


Abstract
An N-terminal domain histidine [corresponding to position 39 of UDP-glucuronosyltransferase (UGT) 1A1] is conserved in all UGT1A and UGT2B subfamily proteins except UGT1A4 (Pro-40) and UGT2B10 (Leu-34). Unlike most UGT1A and UGT2B xenobiotic-metabolizing enzymes, UGT1A4 and UGT2B10 lack the ability to glucuronidate 4-methylumbelliferone (4MU) and 1-naphthol (1NP), both planar phenols, and naproxen (a carboxylic acid). However, only UGT1A4 glucuronidates the tertiary amines lamotrigine (LTG) and trifluoperazine (TFP). In this study, we sought to elucidate the influence of specific N-terminal histidine and proline residues on UGT enzyme substrate selectivity. The conserved N-terminal domain histidine of UGT1A1, UGT1A6, UGT1A9, and UGT2B7 was mutated to proline and leucine 34 of UGT2B10 was substituted with histidine, and the capacity of the wild-type and mutant proteins to glucuronidate 4MU, 1NP, LTG, TFP, and naproxen was characterized. Whereas UGT1A1(H39P), UGT1A6(H38P), and UGT1A9(H37P) lacked the ability to metabolize 4MU, 1NP, and naproxen, all glucuronidated LTG. Km values for UGT1A1(H39P) and UGT1A9(H37P) were 774 and 3812 μM, respectively, compared with 1579 μM for UGT1A4. UGT1A1(H39P) also glucuronidated TFP with a Vmax/Km value comparable to that of UGT1A4. In contrast to the wild-type enzyme, UGT2B10(L34H) glucuronidated 4MU and 1NP with respective Km values of 260 and 118 μM. UGT2B7(H35P) lacked activity toward all substrates. The data confirm a pivotal role for an N-terminal domain proline in the glucuronidation of the tertiary amines LTG and TFP by UGT1A subfamily proteins, whereas glucuronidation reactions involving proton abstraction generally, although not invariably, require a histidine at the equivalent position in both UGT1A and UGT2B enzymes.
UDP-glucuronosyltransferase (UGT) enzymes catalyze the covalent linkage of glucuronic acid, donated by the cofactor UDP-glucuronic acid (UDPGA), to a typically hydrophobic substrate bearing a suitable functional group according to a second-order nucleophilic substitution mechanism. Hydroxyl (aliphatic and aromatic), carboxylic acid, and amine (primary, secondary, and tertiary) functional groups most commonly serve as the nucleophilic “acceptor” for glucuronic acid (Miners and Mackenzie, 1991; Radominska-Pandya et al., 1999). Because a myriad of compounds fulfill these minimal requirements for metabolism by UGT, glucuronidation provides an elimination and detoxification pathway for many drugs, nondrug xenobiotics, and endogenous compounds. Nineteen functional human UGT proteins have been identified to date, and these have been classified in two families (1 and 2) and three subfamilies (UGT1A, UGT2A, and UGT2B) based on sequence identity (Mackenzie et al., 2005). The individual UGT enzymes exhibit distinct, but overlapping, substrate and inhibitor selectivities (Radominska-Pandya et al., 1999; Tukey and Strassburg, 2000; Miners et al., 2004, 2006; Kiang et al., 2005; Sorich et al., 2006).
The UDPGA binding site is highly conserved and located in the C-terminal domain of UGTs (Miley et al., 2007; Patana et al., 2007). Although an X-ray crystal structure for an entire UGT protein is lacking, multiple lines of evidence implicate the N-terminal domain in substrate (aglycone) binding and selectivity. All UGT1A enzymes contain an identical carboxyl terminus of 246 residues but unique N-terminal domains (residues 1 to 285–289) (Mackenzie et al., 2005), indicating that the latter region must determine substrate selectivity. Studies with chimeric UGT2B proteins have similarly associated the N-terminal domain with the substrate selectivities of several human, rabbit, and rat UGT2B enzymes (Mackenzie, 1990; Ritter et al., 1992, 1993; Li et al., 1997; Lewis et al., 2007), whereas site-directed mutagenesis has identified specific N-terminal domain residues involved in the binding of substrates of human UGT1A3, 1A4, 1A6, 1A10, 2B4, 2B15, and 2B17 (Dubois et al., 1999; Senay et al., 2002; Martineau et al., 2004; Xiong et al., 2006; Barre et al., 2007; Kubota et al., 2007; Li et al., 2007).
Particularly striking are differences in the abilities of UGT1A and UGT2B enzymes to glucuronidate tertiary amines and planar phenols. Whereas UGT1A4 and UGT2B10 exhibit little or no activity toward the planar phenols 4-methylumbelliferone (4MU) and 1-naphthol (1NP), Vmax values with UGT1A1, 1A3, 1A6, 1A7, 1A8, 1A9, 2B7, and 2B15 exceed 25 pmol/min · mg (enzymes expressed in HEK293 cells) (Uchaipichat et al., 2004). In contrast, only UGT1A4 has been shown to convert the tertiary amines lamotrigine (LTG) and trifluoperazine (TFP) to their corresponding quaternary ammonium-linked glucuronides (Kubota et al., 2007). Although UGT2B10 lacks activity toward 4MU, 1NP, LTG, and TFP, this enzyme catalyzes the N-glucuronidation of nicotine and cotinine (Chen et al., 2007; Kaivosaari et al., 2007).
Recent studies with hybrid UGT1A3-UGT1A4 proteins demonstrated that the differing selectivities of these enzymes toward planar phenols and tertiary amines were determined by the first 44 residues, which correspond to amino acids 1 to 16 of the mature proteins (Kubota et al., 2007). A histidine in this region is conserved among all UGT1 and UGT2 family enzymes except UGT1A4 and UGT2B10, which have proline and leucine, respectively, at this position (Fig. 1). (The His/Pro/Leu occurs at positions 39, 40, 38, 37, 35, and 34 of UGTs 1A1, 1A4, 1A6, 1A9, 2B7, and 2B10, respectively.) It is noteworthy that the Pro40His mutation in UGT1A4 abolished the glucuronidation of LTG and TFP whereas the reciprocal (i.e., His40Pro) mutation in UGT1A3 conferred tertiary amine glucuronidation (Kubota et al., 2007). Unlike the glucuronidation of compounds containing a hydroxyl (and presumably carboxylate) group, quaternary ammonium glucuronide formation does not require proton abstraction. In this regard, the conserved His has been proposed as the catalytic base in glucuronidation reactions involving proton abstraction (Radominska-Pandya et al., 1999; Miley et al., 2007; Patana et al., 2008).
In this study, we sought to characterize the importance of specific N-terminal domain proline and histidine residues in UGT substrate selectivity. The conserved histidine of UGT1A1, UGT1A6, UGT1A9, and UGT2B7 was substituted with proline, and the glucuronidation of model tertiary amine (LTG and TFP), planar phenolic (4MU and 1NP), and carboxylic acid (naproxen) substrates by each mutant was characterized, as was the effect of the Leu34His mutation on UGT2B10 activity. The results demonstrate that introduction of proline in the N-terminal domain of UGT1A subfamily enzymes favors N-glucuronidation over the glucuronidation of the planar phenols and carboxylic acids. In contrast, activity studies with UGT2B10(L34H) and the UGT1A and UGT2B7 His→ Pro mutants confirms a pivotal role for the near conserved N-terminal domain histidine in the glucuronidation of planar phenols.
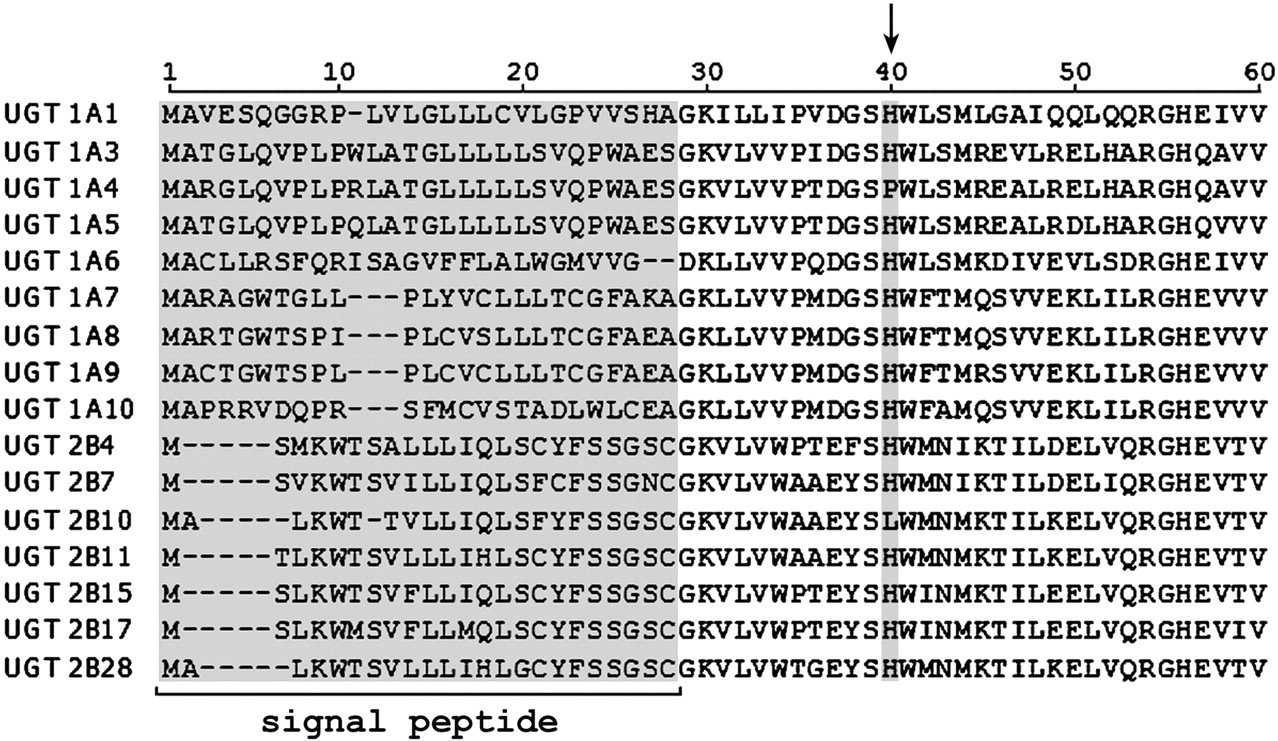
N-Terminal sequence identity of UGT1A and UGT2B proteins. The position of the conserved histidine residue in all proteins except UGT1A4 and UGT2B10 is indicated by the arrow.
Materials and Methods
Materials. Oligonucleotides were synthesized by Sigma Genosys (Castle Hill, Australia). Restriction enzymes were purchased from New England Biolabs (Ipswich, MA), Dulbecco's modified Eagle's medium, minimal essential medium nonessential amino acids solution (10 mM; 100×), and penicillin-streptomycin (5000 units/ml penicillin and 5000 μg/ml streptomycin) were from Invitrogen (Carlsbad, CA). pBluescript II SK(+) was from Stratagene (La Jolla, CA), and the WB-UGT1A human UGT1A subfamily Western blotting kit was from BD Gentest (BD Biosciences, San Jose, CA). LTG and lamotrigine N2-glucuronide were obtained from Wellcome Research Laboratories (Beckenham, UK). Cotinine, 4MU, 4-methylumbelliferone β-d-glucuronide, 1NP, 1-naphthol-β-d-glucuronide, S-naproxen (naproxen), TFP (dihydrochloride salt), and UDPGA (trisodium salt) were purchased from Sigma-Aldrich (Sydney, Australia), and cotinine β-d-glucuronide was from Toronto Research Chemicals (North York, ON, Canada). Naproxen acyl glucuronide was provided by AstraZeneca (Sodertalje, Sweden). All other reagents and solvents were of analytical reagent grade.
Site-Directed Mutagenesis and Expression of UGT Proteins. The wild-type UGT1A1, UGT1A6, UGT1A9, UGT2B7, and UGT2B10 cDNAs in pBluescript II SK(+) were used as templates for mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene). The primers used for mutagenesis are listed in Table 1. Mutations were confirmed by sequencing the entire cDNA (ABI Prism 3100 Genetic Analyzer with BigDye Terminator v3.1 Chemistry; Applied Biosystems, Foster City, CA). UGT1A1, UGT1A1(H39P), UGT1A4, UGT1A6, UGT1A6(H38P), UGT1A9, UGT1A9(H37P), UGT2B7, UGT2B7(H35P), UGT2B10, and UGT2B10(L34H) cDNAs were stably expressed in a human embryonic kidney cell line (HEK293), as described previously (Stone et al., 2003; Uchaipichat et al., 2004). Harvested cells were lysed by sonication using a Vibra Cell VCX130 Ultrasonic Processor (Sonics & Materials, Newtown, CT) set to an amplitude of 40%. Cells were sonicated with four 2-s “bursts,” each separated by 1 min of cooling on ice. Lysed samples were centrifuged at 12,000g for 1 min at 4°C, and the supernatant fraction was separated and stored at –80°C until use.
Primers used for site-directed mutagenesis
Mutated bases are in boldface and underlined. Reverse primers were complementary to the forward primers.
Immunoblotting. Lysate protein from HEK293 cells expressing UGT1A (15 μg), UGT2B7 (30 μg), and UGT2B10 (50 μg) enzymes and mutants was separated by SDS-polyacrylamide gel electrophoresis on a 10% acrylamide gel and transferred onto a nitrocellulose membrane. Blots were probed with a commercial primary antibody specific for the human UGT1A subfamily or with an anti-UGT2B7 antibody (see below), followed by horseradish peroxidase-conjugated goat anti-rabbit antibody. BM Chemiluminescence Blotting Substrate (Roche Diagnostics, Mannheim, Germany) was used for immunodetection. Intensities of the immunoblots were measured with a model GS-700 imaging densitometer (Bio-Rad Laboratories, Hercules, CA). Antibody against UGT2B7 was prepared using amino acids 55 to 165 as the antigen. The peptide was synthesized with a 6-histidine tag at the C terminus in Escherichia coli (BL21-DE3) and purified by affinity chromatography on a nickel-nitrilotriacetic acid column according to the protocol of the manufacturer (QIAGEN, Valencia, CA). Antibody against the purified antigen was raised in rabbits, and its specificity was assessed by immunoblotting against a panel of recombinant human UGTs. Apart from UGT2B7, the antibody weakly recognized UGT2B10 (see Results).
4MU and 1NP Glucuronidation Assay. Incubations contained 0.1 M phosphate buffer (pH 7.4), 4 mM MgCl2, 5 mM UDPGA, HEK293 cell lysate expressing UGT, and substrate (4MU or 1NP) in a total volume of 200 μl. Activity screening studies with 4MU and 1NP were performed at the concentrations corresponding to the known Km or S50 value reported for each UGT enzyme and at 5 times the known Km/S50 (Uchaipichat et al., 2004) (Table 2) or at 100, 1000, and 5000 μM with UGT2B10 and UGT2B10(L34H) as the enzyme source. Nine 4MU and 1NP concentrations in the range 50 to 1000 μM and 25 to 500 μM, respectively, were used in kinetic studies with UGT2B10(L34H). Incubations with 1NP contained 0.5% DMSO. Of the enzymes investigated here, this concentration of DMSO has a significant effect only on the activity of UGT1A9 (27% decrease in control activity) (Uchaipichat et al., 2004). HEK293 cell lysate protein amount and incubation time varied for each enzyme, as reported previously (Uchaipichat et al., 2004), and reaction rates were linear with respect to both of these variables. Incubations conducted with UGT2B10 and UGT2B10(L34H) were performed for 120 min with 1 mg/ml lysate protein. Reactions were initiated by the addition of UDPGA and, after incubation at 37°C for the specified time, were terminated by the addition of 70% perchloric acid (2 μl). Samples were centrifuged at 5000g for 10 min, and a 30-μl aliquot of the supernatant fraction was injected into the high-performance liquid chromatography (HPLC) column. Chromatography conditions were as described by Udomuksorn et al. (2007). 4MU glucuronide and 1NP glucuronide were quantified by comparison of peak areas to those standard curves prepared from the authentic glucuronides over the concentration range 0.5 to 10 μM.
Rates of 4MU, 1NP, naproxen, TFP, and LTG glucuronidation activities by UGT1A1, UGT1A1(H39P), UGT1A6, UGT1A6(H38P), UGT1A9, UGT1A9(H37P), and UGT1A4
Data are means of duplicate estimates (<10% variance).
TFP Glucuronidation Assay. The incubation mixture (200 μl) contained 5 mM UDPGA, 4 mM MgCl2, HEK293 cell lysate expressing UGT (0.25 mg/ml), and TFP in 50 mM Tris-HCl buffer (pH 7.4) (Uchaipichat et al., 2006). Screening for TFP glucuronidation was performed at a TFP concentration of 40 μM, which corresponds to the apparent Km reported for UGT1A4 (Kubota et al., 2007), and at 100 μM. The kinetics of TFP glucuronidation were characterized using 9 to 10 substrate concentrations in the range of 5 to 200 μM for UGT1A4 and 10 to 300 μM for UGT1A1(H39P). Reactions were initiated by the addition of UDPGA, and incubations were performed at 37°C for 20 min. The reactions were terminated by the addition of 200 μl of 4% acetic acid-96% methanol and then were centrifuged at 5000g for 10 min. A 40-μl aliquot of the supernatant fraction was injected into the HPLC column. Chromatography was performed according to Uchaipichat et al. (2006). TFP glucuronide was quantified by comparison of peak areas to those of an external standard curve prepared over the concentration range 0.2 to 10 μM.
LTG Glucuronidation Assay. Incubations (200 μl) contained 5 mM UDPGA, 4 mM MgCl2, HEK293 cell lysate (1 mg/ml), and LTG in 0.1 M phosphate buffer (pH 7.4) (Rowland et al., 2006). Screening for LTG glucuronidation was performed at an LTG concentration of 1500 μM, which corresponds to the apparent Km reported for UGT1A4 (Rowland et al., 2006), and at 3000 μM. The kinetics of LTG glucuronidation were characterized using 12 substrate concentrations in the range of 25 to 3000 μM. Given the limited solubility of LTG, the maximal concentration possible in incubations is 3000 μM. Incubations contained 1% (v/v) acetonitrile, which has an appreciable effect only on the activity of UGT1A6 (22% reduction in control activity) (Uchaipichat et al., 2004). After initiation of the reaction with UDPGA, incubations were performed at 37°C for 75 min. Reactions were terminated by the addition of 70% perchloric acid (2 μl), and then mixtures were centrifuged at 5000g for 10 min. A 40-μl aliquot of the supernatant fraction was injected into the HPLC column. Chromatography was performed according to Rowland et al. (2006). LTG glucuronide was quantified by comparison of peak areas with those of an LTG-glucuronide standard curve prepared over the concentration range 2 to 20 μM.
Naproxen Acyl Glucuronidation Assay. Incubations contained 0.1 M phosphate buffer (pH 7.4), 4 mM MgCl2, 5 mM UDPGA, HEK293 cell lysate expressing UGT (1 mg/ml), and naproxen in a total volume of 200 μl (Bowalgaha et al., 2005). Activity screening studies were performed at naproxen concentrations corresponding to the known Km or S50 value reported for each UGT enzyme and at 5 times the known Km/S50 (Bowalgaha et al., 2005) (Table 2). Samples were incubated at 37°C for 40 min. In addition, incubations with UGT2B7(H35P), UGT2B10, and UGT2B10(L34H) were performed for 120 min with 10 mg/ml lysate protein. Reactions were terminated by the addition of an equal volume of ice-cold 4% (v/v) acetic acid in methanol. Samples were centrifuged at 5000g for 10 min, and a 30-μl aliquot of the supernatant fraction was injected into the HPLC column. Chromatography conditions and quantification of naproxen acyl glucuronide were as described by Bowalgaha et al. (2005).
Cotinine Glucuronidation Assay. Cotinine served as the positive control for UGT2B10 activity. The incubation mixture (200 μl) contained 0.1 M phosphate buffer (pH 7.4), 5 mM UDPGA, 4 mM MgCl2, HEK293 cell lysate expressing UGT2B10 or UGT2B10(L34H) (5 mg/ml), and cotinine (0.5, 1, or 2 mM). Incubations contained 1% (v/v) ethanol, which affects only the activity of UGT1A6 (29% reduction in control activity) (Uchaipichat et al., 2004). Reactions were initiated by the addition of UDPGA, and incubations were performed at 37°C for 3 h. The reactions were terminated by the addition of 3 μl of 70% perchloric acid and then centrifuged at 5000g for 10 min. A 40-μl aliquot of the supernatant fraction was injected into the HPLC column. Cotinine glucuronide was quantified by reverse-phase HPLC using an Agilent 1100 HPLC system (Agilent Technologies, Sydney, Australia). The system consisted of an autosampler, gradient solvent delivery system, and UV detector (set at 254 nm), fitted with a Zorbax Eclipse XBD-C8 (4.6 mm × 150 mm; 5-μm particle size) analytical column (Agilent Technologies) operating at 25°C. The mobile phase consisted of 175 mM acetic acid and 4 mM 1-pentanesulfonic acid in 4% acetonitrile-water (A) and acetonitrile (B) gradient delivered at a flow rate of 1 ml/min. Initial conditions were 100% A held for 6 min, followed by 60% A-40% B over 0.1 min and then held for 2 min, before returning to the starting conditions. Formation of cotinine glucuronide was confirmed by cochromatography with the authentic compound. Unknown concentrations of cotinine glucuronide in incubations were determined by comparison of the peak areas with those of a cotinine β-d-glucuronide standard curve.
Data Analysis. Data points represent the mean of duplicate measurements (<10% variance). Kinetic constants for 4MU, 1NP, TFP, and LTG glucuronidation were obtained by fitting experimental data to the Michaelis-Menten or substrate inhibition equations using EnzFitter (Biosoft, Cambridge, UK). The Michaelis-Menten equation is as follows:  where v is the rate of reaction, Vmax is the maximum velocity, Km is the Michaelis constant (substrate concentration at 0.5 Vmax), and [S] is the substrate concentration. The substrate inhibition is as follows:
where v is the rate of reaction, Vmax is the maximum velocity, Km is the Michaelis constant (substrate concentration at 0.5 Vmax), and [S] is the substrate concentration. The substrate inhibition is as follows:  where Ksi is the constant describing the substrate inhibition interaction.
where Ksi is the constant describing the substrate inhibition interaction.
Goodness of fit to kinetic models was assessed by comparison of the F statistic, coefficient of determination (r2), parameter S.E.s, and 95% confidence intervals. Kinetic constants are reported as the value ± S.E. of the parameter estimate.
Results
His to Pro Substitution in UGT1A Proteins and UGT2B7. UGT enzymes and mutants were expressed in HEK293 cells. The expression of UGT1A1, UGT1A6, and UGT1A9 and their respective His→ Pro mutants relative to UGT1A4 was confirmed by immunoblotting with an antibody specific for human UGT1A subfamily enzymes. The His to Pro substitutions did not appreciably affect expression (Fig. 2A). The relative expression levels of UGT1A1, UGT1A1(H39P), UGT1A6, UGT1A6(H38P), UGT1A9, and UGT1A9(H37P) were 1:1.4:1.1:1.5:1.4:1.1. The expression of UGT1A4 relative to UGT1A1 was 1:1.34.
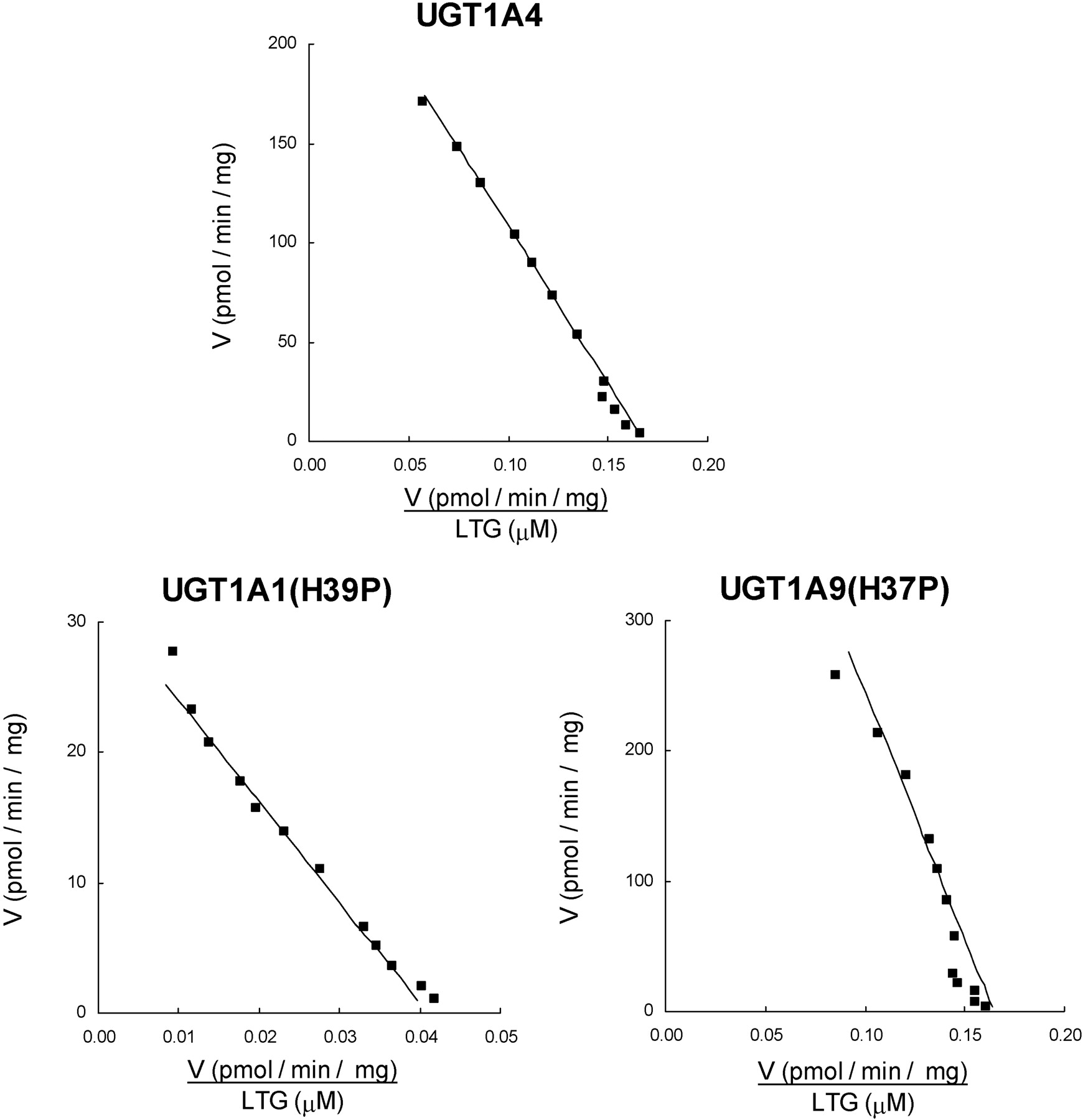
4MU, 1NP, and naproxen activity screening studies were performed at concentrations corresponding to the Km (or S50) and 5 times the Km/S50 for each of the parent enzymes (Uchaipichat et al., 2004; Bowalgaha et al., 2005). Screening studies with LTG were conducted at 1500 μM (the known Km for UGT1A4) and 3000 μM, the maximum concentration achievable in incubations (Rowland et al., 2006). Likewise, screening experiments with TFP were performed at 40 μM (the known Km) and 100 μM, the maximum concentration that avoids decreased activity due to the effects of substrate inhibition (Uchaipichat et al., 2006). UGT1A1, UGT1A6, and UGT1A9 glucuronidated 4MU, 1NP, and naproxen but not LTG and TFP (Table 2). Conversely, UGT1A4 glucuronidated LTG and TFP, but not 4MU, 1NP, or naproxen. Whereas the His to Pro substitution in UGT1A1, UGT1A6, and UGT1A9 abolished activity toward the phenolic compounds 4MU and 1NP and the carboxylic acid-containing substrate naproxen, the mutation conferred LTG glucuronidation activity to all three enzymes (Table 2). Of the three mutants, only UGT1A1(H39P) metabolized TFP (Fig. 3). The low activity of UGT1A6(H38P) precluded full kinetic analysis of LTG glucuronidation. However, like UGT1A4, LTG glucuronidation by UGT1A1(H39P) and UGT1A9(H37P) followed Michaelis-Menten kinetics (Fig. 4). Km values for UGT1A1(H39P) and UGT1A9(H37P) were approximately half and double those of UGT1A4, respectively (Table 3). Vmax/Km values, with Vmax corrected for expression relative to UGT1A4, were 0.168, 0.039, and 0.211 μl/min · mg for UGT1A4, UGT1A1(H39P), and UGT1A9(H37P), respectively. As reported previously for UGT1A4 (Uchaipichat et al., 2006), the kinetics of TFP glucuronidation by UGT1A1(H39P) were best fit to the substrate inhibition equation, although deviation from this model was apparent at low substrate concentrations (Fig. 3). The Km was higher and the Ksi and Vmax values were lower for UGT1A1(H39P) compared with the corresponding parameters for UGT1A4 (Table 3). Vmax/Km (corrected for expression relative to UGT1A4) values for TFP glucuronidation by UGT1A4 and UGT1A1(H39P) were 26.2 and 3.5 μl/min · mg, respectively.
Kinetic parameters for TFP and LTG glucuronidation by UGT1A1(H39P), UGT1A6(H38P), UGT1A9(H37P), and UGT1A4
Data are presented as the parameter ± S.E. of parameter fit. Kinetic parameters are not corrected for relative expression or nonspecific binding.
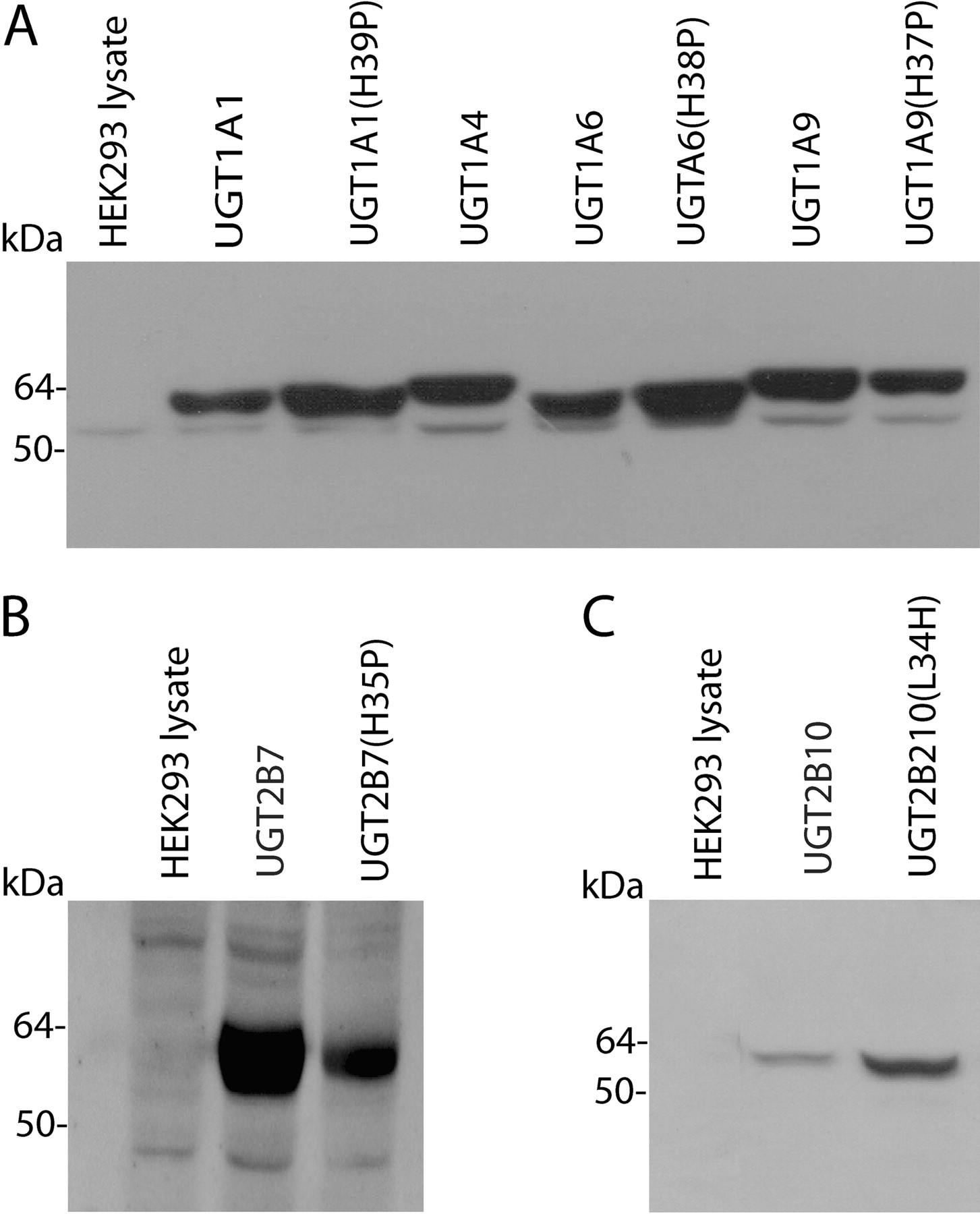
Immunoblots of lysates of HEK293 cells stably expressing UGT1A enzymes and mutants (A), UGT2B7 and UGT2B7(H35P) (B); and UGT2B10 and UGT2B10(L34H) (C). HEK293 protein amounts were 15, 30, and 50 μg of HEK293 for UGT1A, UGT2B7, and UGT2B10 proteins, respectively. Untransfected HEK293 cells served as the negative control.

Eadie-Hofstee plots for trifluoperazine glucuronidation by UGT1A4 (left panel) and UGT1A1(H39P) (right panel). ▪, experimentally determined values (means of duplicate measurements at each concentration); ——, computer-generated curves of best fit.
Expression of UGT2B7(H35P), determined using an antibody raised against a peptide fragment of UGT2B7, was relatively low (1:0.42) (Fig. 2B). Although UGT2B7 glucuronidated 4MU, 1NP, and naproxen at substrate concentrations corresponding to the known Km/S50 and 5 times these values for each reaction (data not shown), UGT2B7(H35P) lacked activity with all substrates investigated here even when the HEK293 cell lysate protein content of incubations was increased 10-fold. It should be noted that the lower limits of quantification for the HPLC assays used in this work translate to rates of glucuronide formation of 0.25 to 0.8 pmol/min · mg.
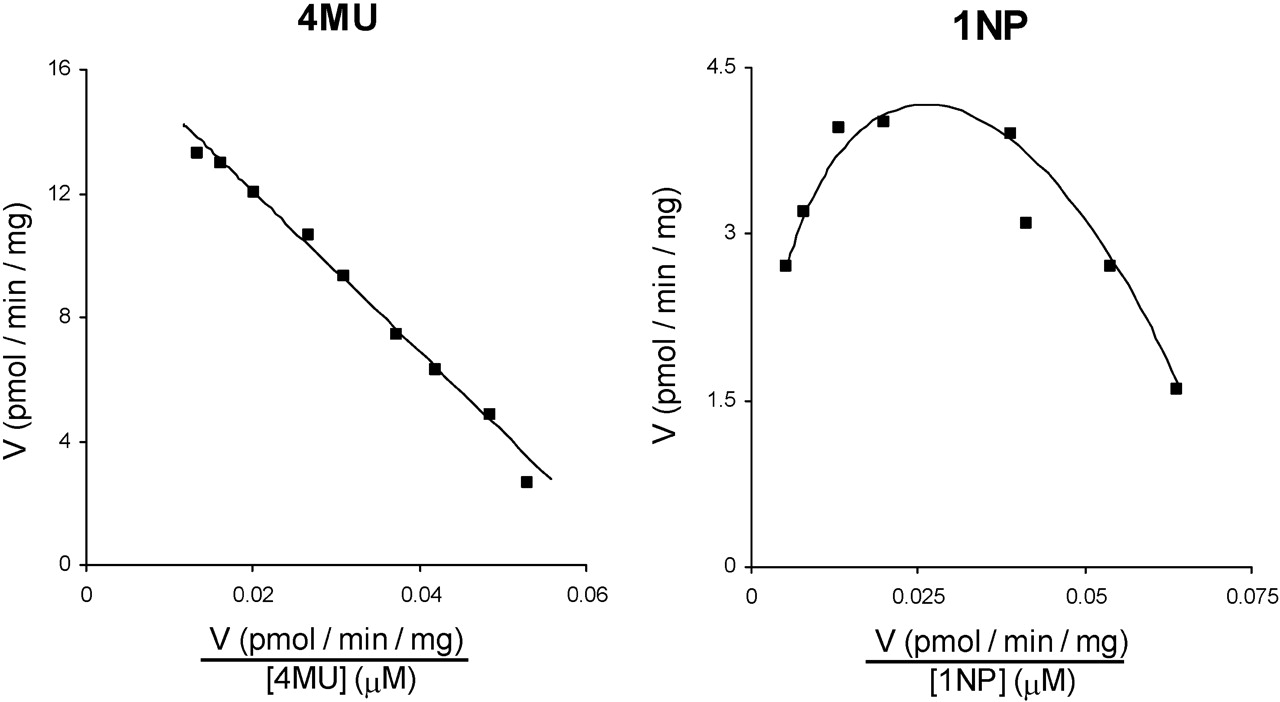
Leu to His Substitution in UGT2B10. In the absence of a commercially available antibody for UGT2B10, expression of this enzyme and the L34H mutant was determined using the anti-UGT2B7 antibody, which cross-reacts, albeit weakly, with UGT2B10. The expression level of UGT2B10 relative to UGT2B10(L34H) was 1:2.1 (Fig. 2C). UGT2B10 activity was confirmed with cotinine as the substrate; rates of cotinine glucuronide formation were 1.2, 1.8, and 1.9 pmol/min · mg of protein at substrate concentrations of 0.5, 1, and 2 mM, respectively. UGT2B10 lacked activity toward 4MU, 1NP, naproxen, LTG, and TFP. Lack of glucuronidation activity persisted when the content of UGT2B10 expressing HEK293 cell lysate present in incubations was increased to 20 mg/ml [to overcome the lower apparent expression compared with UGT2B10(L34H)]. However, UGT2B10(L34H) glucuronidated both 4MU and 1NP (Fig. 5). 4MU glucuronidation by UGT2B10(L34H) followed Michaelis-Menten kinetics (Km 260 ± 6 μM and Vmax 17 ± 0.2 pmol/min · mg), whereas 1NP glucuronidation by UGT2B10(L34H) exhibited substrate inhibition (Km 118 ± 25 μM, Ksi 135 ± 19 μM, and Vmax 14 ± 1.4 pmol/min · mg). UGT2B10(L34H) did not metabolize naproxen, LTG, TFP, or cotinine.
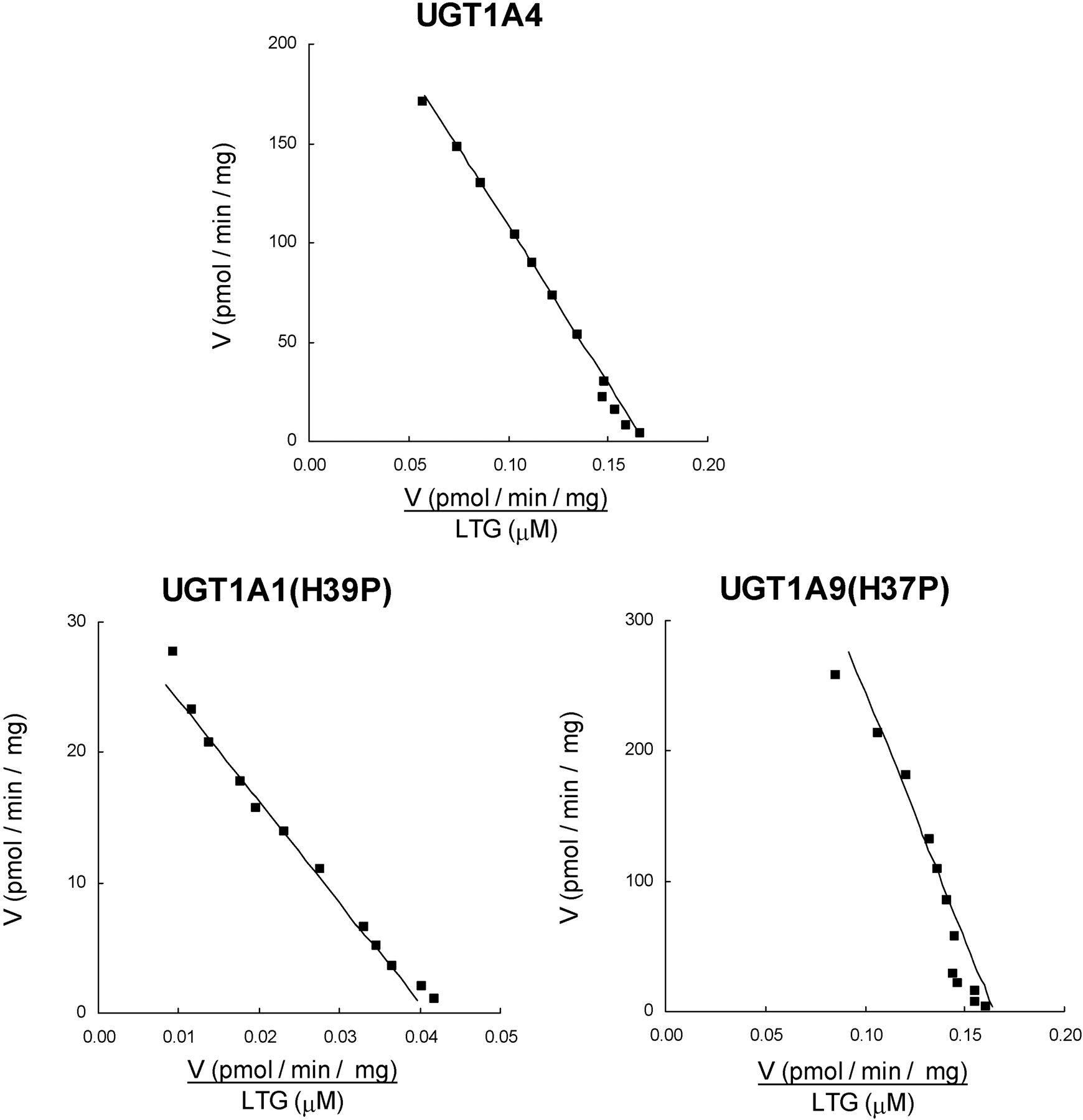
Eadie-Hofstee plots for lamotrigine glucuronidation by UGT1A4 (top panel), UGT1A1(H39P) (bottom left panel), and UGT1A9(H37P) (bottom right panel). ▪, experimentally determined values (means of duplicate measurements at each concentration); ——, computer-generated curves of best fit.
Discussion
With the exception of UGT1A4 and UGT2B10, the N-terminal domain histidine corresponding to position 39 in UGT1A1 is conserved in UGT1A and UGT2B subfamily proteins (Fig. 1). Proline and leucine occur at the corresponding position of UGT1A4 and UGT2B10. Recent work in this laboratory demonstrated that Pro-40 of UGT1A4 is essential for the glucuronidation of the tertiary amines LTG and TFP (Kubota et al., 2007). Furthermore, the His40Pro substitution in UGT1A3 conferred LTG and TFP glucuronidation activity, with concurrent near abolition of planar phenol glucuronidation. Results presented here further demonstrate the importance of the N-terminal domain proline and histidine in tertiary amine and planar phenol glucuronidation, respectively.
UGT1A1(H39P), UGT1A6(H38P), and UGT1A9(H37P) all glucuronidated LTG. Indeed, Vmax/Km values for LTG glucuronidation by UGT1A4 and UGT1A9(H37P) were comparable. However, catalytic efficiencies were lower for UGT1A1(H39P) and UGT1A6(H38P). In contrast, only UGT1A1(H39P) metabolized TFP. Although an X-ray crystal structure for an entire UGT protein is currently not available, a homology model of UGT1A1 based on a plant glycosyltransferase places the conserved histidine in an α-helix (Li and Wu, 2007; Locuson and Tracy, 2007). The structural rigidity of proline is known to disrupt α-helices. Hence, secondary and tertiary structure in the N-terminal domain is likely to differ among UGT1A proteins with the conserved histidine and UGT1A4 (and the His→ Pro mutants generated here). In turn, this may alter the architecture of the active site such that tertiary amine substrates may bind in a catalytically favorable orientation. However, differences between the His→ Pro mutants in their ability to glucuronidate TFP indicate that other regions of the UGT1A active site clearly influence the binding and/or turnover of tertiary amine substrates. Likewise, the ability of UGT2B10 to glucuronidate the tertiary amines cotinine and nicotine is not dependent on an equivalent N-terminal domain proline.
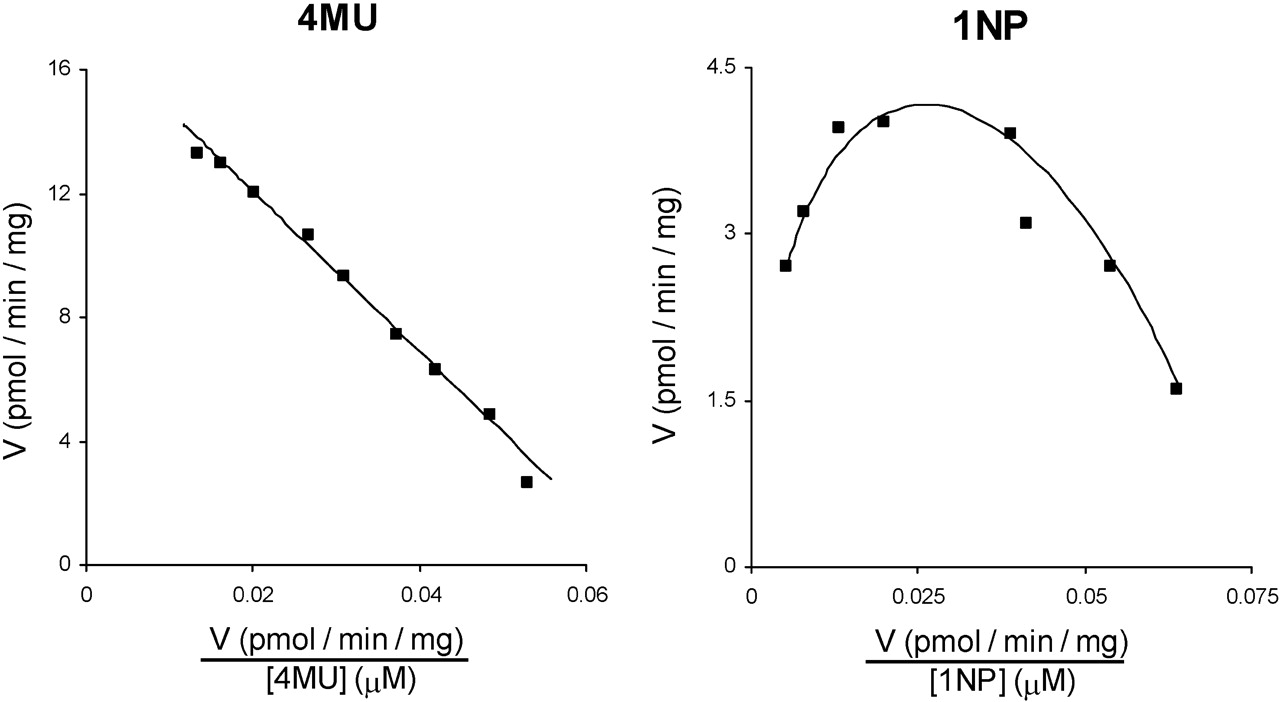
Eadie-Hofstee plots for 4-methylumbelliferone (left panel) and 1-naphthol (right panel) glucuronidation by UGT2B10(L34H). ▪, experimentally determined values (means of duplicate measurements at each concentration); ——, computer-generated curves of best fit.
The glucuronidation reaction proceeds according to a second-order nucleophilic substitution mechanism (Radominska-Pandya et al., 1999). Proton abstraction is considered an obligatory step in the glucuronidation of all aliphatic alcohols, phenols, primary and secondary amines, acidic carbon atoms, and thiols, and the near conserved N-terminal domain histidine has been implicated as the most important catalytic base in this regard (see later discussion). However, the conversion of tertiary amines to quaternary ammonium-linked glucuronides does not involve proton abstraction. Based on the effects of site-directed mutation studies with UGT1A9, it has been suggested that Asp-143 in this protein may stabilize the transition state during the glucuronidation of the primary and secondary amine functional groups of 4-aminobiphenyl and retigabine (Patana et al., 2008). A similar mechanism may conceivably apply to the glucuronidation of tertiary amines.
The His35Pro substitution in UGT2B7 abolished 4MU, 1NP, and naproxen glucuronidation. Moreover, UGT2B7(H35P) lacked activity toward the tertiary amines investigated here as substrates. Miley et al. (2007) reported recently that the His→ Ala substitution at position 35 of UGT2B7 abolished activity toward androsterone, hyodeoxycholic acid and tetrachlorocatechol. This observation and the data presented here are consistent with a pivotal role for His-35 in UGT2B7 activity. However, it should be noted that the UGT2B7 enzyme appears to be “sensitive” to structural change, and most mutant and chimeric proteins generated from UGT2B7 exhibit low or absent activity (Lewis et al., 2007; Miley et al., 2007).
Apart from UGT1A4, UGT2B10 is the only other 1A and 2B subfamily protein that lacks the conserved N-terminal domain histidine (Fig. 1). More importantly, UGT2B10 lacks activity toward the xenobiotic alcohols (including 4MU and 1NP), and carboxylic acids and the hydroxy steroids that typically show overlapping substrate selectivities between UGT enzymes (Jin et al., 1993), although very low activity has been reported with hydroxyeicosatetraenoic acids (Turgeon et al., 2003). More recently, UGT2B10 has been shown to glucuronidate certain tertiary amines, including cotinine, nicotine, and tobacco-specific nitrosamines, but activities are relatively low (Chen et al., 2007, 2008; Kaivosaari et al., 2007). The UGT2B10(L34H) mutant was found here to glucuronidate 4MU and 1NP but not cotinine. These observations suggest that Leu-34 is critical for the glucuronidation of cotinine (and possibly nicotine- and tobacco-specific nitrosamines) by UGT2B10. Although outside the scope of the present work, further studies are warranted to elucidate the role of Leu-34 and other N-terminal domain amino acids on UGT2B10 activity and substrate selectivity.
It has been proposed that the conserved N-terminal histidine acts as the catalytic base in the glucuronidation of planar phenols and other substrates when the reaction mechanism requires proton abstraction (Radominska-Pandya et al., 1999; Li and Wu, 2007; Locuson and Tracy, 2007; Miley et al., 2007; Patana et al., 2008), and data presented here are generally consistent with this proposition. However, exceptions occur. Work in this laboratory showed that the Pro40His mutation in UGT1A4 did not confer 4MU and 1NP glucuronidation (Kubota et al., 2007). Rather, substitution of Thr-36 of UGT1A4 with isoleucine (the corresponding amino acid in UGT1A3) resulted in significant 4MU and 1NP glucuronidation (Vmax values approximately 100 pmol/min · mg). Although Li et al. (2007) reported that the His-40 mutant of UGT1A4 glucuronidated 4MU and 1NP, activities were low (<1 pmol/min · mg) and almost certainly catalytically irrelevant when compared with the Vmax values for 4MU and 1NP glucuronidation by the individual wild-type human UGT enzymes and by UGT1A4(T36I). However, it is interesting to note that Li et al. (2007) further reported that the His38Arg mutation in UGT1A6 subtly altered substrate selectivity. Taken together, these observations suggest that the conserved N-terminal domain histidine may influence UGT substrate selection and a residue(s) elsewhere in the active site can function as the catalytic base.
The His→Pro substitution in UGT 1A1, 1A6, 1A9, and 2B7 abolished naproxen acyl glucuronidation. However, UGT2B10(L34H) also lacked naproxen glucuronidation activity despite its capacity to metabolize planar phenols. Naproxen and other related carboxylic acid-containing nonsteroidal anti-inflammatory drugs that undergo extensive glucuronidation typically have pKa values in the range of 4 to 5. Thus, the carboxylate group is almost completely ionized at pH 7.4. The involvement of a catalytic base would clearly be dependent on whether glucuronidation involved conjugation of the carboxylate ion or the small fraction of the drug present in the neutral state.
It is known that Pro-40 is critical for the UGT1A4-catalyzed conversion of the tertiary amines LTG and TFP to their respective quaternary ammonium-linked glucuronides. This study shows for the first time that substitution of histidine for proline at positions 39, 38, and 37 of UGT1A1, UGT1A6, and UGT1A9, respectively, confers LTG glucuronidation activity to these enzymes. In addition, UGT1A1(H39P) glucuronidated TFP. The data suggest that the His→ Pro substitution induces a conformational change that favors the N-glucuronidation of these tertiary amines, at least in the UGT1A subfamily. It was also demonstrated for the first time that the inability of UGT2B10 to glucuronidate planar aromatic phenols such as 4MU and 1NP is due to the lack of the near conserved histidine in the N-terminal domain of this enzyme. The latter observation and the loss of 4MU and 1NP glucuronidation activity associated with the UGT1A and UGT2B7 His→ Pro mutations are consistent with the hypothesis that the N-terminal domain histidine generally, but not invariably (Kubota et al., 2007; Li et al., 2007), acts as the catalytic base in the metabolism of compounds requiring proton abstraction for glucuronide formation. Taken together with previous reports (Dubois et al., 1999; Senay et al., 2002; Martineau et al., 2004; Xiong et al., 2006; Barre et al., 2007; Kubota et al., 2007; Li et al., 2007), the present study further demonstrates the pivotal roles of individual amino acids in determining the substrate selectivities of human UGT enzymes.
Acknowledgments
Technical assistance from Benjamin Lewis is gratefully acknowledged.
Footnotes
-
This work was supported by the National Health and Medical Research Council of Australia.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.109.028225.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; UDPGA, UDP-glucuronic acid; 4MU, 4-methylumbelliferone; 1NP, 1-naphthol; LTG, lamotrigine; TFP, trifluoperazine; HPLC, high-performance liquid chromatography.
- Accepted May 28, 2009.
- Received April 23, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}