


Abstract
The metabolism of the spleen tyrosine kinase inhibitor N4-(2,2-dimethyl-3-oxo-4-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethyoxyphenyl)-2,4-pyrimidinediamine (R406) and its oral prodrug N4-(2,2-dimethyl-4-[(dihydrogenphosphonoxy)methyl]-3-oxo-5-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethyoxyphenyl)-2,4-pyrimidinediamine disodium hexahydrate (R788, fostamatinib) was determined in vitro and in humans. R788 was rapidly converted to R406 by human intestinal microsomes, and only low levels of R788 were observed in plasma of human subjects after oral administration of 14C-R788. R406 was the major drug-related compound in plasma from human subjects, and only low levels of metabolites were observed in plasma. The plasma metabolites of R406 were identified as a sulfate conjugate and glucuronide conjugate of the para-O-demethylated metabolite of R406 (R529) and a direct N-glucuronide conjugate of R406. Elimination of drug-related material into the urine accounted for 19% of the administered dose, and the major metabolite in urine from all the human subjects was the lactam N-glucuronide of R406. On average, 80% of the total drug was recovered in feces. Two drug-related peaks were observed; one peak was identified as R406, and the other peak was identified as a unique 3,5-benzene diol metabolite of R406. The 3,5-benzene diol metabolite appeared to result from the subsequent O-demethylations and dehydroxylation of R529 by anaerobic gut bacteria because only R529 was converted to this metabolite after the in vitro incubation with human fecal samples. These data indicate that the major fecal metabolite of R406 observed in humans is a product of a hepatic cytochrome P450-mediated O-demethylation and subsequent O-demethylations and dehydroxylation by gut bacteria.
Fc receptor signaling is an important process in the immunological response for macrophages, neutrophils, and mast cells (Turner et al., 2000). Activation of the Fc receptor results in the degranulation and gene transcription of cytokines that play a critical role in inflammation. Binding of ligands to the Fc receptor results in activation of intracellular immunoreceptor tyrosine-based activation motifs that interact with the nonreceptor spleen tyrosine kinase (Syk) for downstream signaling. Because of its critical role in the Fc receptor signal transduction, inhibition of Syk represents a potential target for the treatment of autoimmune diseases, such as rheumatoid arthritis and idiopathic thrombocytopenia purpura (Wong et al., 2004; Singh and Masuda, 2007).
N4-(2,2-Dimethyl-3-oxo-4-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethyoxyphenyl)-2,4-pyrimidinediamine (R406) is a small molecule that is a relatively selective Syk inhibitor and a potent inhibitor of IgE- and IgG-mediated activation of Fc receptor signaling (Braselmann et al., 2006). In a rodent collagen-induced arthritis model, R406 was shown to produce significant improvement in histopathology, clinical scores, and joint radiography (Pine et al., 2007). R406 exhibits low aqueous solubility, which limits the potential for solid dosage form development. Previous reports suggested that use of phosphate prodrugs can produce extensive improvement in bioavailability compared with the parent compound (Vierling and Greiner, 2003; Furfine et al., 2004). Therefore, the methylene phosphate prodrug of R406, N4-(2,2-dimethyl-4-[(dihydrogenphosphonoxy)methyl]-3-oxo-5-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethyoxyphenyl)-2,4-pyrimidinediamine disodium hexahydrate (R788, fostamatinib), was synthesized and tested in preclinical and clinical studies. When tested in animals and human volunteers, R788 in a simple suspension formulation resulted in systemic exposure of R406 that is comparable with, or better than, R406 administration using a solution formulation. In rheumatoid arthritis patients, oral administration of R788 tablets resulted in R406 exposure that is sufficient to show significant improvement of American College of Rheumatology scores in two large clinical trials (Weinblatt et al., 2008, 2009). R788 has also shown promise in the treatment of idiopathic thrombocytopenia purpura (Podolanczuk et al., 2009).
Little is known regarding the metabolism and disposition of R788 in humans. Therefore, in vitro studies using human liver and intestinal microsomes were conducted to evaluate the pathways of R788/R406 metabolism. In addition, a 14C-radiolabeled mass balance study in healthy male subjects was performed in an effort to gain understanding of the metabolic pathways and disposition of the R788/R406 in humans. The findings from these studies are summarized in this article. A metabolic scheme for R788/R406 in humans, including a unique contribution of anaerobic gut bacteria to the overall metabolism, is proposed.
Materials and Methods
Materials.
R788 (fostamatinib) was synthesized by DSM Fine Chemicals (Linz, Austria). R406, the para-O-demethylated metabolite of R406 [N4-(2, 2-dimethyl-3-oxo-5-pyrido[1,4]oxazin-6-yl)-5-fluoro-N2-(4-hydroxy-3,5-dimethoxyphenyl)-2,4-pyrimidinediamine (R529)], N4-{(2,2-dimethyl-4-(1-β-d-glucopyranuronosyl)-3-oxo-5-pyrido[1,4]oxazin-6-yl)}-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine (the lactam N-glucuronide of R406), the putative 3,4-benzene diol metabolite of R406, and the N2-(3,5-dihydroxyphenyl)-N4-(2,2-dimethyl-3-oxo-5-pyrido[1,4]oxazin-6-yl)-5-fluoro-2,4-pyrimidinediamine (3,5-benzene diol metabolite of R406) were synthesized by Rigel, Inc. (South San Francisco, CA). 14C-R788 (55.2 mCi/mmol) was prepared by Aptuit (Kansas City, MO). The 14C was located in the 2-C position of the pyrimidine moiety of R406.
t-Butyl methyl ether (MTBE), alkaline phosphatase from bovine intestinal mucosa, alamethacin, ketoconazole, furafylline, quinidine, sulfaphenazole, and l-cysteine were obtained from Sigma-Aldrich (St. Louis, MO). Bacto brain heart infusion (BHI) medium was obtained from BD Biosciences (Franklin Lakes, NJ). AneroGen compact paper sachets and anaerobic indicator strips were obtained from Oxoid (Basingstoke, UK). Pouch bags were obtained from Remel (Lenexa, KS). Human hepatic and intestinal microsomes were obtained from XenoTech, LLC (Lenexa, KS). Expressed cytochrome P450 isoforms and (S)-(+)-N-3-benzylnirvanol were obtained from BD Biosciences (San Jose, CA). All the other chemicals were the highest grade available.
In Vitro Metabolism of R406 and R788.
The stability of R406 was examined using hepatic human microsomes and expressed human CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Reactions were performed at 37°C in plastic vials at a final volume of 502 μl. The reaction mixture consisted of 445 μl of 0.1 M phosphate buffer, pH 7.4, 5 μl of hepatic microsomes (0.1 mg of protein) or expressed enzymes (20 pmol), 2 μl of R406 (final concentration 1 μM) in dimethyl sulfoxide (DMSO), and 50 μl of NADPH (final concentration, 1 mM). Metabolic reactions were initiated by the addition of the NADPH. For microsomal glucuronidation studies, 0.4 mg/ml human liver microsomes, 0.025 mg of alamethicin, 10 mM MgCl2, 100 mM sodium phosphate buffer, pH 7.5, 10 μM R406, and 5 mM UDP-glucuronic acid (UDPGA) were incubated in a total volume of 200 μl. Before addition of the UDPGA, the microsomes were activated by alamethicin for 15 min on ice. Metabolic reactions were stopped at 0, 15, 30, and 50 min by removing 50 μl of the reaction mixture and adding this to 100 μl of a stop solution consisting of acetonitrile/DMSO/ethanol/100 mM ammonium acetate/0.5 μM verapamil internal standard (7.5:0.6:0.6:2.5:1, v/v). For the hepatic microsomal NADPH-dependent metabolism studies, the effect of the chemical inhibitors ketoconazole (1 μM), furafylline (10 μM), quinidine (10 μM), sulfaphenazole (10 μM), and 3-N-benzylnirvanol (5 μM) was also examined. R406 concentrations were determined using an AB Sciex (Foster City, CA) QTrap mass spectrometer in the multiple-reaction monitoring mode. Separations were performed using a Dash-18 (Thermo Fisher Scientific, Waltham, MA) C18 column (20 × 2.1 mm; 3 μm) and a Shimadzu (Kyoto, Japan) SCL-10A VP high-performance liquid chromatography (HPLC) system. Flow rate was 0.25 ml/min. Mobile phase A was 10 mM ammonium acetate, pH 6.5, in water, and mobile phase B was 100% acetonitrile. Initial conditions were 10% mobile phase B, and after 0.75 min, a linear gradient to 90% mobile phase B was achieved at 2.5 min and held at these conditions until 3.5 min.
Liquid chromatography/mass spectrometry (LC/MS) analysis with UV (254 nm) detection was used to determine the pattern of the NADPH-dependent R406 metabolites formed in human hepatic microsomes. The microsomal incubation conditions were similar to those described above, but with a final concentration of R406 at 10 μM. Samples from the in vitro glucuronidation assay were also examined. Reactions were terminated after 15 min for the NADPH-dependent reaction and at 50 min for the glucuronidation assay by removing 50 μl of the reaction mixture and adding this to an equal volume of acetonitrile. After centrifugation, the supernatants were analyzed using a Shimadzu SCL-10A VP HPLC system equipped with an Agilent Technologies (Santa Clara, CA) 1100 series diode array detector and AB Sciex QTrap mass spectrometer. A Betasil (Thermo Fisher Scientific) C18 column (150 × 1 mm; 3 μm) was used for the analysis. Mobile phase A was 1% ammonium acetate in water, whereas mobile phase B was 100% acetonitrile. Initial conditions were 5% B, and a linear gradient to 75% B was performed over 35 min and held at these conditions for 10 min. Flow rate was 0.20 ml/min.
R788 (10 μM) stability in human intestinal microsomes (0.5 mg/ml) or alkaline phosphatase (42 units/ml) was examined at 37°C in a 0.2 M Tris buffer, pH 7.4, containing 0.05 M MgCl2. Reactions were terminated at 0, 15, and 30 min by removing 50 μl and adding to an equal volume of acetonitrile. Analyses were performed by LC/MS with UV (254 nm) detection as described above.
Human Mass Balance Study.
The human mass balance study was performed at the Covance Clinical Research Unit, Inc. (Madison, WI). Six healthy male subjects (19–35 years old) with a body weight between 63.8 and 85.3 kg participated in the study according to an Institutional Review Board-approved protocol. Participants were housed in the clinical unit until the end of the study (up to 8 days). The target dose of 14C-R788 (150 mg, 100 μCi) was administered orally as a solution in 0.01 N citrate buffer, pH 6, using a syringe. Subjects were fasted for 10 h before R788 administration.
Blood samples were obtained predose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 32, 48, 72, 96, 120, 144, and 168 h postdose, and then at 24-h intervals until discharged. Blood samples were collected into lavender-top Vacutainer tubes (BD Biosciences) containing K2EDTA and were kept on ice until plasma was prepared by centrifugation. Urine samples were obtained predose, 0 to 4, 4 to 8, 8 to 12, 12 to 24 h and at 24-h intervals until the subjects were discharged. Feces were obtained predose and at 24-h intervals until the subjects were discharged. Blood, plasma, and urine samples were stored at −20°C until analyzed. Feces were kept at −70°C until analyzed.
Analysis of Samples Obtained from the Human Mass Balance Study.
Radioanalysis was performed at Covance Laboratories (Madison, WI). Duplicate samples of plasma and urine (0.2 ml) were combusted in a model 307 sample oxidizer (PerkinElmer Life and Analytical Sciences, Waltham, MA), and the 14CO2 was trapped in a mixture of Perma Fluor and Carbo-Sorb (PerkinElmer Life and Analytical Sciences). Fecal samples were diluted with an equal weight of methanol and frozen overnight. The fecal mixtures were thawed and then homogenized using a Polytron PT 10/35 (Kinematica, Littau-Lucerne, Switzerland) equipped with a 36-mm generator. Duplicate weighed aliquots (approximately 0.2 g) were combusted. All the combusted samples were analyzed for radioactivity in model 2900TR liquid scintillation counters (PerkinElmer Life and Analytical Sciences) for at least 5 min or 100,000 counts. Metabolite profiles were determined for extracts of the plasma, urine, and fecal samples.
Two milliliters of the plasma samples from each subject collected between 0.5 and 24 h postdose was pooled by time point, and 5 ml of each pooled plasma sample was extracted twice with acetonitrile (1:3, v/v). The combined supernatants were evaporated to dryness and reconstituted in 0.01 M ammonium acetate in 50% acetonitrile. Urine samples were pooled by subject to generate a single 0- to 72-h sample. Samples were centrifuged before analysis. Fecal samples (4 g) collected between 0 and 24 h from each subject were extracted twice with methanol. The combined fecal extracts were evaporated to dryness and reconstituted in 0.01 M ammonium acetate in 50% acetonitrile/MeOH (1:0.1, v/v). Duplicate aliquots of the urine and extracts of plasma and feces were analyzed by liquid scintillation counting to determine extraction recoveries. Recovery of radioactivity was 71.4 to 111.0% from plasma, 63.8 to 77.6% from feces, and 94.1 to 104.0% for urine.
Radioactivity profiles of urine, plasma, and feces were performed using a Hewlett-Packard (Palo Alto, CA) HP1000 series HPLC system equipped with a Waters (Milford, MA) Nova-Pak C18, 3.9 × 300-mm column (4 μm) and a Phenomenex (Torrance, CA) C18, 3 × 4-mm guard column. Columns were heated to 30°C. Mobile phase A consisted of 0.02 M ammonium acetate in water, pH 7, and mobile phase B was acetonitrile. Initial conditions were 3% B, and after 3 min, the contribution of the mobile phase B was linearly increased to 20% (20 min), 24% (30 min), 55% (30 min), and 80% (43 min), where it was held for 5 min. Flow rate was 1 ml/min. Fractions were collected at 10-s intervals and analyzed by TopCount solid scintillation counting (PerkinElmer Life and Analytical Sciences).
Selected samples of human plasma, urine, and feces were analyzed by LC/MS using a Shimadzu/Prominence HPLC system consisting of an SIL-20AC HT (10°C) autosampler, a CBM-20A controller, two LC-20AD pumps, and a CTO-20AC column oven (30°C). Mass spectrometry was performed using LTQ Orbitrap XL with electrospray ionization in the positive mode and Xcalibur 2.0 software (both Thermo Fisher Scientific). Separations were performed using a Waters Nova-Pak C18 column (3.9 × 300 mm; 4 μm) equipped with a Phenomenex C18 guard column (3 × 4 mm). Mobile phase A was 20 mM ammonium acetate, and mobile phase B was acetonitrile. Flow rate was 1 ml/min. Initial conditions were 97% mobile phase A, and the percentage of mobile phase B was increased linearly to 20% (20 min), 24% (30 min), 55% (38 min), and 80% (43 min), where it was held for 5 min.
Purification and NMR Analysis of Major Fecal Metabolite.
Twenty grams of human feces was mixed with an equal volume of water. Twice the volume of MTBE was added for liquid-liquid extraction. After 15 min of shaking, the mixture was centrifuged at 750 rpm for 5 min in a Beckman Coulter (Brea, CA) Allegra 6 centrifuge. The MTBE solvent layer was transferred to glass tubes and dried down under nitrogen using a TurboVap LV evaporator (Zymark, Hopkinton, MA). After repeating the liquid-liquid extraction four times, all the MTBE extracts were dried down and resuspended with 200 μl of H2O/acetonitrile/formic acid (70:30:0.1). The major fecal metabolite was purified by an isocratic HPLC method on an Agilent Technologies 1100 HPLC system. Solvent A was HPLC water with 5% acetonitrile and 10 mM ammonium acetate. Solvent B was 100% acetonitrile. The isocratic method was run at 30% B. An Alltima C8 HPLC column (100 × 4.6 mm; Grace, Deerfield, IL) was used with a flow rate of 1.0 ml/min. The elution time for this metabolite was approximately 6 min. The collected fractions were dried down in the TurboVap evaporator and then resuspended in deuterated DMSO for NMR analysis. NMR analysis was run on a Varian, Inc. (Palo Alto, CA) Unity-300 NMR instrument. Because of the low amount of purified metabolite, the 1H signal was collected for 3 days.
In Vitro Fecal Incubation Studies.
Human fecal incubations were performed using a sterile BHI medium (3.7 g/100 ml of water) containing 5 mg of l-cysteine/100 ml. Under aseptic conditions, 2.8 ml of the BHI medium was added to a six-well flat-bottom plate (Costar 3516; Corning Life Sciences, Lowell, MA). R788 (100 μM), R406 (100 μM), R529 (100 μM), or the N-glucuronide of R406 (50 μM) was added to 10 μl of DMSO. Fresh human fecal material (6 g) was placed into a 50-ml Falcon tube (BD Biosciences), and 20 ml of the BHI medium was added. The fecal mixture was vortexed for 2 min until homogenous. The human fecal mixture (0.2 ml) was added to the plate, after which the plate was placed into a pouch bag. Two AnaeroGen (Oxoid) compact paper sachets were added along with two Oxoid anaerobic indicator strips. The excess air was expelled from the pouch bag; the bag was sealed using a Hualion model FS-205 impulse sealer and placed onto a Lab-line three-dimensional rotator (Barnstead, Melrose Park, IL) in a 37°C Yamato Scientific America Inc. (Santa Clara, CA) IC600 incubator. The samples were rotated continuously during the incubation. After approximately 36 h, the samples were removed; the pouch bag was opened; and 0.5 ml of sample from each well was added to an equal volume of acetonitrile. The samples were vortexed, centrifuged, and then analyzed by LC/MS with UV detection as described above for the R406 in vitro metabolism studies.
Results
In Vitro Metabolism.
R788 was completely hydrolyzed to R406 by human intestinal microsomes within 15 min (Fig. 1). Complete hydrolysis to R406 was also achieved with alkaline phosphatase (data not shown). With human hepatic microsomes and NADPH, a time-dependent decrease in the R406 peak area was observed (t1/2, 24 min). One major metabolite of R406 was observed by UV detection (Fig. 2), and the protonated molecular ion of this peak (m/z 457) was 14 mass units less than R406. Based on coelution with an authentic standard, the metabolite was identified as the para-O-demethylated product of R406 (R529). A smaller metabolite peak, representing the meta-O-demethylated metabolite (retention time, 35.78 min), was also observed in the microsomal studies. Among the cytochrome P450 inhibitors tested, ketoconazole was the only inhibitor that markedly (>90%) reduced the formation of this metabolite. In addition, expressed CYP3A4 metabolized R406 to the greatest extent of any of the expressed isoforms examined. No appreciable loss of R406 peak area was observed after incubation with human hepatic microsomes and UDPGA. However, three distinct direct glucuronide conjugates of R406 could be observed by LC/MS (m/z 647) (Fig. 3).
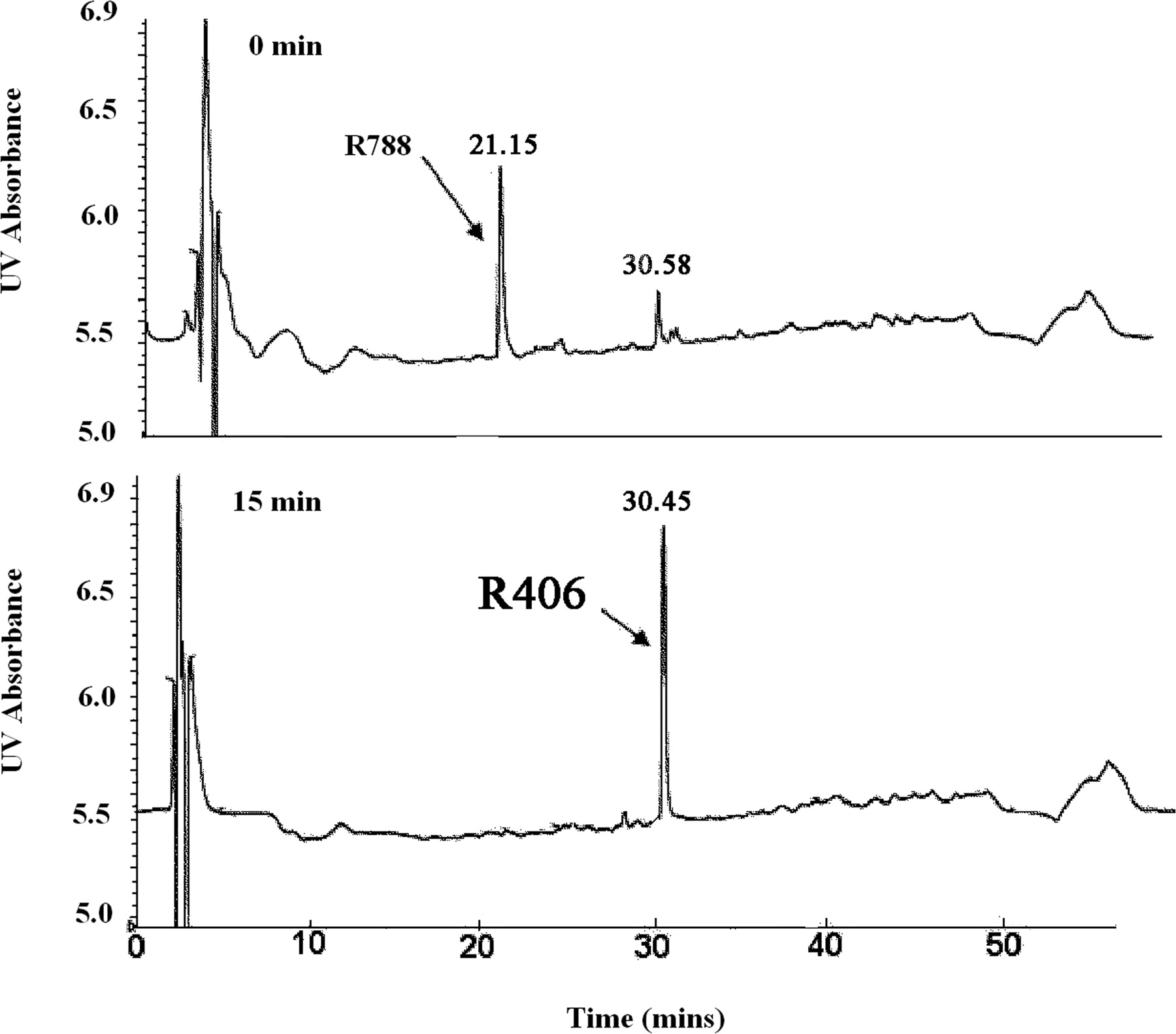
Hydrolysis of R788 to R406 by human intestinal microsomes. The top panel shows the UV (254 nm) chromatogram of the 0-min sample, whereas the bottom panel shows a sample obtained after a 15-min incubation at 37°C.

UV profile of sample obtained 15 min after incubation of R406 (10 μM) with human hepatic microsomes and NADPH. The mass spectra for R406 and the para-O-demethylated metabolite of R406 (R529) are shown in the middle and bottom panels, respectively.
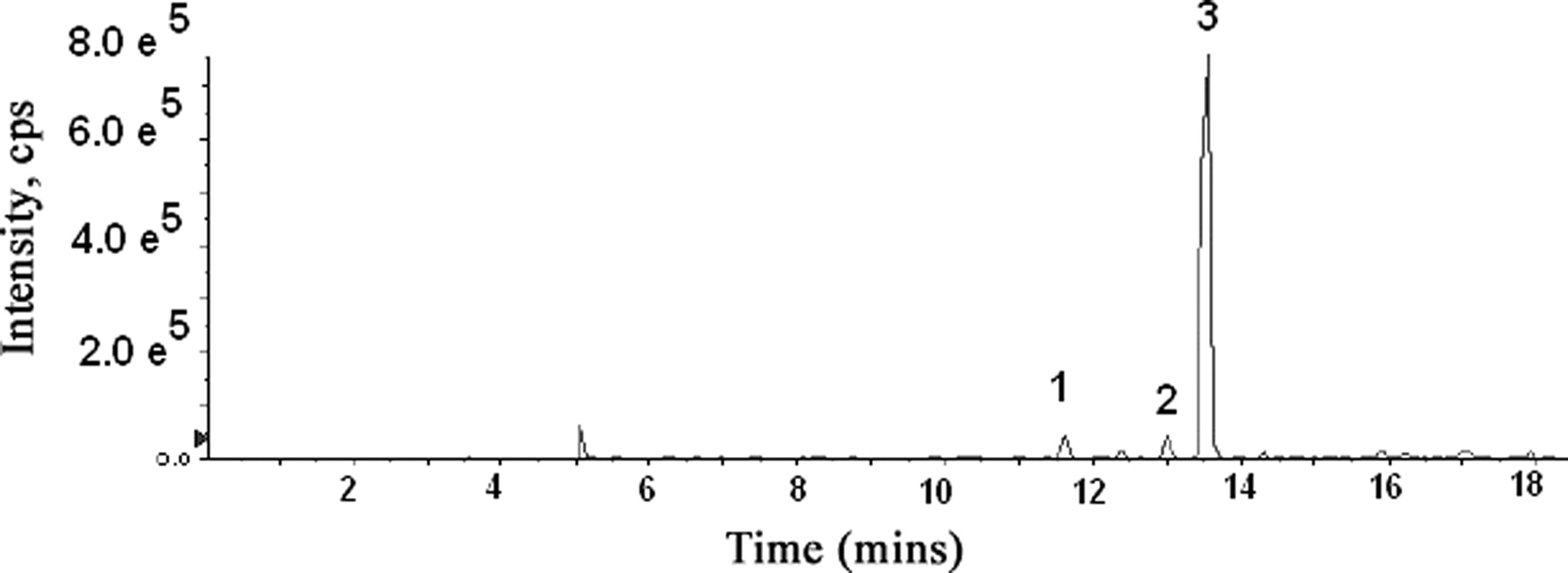
HPLC profile of sample obtained 50 min after incubation of R406 with human hepatic microsomes and UDPGA using selective ion monitoring (m/z 647) to detect the formation of three distinct direct N-glucuronides of R406.
Plasma Radioactivity Time Course.
The time course of plasma radioactivity in subjects who received a single oral dose of 14C-R788 is shown in Fig. 4. Peak plasma radioactivity levels were observed at 1 h after dosing in all the individuals and steadily declined thereafter. Cmax for plasma radioactivity levels ranged from 1770 to 2630 dpm/ml, whereas the plasma half-life ranged from 10.8 to 15.7 h. Subject 4 had the shortest plasma half-life and the lowest plasma radioactivity exposure (10,493 dpm · h/ml) of all the individuals in the study. Subject 2 had the longest half-life and the highest exposure (25,343 dpm · h/ml) of all the subjects.

Time course of plasma radioactivity in human subjects after oral administration of 14C-R788.
Plasma Metabolites.
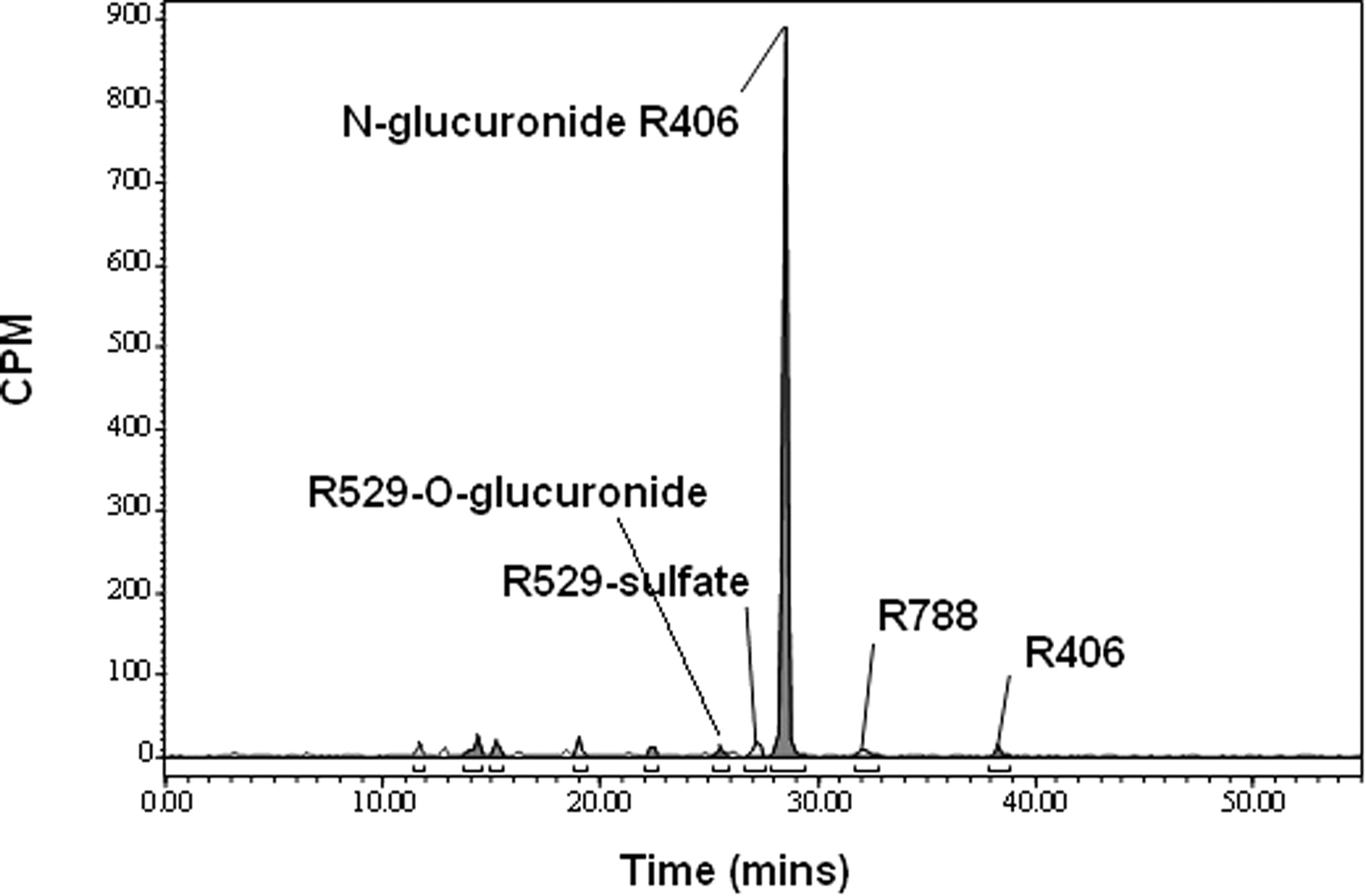
The major peak observed in plasma at all the time points was identified by LC/MS as R406 based on the protonated molecular ion at m/z 471. Representative radioactivity profiles for the pooled plasma obtained at 0.5 and 4 h after dosing are shown in Fig. 5. The prodrug R788 (m/z 581) was also observed in plasma, with higher levels being observed in the early time point samples. All the other radioactive peaks in plasma were minor at every time point examined. The plasma metabolites were identified by LC/MS as the O-glucuronide conjugate R529 (m/z 633) based on the 176-atomic mass unit (amu) loss to generate a product ion at m/z 457, a sulfate conjugate of R529 (m/z 537) based on the 80-amu loss to a product ion at 457 m/z, and direct N-glucuronides of R406 (m/z 647) based on the 176-amu loss to generate a product ion at m/z 471.

Plasma radioactivity profiles of the pooled 0.5- and 4-h plasma samples from human subjects orally administered 14C-R788.
Profile of Metabolites in Urine and Feces.
Overall mean recovery of radioactivity from the human subjects was 99.3%. The majority of the dose (80%) was recovered in feces within 96 h, whereas urinary recovery (19.3%) was nearly complete within 72 h (Table 1). One major radioactive peak in urine, accounting for greater than 75.2% of the total urinary radioactivity, was observed in the 0- to 72-h sample of all the patients. A representative chromatogram for urine is shown in Fig. 6. The major urinary metabolite was identified by LC/MS as an N-glucuronide of R406 (m/z 647) based on the characteristic loss of 176 mass units to the m/z 471 ion of R406. This N-glucuronide conjugate of R406 observed in urine coeluted with the major R406 N-glucuronide that was formed in hepatic microsomes, and was identified as the lactam N-glucuronide of R406 based on coelution with the chemically synthesized standard.
Mean (±S.D.) percentage of radioactive dose recovered in urine and feces at specified time intervals after administration of 14CR-788 to six healthy male subjects

Representative profile of radioactivity in urine (0–72 h) from a human subject orally administered 14C-R788.
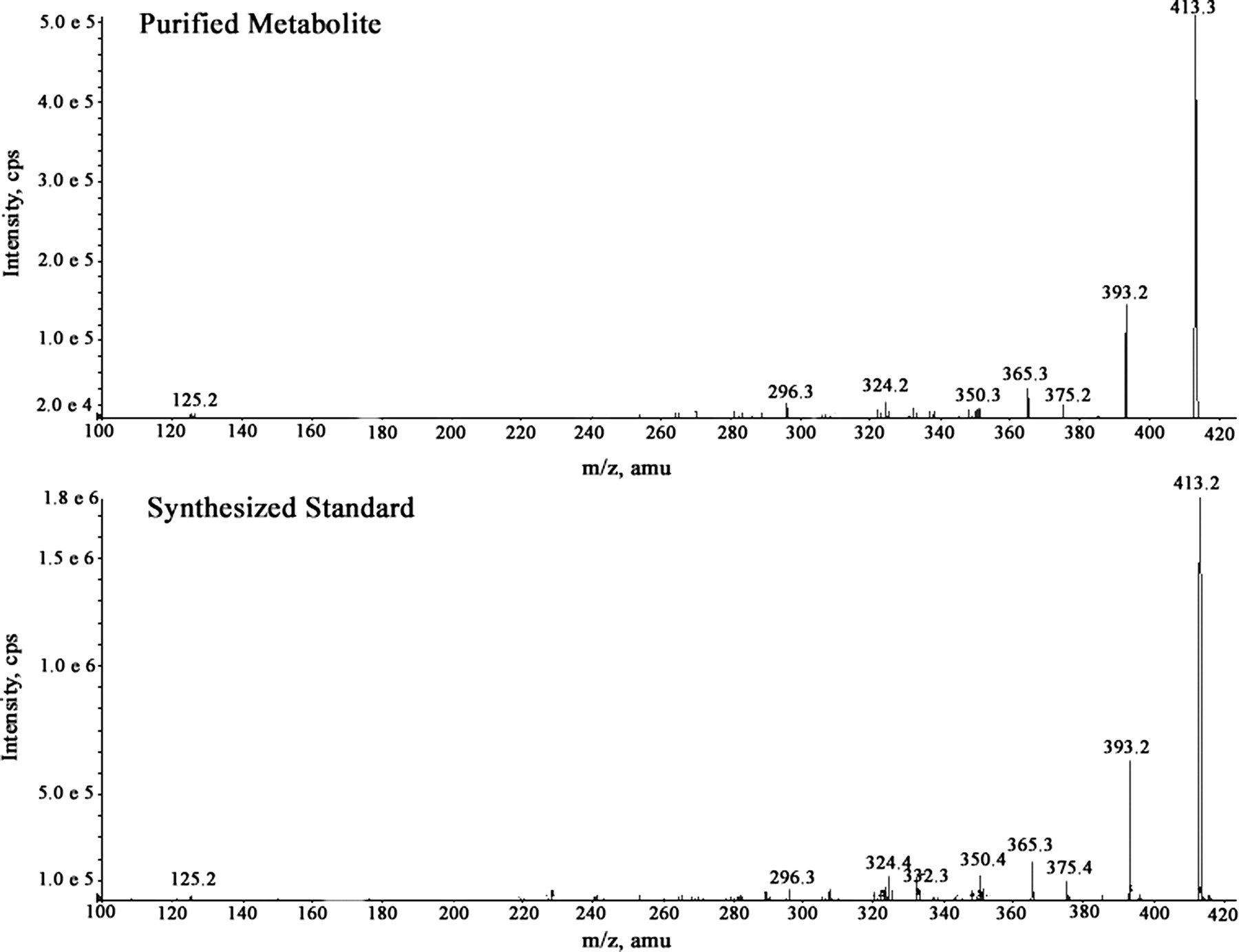
Two major radioactive peaks were observed in the 0- to 120-h pooled feces from all the subjects, but the relative amount of each of these peaks varied between individuals (Fig. 7). Subject 4 had a greater proportion (11.6:1) of the earlier (35 min) eluting radioactive peak, whereas Subject 2 had a greater proportion (5.5:1) of the later eluting radioactive peak (40 min). Three of the additional subjects had a greater proportion (1.7–3.9:1) of the later eluting peak, whereas the remaining subject had more (1.9:1) of the earlier eluting peak. The later eluting peak was identified by LC/MS as R406 (m/z 471). The protonated molecular ion of the earlier eluting peak was observed at m/z 413, which was 58 Da lower than R406. The accurate mass measurement (413.13659) suggested a formula of C19H18FN6O4+ (loss of C3H6O from R406) with a mass error of ±0.5 ppm. The mass spectrum of the fecal metabolite showed product ions at m/z 393 (loss of hydrogen fluoride), 385 (loss of carbon monoxide), 375 (loss of hydrogen fluoride and water), and 365 (loss of hydrogen fluoride and carbon monoxide). The metabolite was purified by HPLC for structural elucidation by NMR. 1H NMR analysis of the purified metabolite did not show any singlet in the aliphatic region corresponding to OCH3 signals, indicative of the loss of CH3 groups present on the trimethoxyphenyl ring of R406 (Fig. 8). Although the spectra showed multiple peaks downfield of δ 2.0, a characteristic peak singlet resonance observed at δ 1.49 in the aliphatic region corresponding to gem-dimethyl signals, as observed for R406, indicated conservation of the 6,6-bicyclic system of R406 in the metabolite. The protons observed at δ 6.59 and 5.80 of the metabolite are attributable to chemical shifts of the aromatic ring protons of the aryl ring. There was no pattern of splitting observed for these two protons, thereby precluding the formation of 3,4-benzene diol because the ortho-aromatic protons are expected to show a coupling constant at least >7 Hz. The mass spectrum of the chemically synthesized 3,5-benzene diol metabolite was similar to that of the purified metabolite (Fig. 9). Therefore, based on the NMR data, the HPLC coelution with an authentic chemically synthesized standard (data not shown), and the tandem mass spectrometry data, the fecal metabolite was identified as the 3,5-benzene diol metabolite of R406.

Profile of radioactivity in the 0- to 48-h fecal sample of Subject 4 and Subject 2 obtained after the oral administration of 14C-R788.

1H NMR of the major human fecal metabolite R406 and chemically synthesized standard. The numbers represent the assignment of protons in the 3,5-benzene diol metabolite.
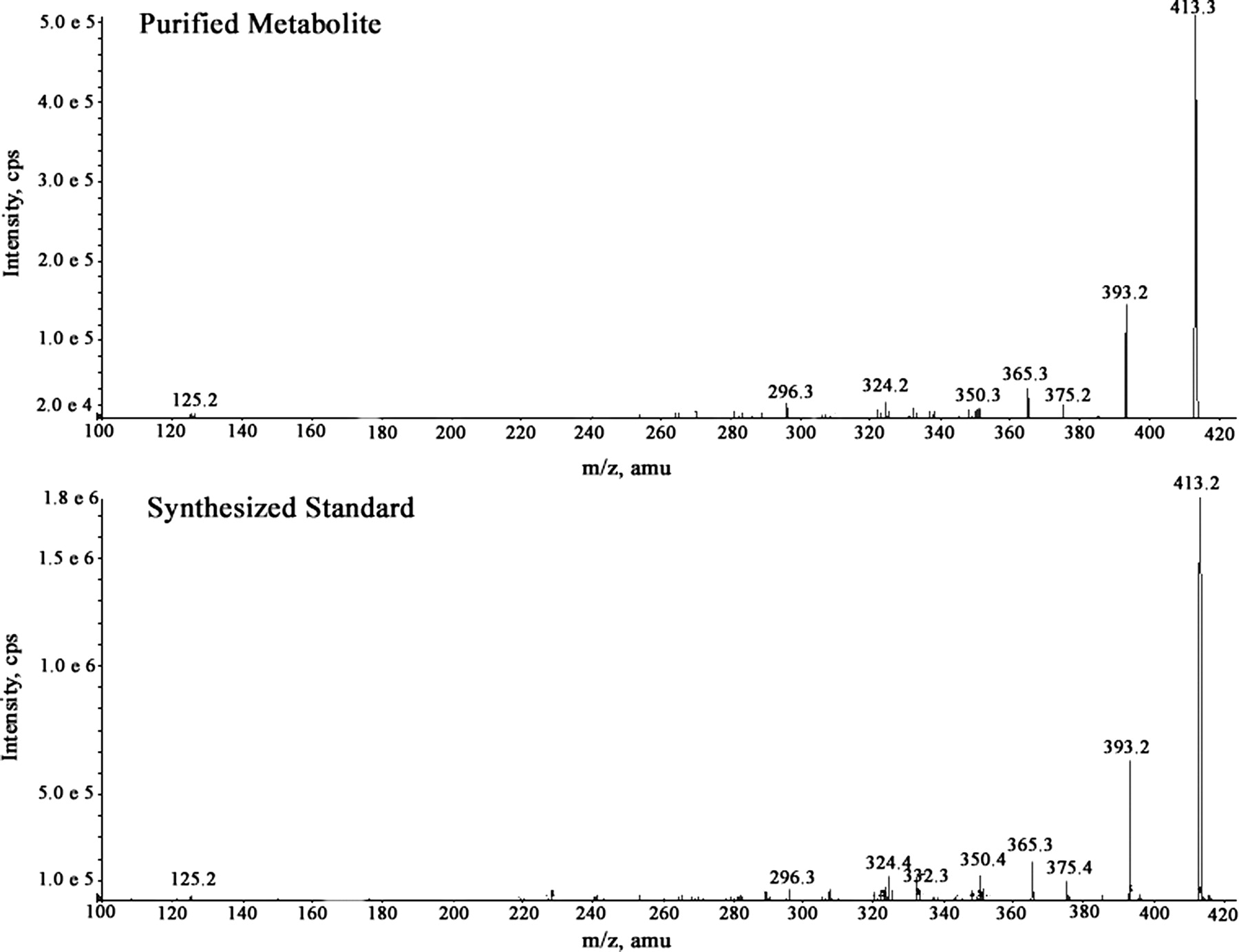
Tandem mass spectrometry spectra of purified M413 and synthesized 3,5 benzene diol metabolite.
Formation of Fecal Metabolite In Vitro.
The unusual 3,5-benzene diol metabolite of R406 was only observed in feces; therefore, the role for gut microflora in the formation of this metabolite was examined. R406, R529, R788, and the N-glucuronide of R406 were incubated with a human fecal sample under anaerobic conditions to determine whether any of these compounds could be converted to the 3,5-benzene diol metabolite. After a 36-h incubation, the only compound that was converted to the 3,5-benzene diol metabolite was R529 (Fig. 10). R406 was stable under these incubation conditions, whereas R788 and the lactam N-glucuronide of R406 were both converted to R406.

HPLC UV profile of the R529 samples incubated for 36 h under anaerobic conditions in the absence and presence of fresh human feces.
Discussion
Because of its poor pharmaceutical properties, R406 is orally administered as the methylene phosphate prodrug R788. This oral prodrug was designed to be cleaved to R406 by alkaline phosphatase(s) that are present on the apical brush-border membranes of the intestinal enterocytes (Fleisher et al., 1985), after which the more hydrophobic R406 would be readily absorbed. The R788 prodrug appears to perform as designed because only low levels of R788 were observed in plasma of the human subjects. Moreover, drug-related material was rapidly absorbed, and R406 was the major radioactive peak in plasma at all the time points. Although R406 was extensively metabolized before elimination, only low levels of circulating R406 metabolites were observed in plasma, indicating the rapid clearance of these metabolites. The plasma metabolites were identified as the glucuronide and sulfate conjugates of the para-O-demethylated metabolite (R529) and direct N-glucuronides of R406. Overall, these metabolites represented less than 3% of total radioactivity in plasma.
The rapid O-demethylation of R406 in human hepatic microsomes together with the absence of significant R406 disappearance in the presence of UDPGA suggested that oxidative metabolism of R406 may have predominated in vivo. However, glucuronidation of R406 was evident in vivo because the lactam N-glucuronide of R406 was the major metabolite in urine. The lactam N-glucuronide in urine represented 14.9% of the administered dose. However, the contribution of N-glucuronidation to the metabolism of R406 in vivo may be higher because the biliary-secreted N-glucuronide conjugate may not be observed in feces as a result of hydrolysis to R406 by gut bacteria. The underestimation of the in vivo glucuronidation of R406 may be in part because of the poor solubility of R406 limiting the concentration (10 μM) used in the microsomal studies because the Kms of many substrates that undergo microsomal glucuronidation are reported in the high micromolar range (Williams et al., 2004). However, other studies using hepatic microsomes have also underestimated the extent of glucuronidation in vivo (Mistry and Houston, 1987; Miners et al., 2004), and it had been suggested that microsomes are not a useful model for predicting the clearance of drugs that undergo glucuronidation (Engtrakul et al., 2005).
The majority of radioactivity (80%) was eliminated in feces, and two major radioactive peaks were observed. One of these radioactive peaks was R406, which may have resulted either from the hydrolysis of unabsorbed R788 or from the hydrolysis of the R406 N-glucuronide secreted into bile because both of these compounds were shown to be converted to R406 after incubation with human feces. The other major fecal radioactive peak was identified as the 3,5-benzene diol metabolite of R406. This unique metabolite was only present in feces and was conceivably formed by the removal of the three methoxyl groups of R406 and the subsequent dehydroxylation of the para-hydroxyl group. A role for gut bacteria in the formation of this metabolite was examined because anaerobic gut bacteria have been shown to carry out the O-demethylation and dehydroxylation of the dietary lignans (Adlercreutz, 2007; Eeckhaut et al., 2008). Secoisolaricirresinol is the most widely studied plant lignan and depends on conversion by colonic bacteria into the biologically active mammalian lignans enterodiol and enterolactone (Wang et al., 2001; Clavel et al., 2005). Multiple strains of anaerobic bacteria are responsible for this metabolism, with the O-demethylation by acetogenic bacteria being reported to occur before the dehydroxylation process (Heider and Fuchs, 1997). In contrast to the aerobic environment, where the O-demethylation reactions are catalyzed by monooxygenases, in the anaerobic gut environment the O-demethylation reactions are mediated by corrinoid-dependent methyl transferases, with tetrahydrofolate serving as ultimate methyl acceptor (Berman and Frazer, 1992). After O-demethylation, the plant lignans are dehydroxylated by a reductive process by additional strains of anaerobic bacteria (Wang et al., 2001).
Because R788, R406, the direct N-glucuronide, or conjugates of R529 may all present in the intestinal tract, any of these compounds could have been the precursor to the 3,5-benzene diol metabolite. However, after in vitro incubations with human fecal bacteria, only R529 was converted to the 3,5-benzene diol metabolite. Because R406 did not undergo O-demethylation by gut bacteria, the presence of the free para-hydroxyl group in R529, like that in the plant lignan secoisolaricirresinol, may be one structural requirement for meta-O-demethylation of R529 to ensue. In support of this supposition, in the sequential O-demethylation of 3,4,5-trimethoxybenzoate by Eubacterium limosum, the initial removal of the para-methoxyl group was shown to be necessary for the meta-O-demethylations to proceed (Cocaign et al., 1991).
The presence of R529 in the gut most likely resulted from the hepatic O-demethylation of R406 and subsequent elimination of the R529 conjugates into bile. Once in the gut, the conjugates could be hydrolyzed back to R529, which was then converted to the 3,5-benzene diol metabolite by gut bacteria. In support of this hypothesis, recent studies in the monkey have shown that R529 conjugates are the major metabolites of R406 in bile and that incubation of these biliary metabolites with monkey feces led to the conversion of the R529 conjugates to the 3,5-benzene diol metabolite (Sweeny et al., 2010). As observed in humans, the 3,5-benzene diol metabolite was also the major metabolite of R788 in the monkey feces.
The identification of the 3,5-benzene diol metabolite of R406 shows a novel sequence of drug metabolism in which an O-demethylation mediated by a hepatic monooxygenase is followed by subsequent O-demethylations and dehydroxylation by anaerobic gut bacteria. These results indicate that gut bacteria are capable of modifying hepatic-derived drug metabolites by pathways other than the commonly recognized reductive (Zachariah and Juchau, 1974) and hydrolytic pathways (Christopher et al., 2008).
The fecal levels of the 3,5-benzene diol metabolite varied markedly among subjects. Subject 4 had very high levels of the 3,5-benzene diol metabolite, whereas low levels of this metabolite were observed in Subject 2. One possible explanation for the variation could be differences in gut bacterial populations between subjects because the activity of the gut microflora has been suggested to be a major determinant of the human lignan levels in plasma (Lampe and Chang, 2007). However, because no intact R529 was observed in the feces from any of the subjects, the differences in the fecal 3,5-bezene diol metabolite levels probably did not reflect variations in the colonic bacterial populations. More likely, the variation in the 3,5-benzene diol levels in feces represents differences in formation of R529, which from the in vitro phenotyping experiments is mediated by CYP3A4. Higher levels of CYP3A4 in Subject 4 may possibly be responsible for the more rapid clearance of R406 and the higher portion of the 3,5-benzene diol metabolite in feces from this individual.
In summary, the metabolic fate of R788 was delineated in the human mass balance study (Scheme 1) and additional in vitro studies. The prodrug R788 was most likely cleaved by intestinal alkaline phosphates to R406, which was the major drug-related product observed in plasma. R406 underwent both direct glucuronidation and a CYP3A4-mediated para-O-demethylation to R529. R529 was most likely conjugated by sulfation and glucuronidation, and these conjugates were secreted into bile. After secretion into the gut, these conjugates were hydrolyzed to R529, which was subsequently O-demethylated and dehydroxylated by anaerobic gut bacteria to the 3,5-benzene diol metabolite, which was the major metabolite fecal of R788 in humans. Overall, the formation of the unique 3,5-benzene diol metabolite resulted from a sequential process involving a hepatic monooxygenase and subsequent metabolism by multiple strains of anaerobic gut bacteria.
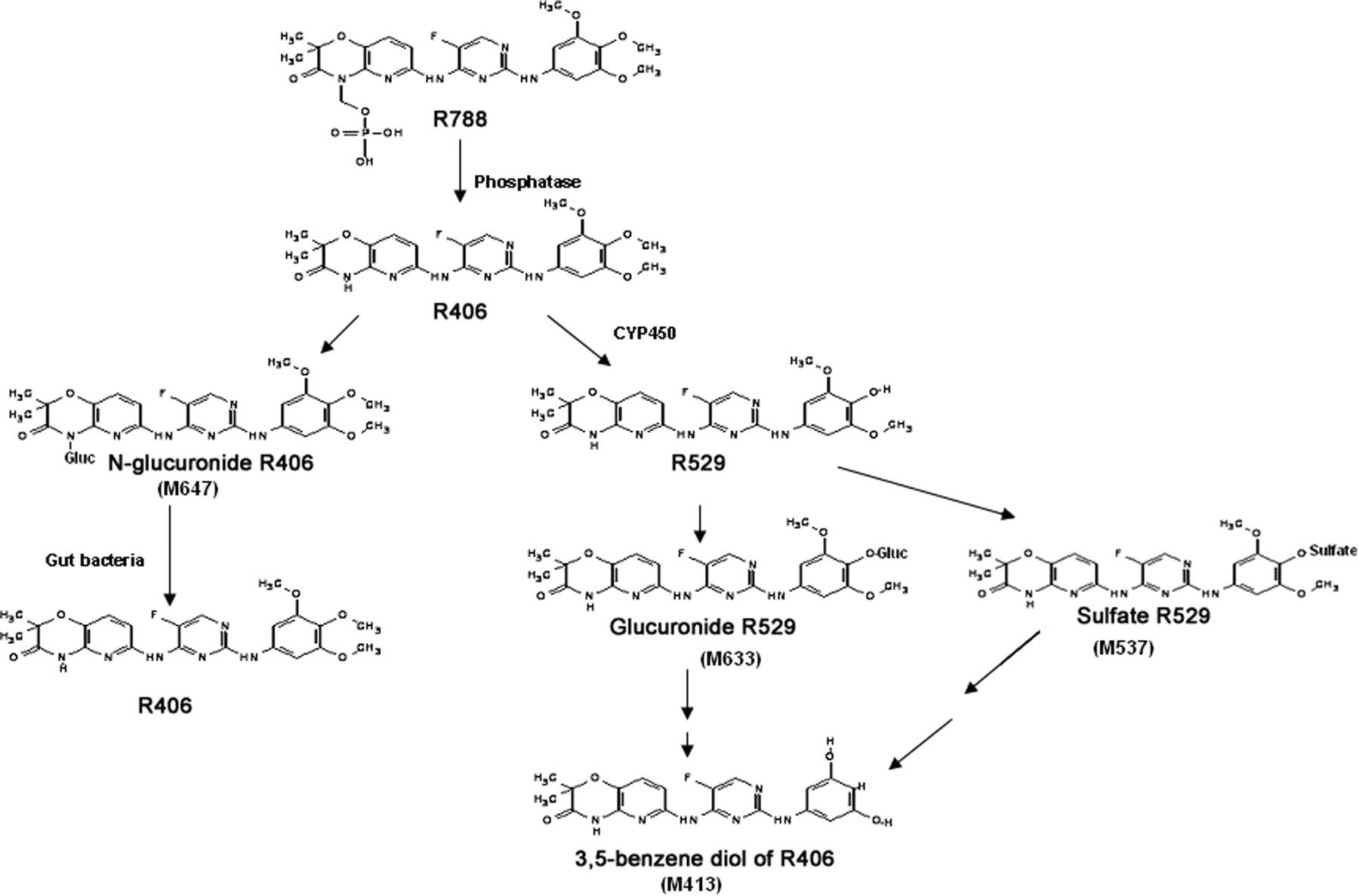
Proposed scheme of R788 metabolism in humans.
Acknowledgments.
We thank Dr. Mohammad Bashir, Dr. Jie Ding, and Claudine Oelke (Covance) for expertise in analyzing samples from the human mass balance study. We also thank members of the Rigel clinical department, Theresa Musser and Aaron Siek, for involvement in the study.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.032151.
-
ABBREVIATIONS:
- Syk
- spleen tyrosine kinase
- R406
- N4-(2,2-dimethyl-3-oxo-4-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3,4,5-trimethyoxyphenyl)-2,4-pyrimidinediamine
- R788
- fostamatinib, N4-(2,2-dimethyl-4-[(dihydrogenphosphonoxy)methyl]-3-oxo-5-pyrid[1,4]oxazin-6-yl)-5-fluoro-N2-(3, 4,5-trimethyoxyphenyl)-2,4-pyrimidinediamine disodium hexahydrate
- R529
- N4-(2,2-dimethyl-3-oxo-5-pyrido[1,4]oxazin-6-yl)-5-fluoro-N2-(4-hydroxy-3,5-dimethoxyphenyl)-2,4-pyrimidinediamine
- lactam N-glucuronide of R406
- N4-{(2,2-dimethyl-4-(1-β-d-glucopyranuronosyl)-3-oxo-5-pyrido[1,4]oxazin-6-yl)}-5-fluoro-N2-(3,4,5-trimethoxyphenyl)-2,4-pyrimidinediamine
- 3,5-benzene diol metabolite of R406
- N2-(3,5-dihydroxyphenyl)-N4-(2,2-dimethyl-3-oxo-5-pyrido[1,4]oxazin-6-yl)-5-fluoro-2,4-pyrimidinediamine
- MTBE
- t-butyl methyl ether
- BHI
- brain heart infusion medium
- DMSO
- dimethyl sulfoxide
- UDPGA
- UDP-glucuronic acid
- HPLC
- high-performance liquid chromatography
- LC/MS
- liquid chromatography/mass spectrometry
- amu
- atomic mass unit(s).
- Received January 12, 2010.
- Accepted April 6, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}