receptor (CAR) are important for examining the induction of CYP3A4 (Timsit and Negishi, 2007). PXR is activated by binding of an inducer to PXR itself, whereas CAR is not necessarily activated by inducer binding (Kawamoto et al., 1999). Because the molecular mechanism of induction is not yet fully understood, it has been assumed that it may be difficult to quantitatively predict in vivo enzyme induction profiles from the results of in vitro experiments with the reporter gene assay. In the present study, we have demonstrated the usefulness of the physiologically based prediction method of in vivo induction of CYP3A4 in humans in a quantitative manner from the results of in vitro experiments with the reporter gene assay.
Materials and Methods
Analysis of in Vivo CYP3A4 Induction. First, we collected data on the CYP3A4 induction caused by five prototypical inducers (rifampicin, phenobarbital, phenytoin, carbamazepine, and omeprazole) from previous reports (Table 2). As an index of enzyme induction, the ratio of the hepatic intrinsic clearance of probe drugs after the administration of an inducer (CLint,after) to that before administration (CLint,before) was defined by eq. 1: 
Rin vivo values of inducers
Previous reports on the in vivo disposition of probe substrate in humans before and after administration of inducers were analyzed to determine Rin vivo. CR3A4 values were taken from our previous report.
For the calculation of the CLint of probe drugs listed in Table 2, the following methods were used. For probe drugs administered intravenously, their total body clearance (CL) was calculated from the reported dose (D) and area under the plasma concentration-time curve (AUCi.v.) with eq. 2: 
For probe drugs administered orally, CL was calculated from the data reported in Goodman and Gilman's textbook (Hardman et al., 2001) and drug package inserts on the elimination half-life (t1/2) and volume of distribution (Vd) with eq. 3, to focus on the induction in hepatic enzymes by minimizing the first-pass effect after oral administration of drugs: 
In these analyses, a one-compartment model was used for the analysis of the pharmacokinetics of probe drugs, and either the volume of distribution or renal clearance was assumed not to be affected by coadministration of an inducer. The hepatic clearance (CLh) of probe drugs was then calculated from CL, and the urinary excretion ratio (U) was calculated according to eq. 4: 
In these calculations, the U values for probe drugs were taken from Goodman and Gilman's textbook (Hardman et al., 2001) and drug package inserts. Based on the well stirred model, the CLint of probe drugs was then calculated from CLh with eq. 5,  where Qh,RB, and fP represent the hepatic blood flow rate, blood/plasma concentration ratio, and the plasma unbound fraction of probe drugs, respectively. Qh and RB were assumed to be 96.6 l/h and 1, respectively. fP values were from Goodman and Gilman's textbook (Hardman et al., 2001) and drug package inserts. By using these calculated CLint values of probe drugs before and after inducer treatment, Rapp was determined for probe drugs with eq. 1.
where Qh,RB, and fP represent the hepatic blood flow rate, blood/plasma concentration ratio, and the plasma unbound fraction of probe drugs, respectively. Qh and RB were assumed to be 96.6 l/h and 1, respectively. fP values were from Goodman and Gilman's textbook (Hardman et al., 2001) and drug package inserts. By using these calculated CLint values of probe drugs before and after inducer treatment, Rapp was determined for probe drugs with eq. 1.
Finally, for the ratio of the contribution of CYP3A4 to the hepatic intrinsic clearance of the probe substrates before inducer treatment (CR3A4), which was taken from our previous report (Ohno et al., 2007), the in vivo intrinsic induction ratio of CYP3A4 (Rin vivo) was calculated from Rapp with eq. 6: 
For the analysis of clinical studies evaluating CYP3A4 induction using the endogenous cortisol metabolism, Rin vivo, values were calculated simply by dividing the urinary metabolic ratio (6β-hydroxycortisol/cortisol or 17-hydroxycorticosteroids) after an inducer treatment by that before an inducer treatment. Rin vivo values, which depend on the kind, the dose, and the administration duration of inducers, were used for the subsequent analyses.
Analysis of in Vitro CYP3A4 Induction. For the studies with the reporter gene assay for PXR-mediated induction of CYP3A4, the induction potency of an inducer was given by a reporter activity at each inducer concentration. In the case of the reporter gene assay, the absolute value of the induction ratio, which is obtained by dividing the reporter activity after an inducer treatment by that before an inducer treatment, does not carry a physiological meaning. Therefore, we chose rifampicin as a reference compound and evaluated the induction potency of other inducers by normalizing the extent of induction by inducers in terms of that of rifampicin. For these analyses, we adopted the Emax model, which is often used for describing the characteristics of the induction potency of an inducer. For rifampicin treatment, the induction ratio (Rin vitro,RIF) can be described as follows (eq. 7):  where Emax,RIF, EC50,RIF, fm,RIF, and Cm,RIF represent the maximum activity of rifampicin to induce CYP3A4 in the reporter gene assay, the unbound concentration of rifampicin at the half-maximal activity of its induction potency, the unbound fraction of rifampicin in the incubation medium, and the total concentration of rifampicin in the medium, respectively. In the same way, the induction ratio for other inducers (Rin vitro) can be described as follows (eq. 8):
where Emax,RIF, EC50,RIF, fm,RIF, and Cm,RIF represent the maximum activity of rifampicin to induce CYP3A4 in the reporter gene assay, the unbound concentration of rifampicin at the half-maximal activity of its induction potency, the unbound fraction of rifampicin in the incubation medium, and the total concentration of rifampicin in the medium, respectively. In the same way, the induction ratio for other inducers (Rin vitro) can be described as follows (eq. 8):  where Emax, Emax,relative, EC50, fm, and Cm represent the maximum activity of an inducer to induce CYP3A4 in the reporter gene assay, the maximal induction potential of an inducer defined as a value relative to that of rifampicin, the unbound concentration of an inducer at the half-maximal activity of its induction potency, the unbound fraction of inducers in the incubation medium, and the total concentration of inducers in the medium, respectively. The introduction of Emax,relative enabled us to compare the data from different reports despite the difference in the absolute values of the induction ratio, which depend largely on the experimental conditions.
where Emax, Emax,relative, EC50, fm, and Cm represent the maximum activity of an inducer to induce CYP3A4 in the reporter gene assay, the maximal induction potential of an inducer defined as a value relative to that of rifampicin, the unbound concentration of an inducer at the half-maximal activity of its induction potency, the unbound fraction of inducers in the incubation medium, and the total concentration of inducers in the medium, respectively. The introduction of Emax,relative enabled us to compare the data from different reports despite the difference in the absolute values of the induction ratio, which depend largely on the experimental conditions.
To determine the EC50 values for the analysis, we surveyed previous reports in which the reporter construct, including both the proximal promoter region and the distal xenobiotic responsive enhancer module, was used; these elements are necessary for the maximal induction response of the CYP3A4 gene caused by PXR. In Table 3, the EC50 values of rifampicin obtained from the reporter gene assay are summarized. The algebraic mean of the EC50 values (0.92 ± 0.72 μM) was used for subsequent analysis. The Emax,relative, and EC50 values for other inducers are summarized in Table 4.
EC50 values of rifampicin to induce CYP3A4 determined by in vitro reporter gene assay
Previous reports on the in vitro reporter gene assay were surveyed to summarize the EC50 values of rifampicin.
Emax,relative and EC50 values of inducers determined by in vitro reporter gene assay
Previous reports on the in vitro reporter gene assay were surveyed to summarize the Emax,relative and EC50 values of phenobarbital, phenytoin, carbamazepine, and omeprazole.
For the calculation of fm in eqs. 7 and 8, it was assumed that there is no difference in the binding of inducers to human and bovine serum albumin, so that the binding capacity of an inducer to albumin in the medium can be calculated from the unbound fraction in human plasma. Based on the Langmuir equation, eq. 9 holds,  where n, Pt, and Kd represent the number of binding sites, albumin concentrations and dissociation constant, respectively. The fm values were calculated by correcting human nPt/Kd by the ratio (a) of the bovine serum albumin concentration in the medium to the human albumin concentration in plasma (eq. 10):
where n, Pt, and Kd represent the number of binding sites, albumin concentrations and dissociation constant, respectively. The fm values were calculated by correcting human nPt/Kd by the ratio (a) of the bovine serum albumin concentration in the medium to the human albumin concentration in plasma (eq. 10): 
Construction of the Prediction Model. Then, we derived equations with which Rin vivo can be predicted. The mass balance equation for the amount of CYP3A4 mRNA in the liver [m(t)] after an inducer treatment is given by eq. 11:  where vconst represents synthetic velocity of constitutive expression of CYP3A4 mRNA, correction coefficient (CC) represents the coefficient to correlate the extent of in vitro induction to in vivo induction, kdeg,m represents the degradation rate constant of the mRNA, and Ch,u(t) represents the time profiles of the unbound concentration of inducers in the liver. In the following calculation with eq. 11, the values of Emax,relative and EC50 were taken from Table 4. For kdeg,m, although the half-life of human CYP3A4 mRNA is not available, we assumed the half-life of CYP3A4 mRNA to be 10 h based on a previous report (Table 5). For each calculation, the vconst value was defined to be equal to kdeg,m multiplied by the amount of mRNA before induction [m(0)].
where vconst represents synthetic velocity of constitutive expression of CYP3A4 mRNA, correction coefficient (CC) represents the coefficient to correlate the extent of in vitro induction to in vivo induction, kdeg,m represents the degradation rate constant of the mRNA, and Ch,u(t) represents the time profiles of the unbound concentration of inducers in the liver. In the following calculation with eq. 11, the values of Emax,relative and EC50 were taken from Table 4. For kdeg,m, although the half-life of human CYP3A4 mRNA is not available, we assumed the half-life of CYP3A4 mRNA to be 10 h based on a previous report (Table 5). For each calculation, the vconst value was defined to be equal to kdeg,m multiplied by the amount of mRNA before induction [m(0)].
Half-lives of human CYP3A4
The half-lives of human CYP3A4 mRNA and proteins were summarized from previous reports.
In the calculation with eq. 10, we assumed that Ch,u(t) is equal to the unbound concentration in the portal vein [Cpv,u(t)]. Because in the present study, we analyzed the data from previous reports on the effects of orally administered inducers, Cpv,u(t) was calculated using eq. 12. In this calculation, we assumed an absorption lag-time (Tlag) of 0.5 h after drug administration:  where ka and ke represent the absorption and elimination rate constants of an inducer, respectively, and Qpv represents the blood flow rate in the portal vein. The values of ka, ke, fp, and Vd of inducers were taken from Goodman and Gilman's textbook (Hardman et al., 2001) and drug package inserts.
where ka and ke represent the absorption and elimination rate constants of an inducer, respectively, and Qpv represents the blood flow rate in the portal vein. The values of ka, ke, fp, and Vd of inducers were taken from Goodman and Gilman's textbook (Hardman et al., 2001) and drug package inserts.
Then, based on the m(t) calculated by eq. 11, we calculated the mass balance equation for the amount of CYP3A4 protein in the liver [p(t)] given by eq. 13:  where ktrans and kdeg,e represent the production rate constant of enzyme by translation of the mRNA and degradation rate constant of CYP3A4 protein, respectively. The values for the half-life of human CYP3A4 protein were obtained from in vivo and in vitro studies (Table 5). The reported half-lives ranged from 1 to 6 days. We performed calculations by assuming that the half-life of CYP3A4 protein is 24, 72, and 144 h.
where ktrans and kdeg,e represent the production rate constant of enzyme by translation of the mRNA and degradation rate constant of CYP3A4 protein, respectively. The values for the half-life of human CYP3A4 protein were obtained from in vivo and in vitro studies (Table 5). The reported half-lives ranged from 1 to 6 days. We performed calculations by assuming that the half-life of CYP3A4 protein is 24, 72, and 144 h.
In the present analysis, we defined Rpredict(t) by eq. 14:  where p(0) represents the amount of CYP3A4 in the liver before inducer administration. Rpredict(t) was calculated in a numerical manner by substituting the required parameter values. To calculate the value of CC, the Rpredict(t) values were fitted to the reported data of Rin vivo values due to rifampicin treatment (Table 2) using the SAAM II program (SAAM Institute, Seattle, WA). Rifampicin was used as a compound to determine CC values, because it is a strong PXR-selective inducer used as a positive control in numerous in vivo and in vitro studies (Lemaire et al., 2004; Faucette et al., 2007). In the analysis, we used some physiological parameters, the absolute values of which are listed in Table 6. The degradation rate constants of CYP3A4 mRNA and protein (kdeg,m and kdeg,e) were 0.0693 and 0.00963 h–1, respectively, and both of these were within the reported ranges (Table 5). The CC value was determined to be 11.1. Then, by using this calculated CC value, we calculated the Rpredict(t) values for other inducers, such as phenobarbital, phenytoin, carbamazepine, and omeprazole and compared the results with their observed Rin vivo values.
where p(0) represents the amount of CYP3A4 in the liver before inducer administration. Rpredict(t) was calculated in a numerical manner by substituting the required parameter values. To calculate the value of CC, the Rpredict(t) values were fitted to the reported data of Rin vivo values due to rifampicin treatment (Table 2) using the SAAM II program (SAAM Institute, Seattle, WA). Rifampicin was used as a compound to determine CC values, because it is a strong PXR-selective inducer used as a positive control in numerous in vivo and in vitro studies (Lemaire et al., 2004; Faucette et al., 2007). In the analysis, we used some physiological parameters, the absolute values of which are listed in Table 6. The degradation rate constants of CYP3A4 mRNA and protein (kdeg,m and kdeg,e) were 0.0693 and 0.00963 h–1, respectively, and both of these were within the reported ranges (Table 5). The CC value was determined to be 11.1. Then, by using this calculated CC value, we calculated the Rpredict(t) values for other inducers, such as phenobarbital, phenytoin, carbamazepine, and omeprazole and compared the results with their observed Rin vivo values.
List of parameter values used in the present study
Criteria for the use of other parameter values are described in the text.
Results
In Vivo Data.Table 2 shows the in vivo induction ratio of CYP3A4 (Rin vivo) calculated from the hepatic intrinsic clearances of CYP3A4 substrates, before and after inducer treatment. The Rin vivo values vary, depending on the differences in dose and treatment duration (Table 2). Rin vivo values with 600 mg/day rifampicin treatment for more than 7 days were in the range of 2.6- to 5.2-fold (Table 2). The Rin vivo values with phenobarbital were almost identical to those with carbamazepine and phenytoin, although the dose of phenobarbital was less (Table 2). All inducers except for rifampicin caused less than a 3-fold induction. In particular, the Rin vivo values of omeprazole were very small (Table 2).
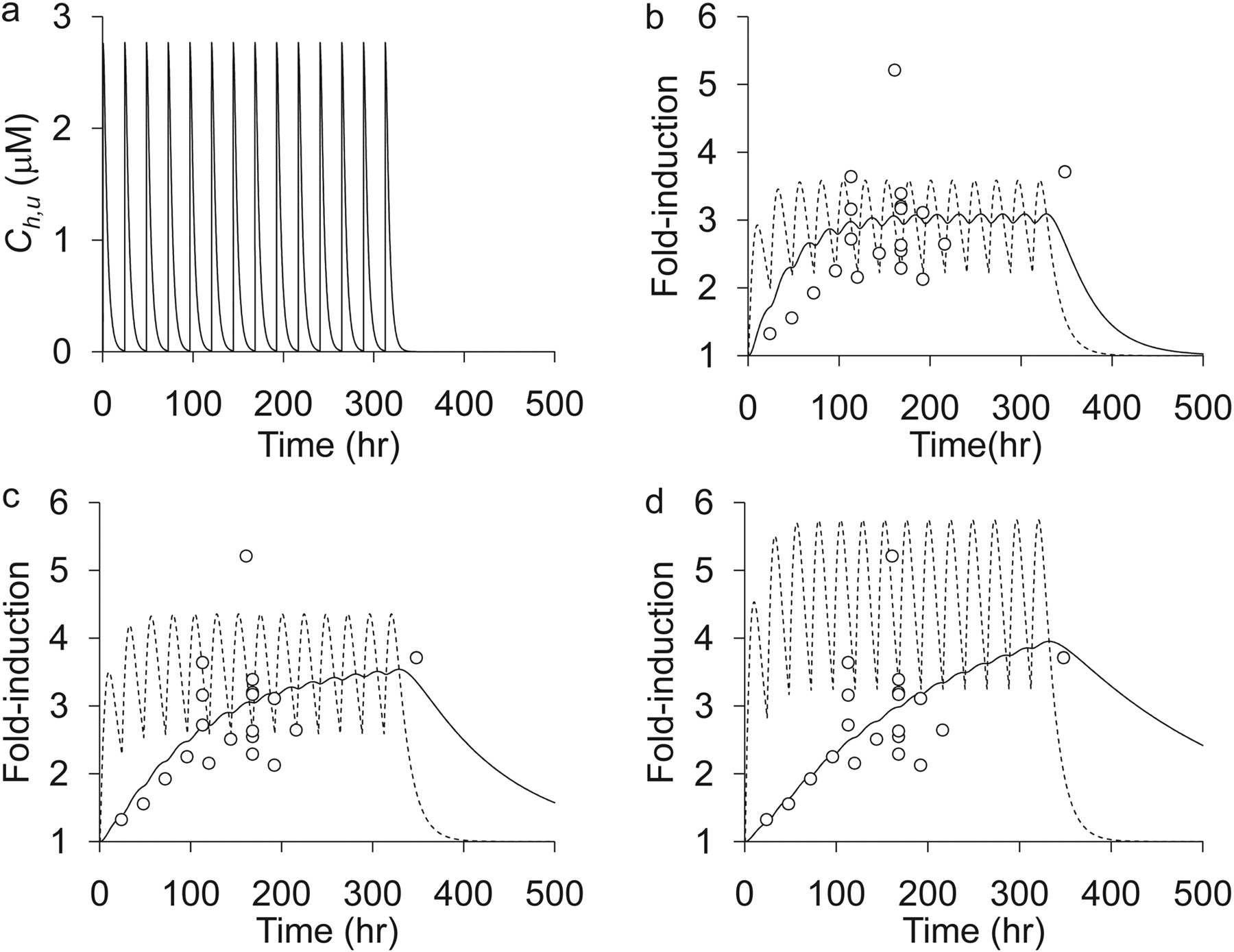
Prediction ofRin vivo of Rifampicin from in Vitro Data with the Reporter Gene Assay.Figure 1a shows the time profiles of the unbound concentration of rifampicin in the liver [Ch,u(t)] after administration of an oral dose of 600 mg/day. Using this calculated Ch,u(t), we determined the CC, which correlates the extent of in vitro induction to in vivo induction, by fitting the predicted values of CYP3A4 induction ratio (Rpredict) to the observed values of Rin vivo for rifampicin. Because the half-life of CYP3A4 protein is necessary to determine CC values, we performed the fitting by assuming that the half-life of CYP3A4 protein is 24, 72, and 144 h (Fig. 1, b, c, and d, respectively). In this figure 1 the increases in mRNA levels and Rpredict after oral administration of rifampicin (600 mg/day) are shown by dotted and solid lines, respectively. By comparing the Rpredict values with the observed Rin vivo values, it was found that Rin vivo can be predicted by assuming that the half-life of CYP3A4 is 72 h (Figs. 2, a–c), and in that case, the CC value was calculated to be 11.1. For the following calculation, the half-life of CYP3A4 and CC were assumed to be 72 h and 11.1, respectively. Then, we checked whether the Rin vivo values could be predicted for different doses of rifampicin by using the above parameter values (Kanebratt et al., 2008). The reported Rin vivo values after the administration of 20, 100, and 500 mg of rifampicin daily for 14 days were 1.5, 2.5, and 4.0, respectively, whereas the Rpredict values were 1.2, 1.9, and 3.5 (Table 7). We think that the accuracy of the above prediction is acceptable, considering that the data obtained for much higher doses (600 and 1200 mg of rifampicin daily) were used to construct the prediction model.
Prediction of Rin vivo values for different doses of rifampicin
Prediction of CYP3A4 Induction Profiles by Inducers Other Than Rifampicin.Table 4 summarizes the Emax,relative, and EC50 values of each inducer determined in vitro, which represent the maximal induction potential of an inducer defined as a value relative to that of rifampicin and the unbound concentration of an inducer at the half-maximal activity of its induction potency, respectively. There was only one report investigating the EC50 of phenytoin and omeprazole (Table 4). By using these parameter values, the Rin vivo values were numerically predicted for other inducers. Figure 3 shows the time profiles of Ch,u(t), the increase in mRNA levels, and Rpredict after treatment with phenobarbital, phenytoin, carbamazepine, and omeprazole. In addition, Fig. 4 shows the correlations between the observed and predicted induction ratios with these drugs. Although excellent correlations were observed for the induction by phenobarbital, phenytoin, and omeprazole, the prediction of induction by carbamazepine was unsuccessful (Fig. 4, a–d).
Discussion
The induction of drug-metabolizing enzymes in the liver possibly affects the disposition of coadministered drugs. Although a number of metabolic enzymes, such as CYP2B, CYP2C, and CYP3A, are known to be induced with drugs, CYP3A4 is the most abundant cytochrome P450 isoform in the liver and is involved in the metabolism of approximately 50% of drugs used in clinical situations (Zuber et al., 2002). In the present study, we focused on CYP3A4 induction in the liver and established a method to predict in vivo CYP3A4 induction from in vitro data.
A reporter gene assay, which uses a mammalian cell line transfected with both a target nuclear receptor and a construct including a reporter gene downstream of target gene promoter regions, is a common tool to evaluate the function of a nuclear receptor in a quantitative manner. By standardizing experimental conditions, such as the cell line, constitution of the gene constructs, and duration of an inducer treatment, and by normalizing the results of experiments with those of rifampicin, the experimental results are considered to be reproducible, and the variation of results among laboratories can be minimized. Moreover, it is suitable for use in high throughput screening because of the availability of materials. In the present study, we focused on the PXR-mediated induction of CYP3A4.

Determination of CC values from the data on rifampicin. a, predicted unbound concentration in the liver (Ch,u) of rifampicin after administration of 600 mg/day. b, c, and d, comparison between the observed Rin vivo and Rpredict calculated by assuming the half-life of CYP3A4 to be 24, 72, and 144 h, respectively. ○, observed Rin vivo values taken from Table 2. Dotted and solid lines represent the predicted increase in mRNA for CYP3A4 and CYP3A4 proteins (Rpredict values), respectively.
First, to determine the CC value, the in vivo induction of CYP3A4 by rifampicin in humans was analyzed on the basis of the EC50 value determined in vitro and pharmacokinetic parameters of rifampicin. With the calculated CC value, we found that in vivo induction by other inducers can be predicted from in vitro results obtained in reporter gene assays (Figs. 3 and 4), except for carbamazepine (Figs. 3 and 4). For carbamazepine, Rpredict values were underestimated compared with the observed Rin vivo values (Figs. 3 and 4). In this study, we assumed that we can compare the unbound concentration of an inducer in the portal vein (in vivo) and the unbound concentration of that in the medium (in vitro). Therefore, the existence of a marked difference in the situation between in vitro and in vivo studies regarding the distribution of an inducer into hepatocytes may result in the underestimation or overestimation of the in vivo induction ratio. Alternatively, we neglected the contribution of metabolites of an inducer to the CYP3A4 induction. For metabolites having induction ability, this may also lead to the underestimation of the in vivo induction ratio. Oscarson et al. (2006) reported that carbamazepine-10,11-epoxide, which is a major metabolite of carbamazepine catalyzed by CYP3A4 with a usual plasma concentration of 10 to 50% of that of carbamazepine, exhibits the same degree of PXR activation potency as carbamazepine itself. Consequently, this discrepancy between Rin vivo and Rpredict for carbamazepine may be accounted for by considering the contribution of its metabolite. Conversely, when the prediction of the in vivo induction of CYP3A4 was made for other inducers and the Rpredict values were still underestimated after the calculation was corrected considering the hepatic uptake of an inducer, it is necessary to consider the possibility that the metabolites also have the ability to induce CYP3A4.
Then, the difference in the induction activity between in vitro and in vivo studies should be discussed. Because the Emax,relative, of omeprazole was 147%, it appears that this compound has a higher activation potency of PXR than rifampicin, whereas its Rin vivo was the lowest of the inducers examined in the present study (Tables 3 and 4). This result may be accounted for by considering the disposition of omeprazole. As shown in Fig. 3, the mean Ch,u(t) of omeprazole (0.0402 μM) was much lower than its EC50 value determined by the in vitro reporter gene assay (3.54 μM) (Table 4). In contrast, the mean Ch,u(t) values for other compounds (0.566, 22.7, and 1.70 μM for rifampicin, phenobarbital, and phenytoin, respectively) were closer to their EC50 values (0.92, 251, and 4.02, respectively) (Tables 3 and 4). In addition, despite the fact that the ratio of Ch,u(t) to the EC50 of phenobarbital was smaller than that of phenytoin, their observed Rin vivo values were similar. These results may be accounted for by considering the difference in Emax,relative between these two inducers (phenobarbital > phenytoin) (Table 4).

Correlations between Rin vivo and Rpredict for rifampicin. a, b, and c, comparison between the observed Rin vivo and Rpredict calculated by assuming the half-life of CYP3A4 to be 24, 72, and 144 h, respectively. ▵, data within 5 days of initiation of rifampicin administration; ○, data 5 days after initiation of rifampicin administration; □, data after stopping the administration of rifampicin.
Although we have proposed a method by means of which Rin vivo can be predicted from the results of a reporter gene assay, the limitation of the reporter gene assay is such that the effect of only one nuclear receptor can be evaluated. For the induction of CYP3A4, in addition to PXR, CAR may be involved at least partly. The success of our prediction for rifampicin and omeprazole may be supported by the fact that these two drugs are selective PXR activators (Faucette et al., 2007), and, consequently, the induction effect was considered to be caused predominantly through the activation of PXR. In contrast, phenobarbital, phenytoin, and carbamazepine also activate CAR along with PXR (Wang et al., 2004). Although phenytoin and carbamazepine, in particular, are regarded as preferential CAR activators (Faucette et al., 2007), the contribution to the induction is considered to be smaller than that for PXR (Moore et al., 2000, 2002). Indeed, Faucette et al. (2006) reported that the binding affinity of human CAR to the CYP3A4 proximal promoter region was much lower than that of PXR, suggesting that the contribution of human CAR to CYP3A4 induction may be minimal. Our results are also in agreement with these previous reports, because we could satisfactorily predict the Rin vivo values of phenobarbital and phenytoin from the reporter gene assay with PXR (Figs. 3 and 4).
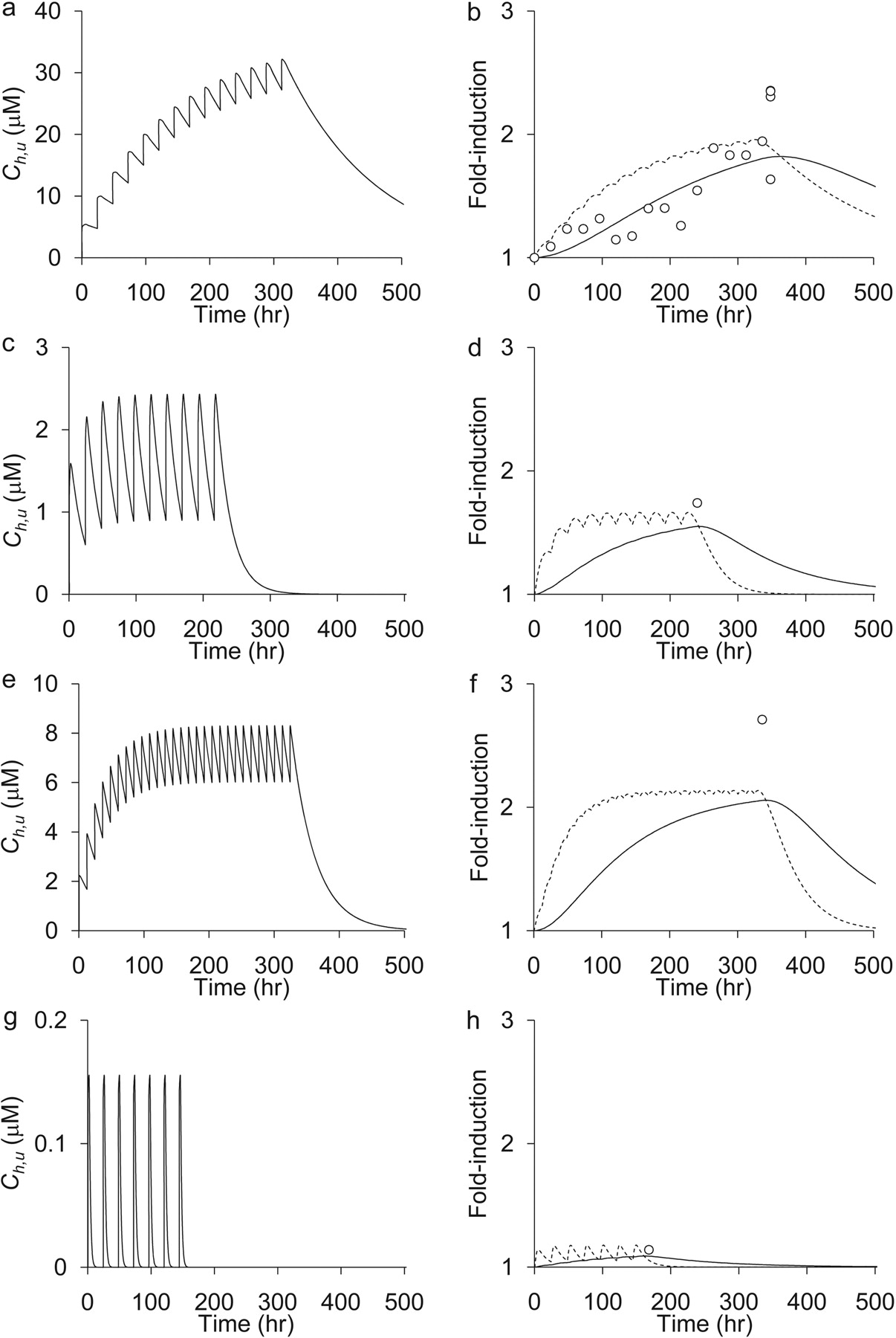
Prediction of Rin vivo for phenobarbital, phenytoin, carbamazepine, and omeprazole. The Rin vivo values for phenobarbital, phenytoin, carbamazepine, and omeprazole were predicted from the results of the in vitro reporter gene assay. In this calculation, the half-life of CYP3A4 protein was assumed to be 72 h. The left panel represents Ch,u for each drug after administration of phenobarbital (100 mg/day) (a), phenytoin (200 mg/day) (c), carbamazepine (200 mg twice a day) (e), and omeprazole (120 mg/day) (g), respectively. The right panel shows the comparison between the observed Rin vivo and Rpredict for phenobarbital (b), phenytoin (d), carbamazepine (f), and omeprazole (h), respectively. ○, represent the observed Rin vivo values taken from Table 2. Dotted and solid lines represent the predicted increase in mRNA for CYP3A4 and CYP3A4 proteins (Rpredict values), respectively.
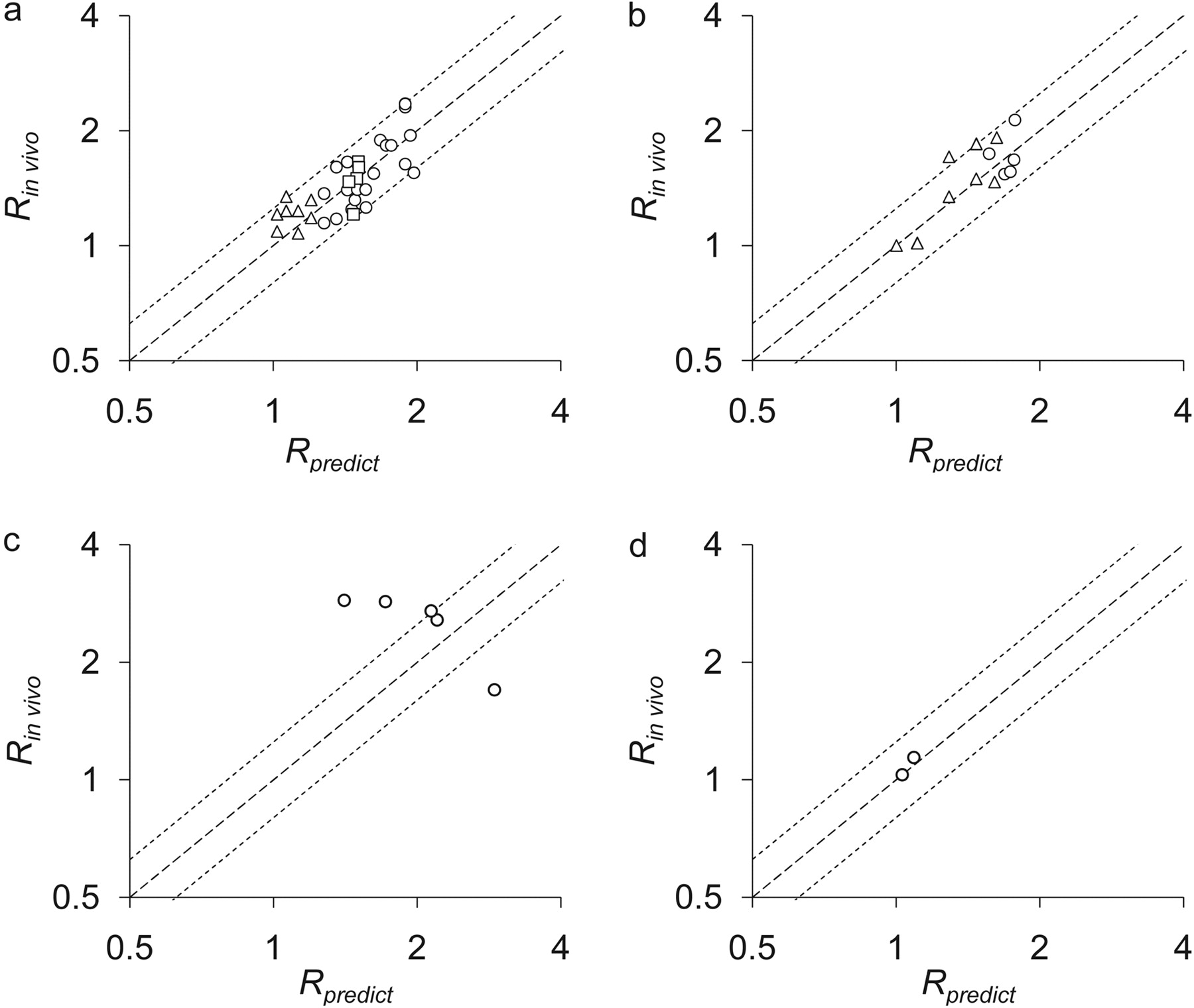
Correlations between Rin vivo and Rpredict for phenobarbital, phenytoin, carbamazepine, and omeprazole. The correlations between Rin vivo and Rpredict for phenobarbital (a), phenytoin (b), carbamazepine (c), and omeprazole (d) are shown. ▵, data within 5 days of initiation of drug administration; ○, data 5 days after initiation of rifampicin administration; □, data after stopping the administration of rifampicin.
The methodology proposed in the present study may be useful in drug development. On the basis of the results of the present study, the following procedure may be proposed to quantitatively predict the extent of in vivo induction by drug candidates from an in vitro data. First, a reporter gene assay with PXR will be performed using rifampicin as a standard inducer. From these in vitro results, the Emax,relative and EC50 values of drug candidates will be calculated. Then, based on the Ch,u(t) predicted from in vitro experiments (Theil et al., 2003), Rpredict will be calculated from eqs. 11 to 14 described under Materials and Methods. In this calculation, the parameter values listed in Table 6 will be used.
In conclusion, we have produced the first model that is useful for predicting the time profiles of in vivo CYP3A4 induction in humans from the results of an in vitro reporter gene assay. Using this model, the effect of the coadministration of inducers can be predicted before the extent of induction reaches steady-state and after the coadministration of inducers is stopped. Our results also support the previous conclusion that the contribution of PXR to CYP3A4 induction is much greater than that of CAR, and the reporter gene assay for PXR activation is a useful tool for predicting CYP3A4 induction by drug candidates.
Footnotes
-
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
-
M.K. and M.H. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.025734.
-
ABBREVIATIONS: DDI, drug-drug interaction; PXR, pregnane X receptor; CAR, constitutive androstane receptor; AUC, area under the curve; CC, correction coefficient; CL, clearance.
-
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material. - Received November 19, 2008.
- Accepted March 9, 2009.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}