


Abstract
Organic anion-transporting polypeptides (OATPs), members of the SLCO/SLC21 family, mediate the transport of various endo- and xenobiotics. In human liver, OATP1B1, 1B3, and 2B1 are located at the basolateral membrane of hepatocytes and are involved in hepatic drug uptake and biliary elimination. Clinically significant drug-drug interactions (DDIs) mediated by hepatic OATPs have drawn great attention from clinical practitioners and researchers. However, there are considerable challenges to prospectively understanding the extent of OATP-mediated DDIs because of the lack of specific OATP inhibitors or substrates and the limitations of in vitro tools. In the present study, a novel RNA interference knockdown sandwich-cultured human hepatocyte model was developed and validated. Quantitative polymerase chain reaction, microarray and immunoblotting analyses, along with uptake assays, illustrated that the expression and transport activity of hepatic OATPs were reduced by small interfering (siRNA) efficiently and specifically in this model. Although OATP siRNA decreased only 20 to 30% of the total uptake of cerivastatin into human hepatocytes, it caused a 50% reduction in cerivastatin metabolism, which was observed by monitoring the formation of the two major metabolites of cerivastatin. The results suggest that coadministration of a drug that is a hepatic OATP inhibitor could significantly alter the pharmacokinetic profile of cerivastatin in clinical studies. Further studies with this novel model demonstrated that OATP and cytochrome P450 have a synergistic effect on cerivastatin-gemfibrozil interactions. The siRNA knockdown sandwich-cultured human hepatocytes may provide a new powerful model for evaluating DDIs.
Introduction
Organic anion-transporting polypeptides (known as OATPs in human and Oatps in rodents) are members of the SLCO/SLC21 family categorized in the solute carrier class gene superfamily (Hagenbuch and Meier, 2004). OATPs have substrate specificities and broad tissue distributions. They mediate the transport of a variety of structurally diverse endogenous compounds (such as bile acids, prostaglandins, thyroid hormones, and conjugated steroids) and xenobiotics (such as anticancer drugs, antibiotics, cardiac glycoside, and some peptides). Some OATPs are expressed in multiple organs, such as liver, brain, intestine, and kidney, whereas others have limited tissue distribution, for example, OATP1B1 and OATP1B3 are specific to liver. Therefore, OATPs are engaged in drug absorption, distribution, and elimination. The alteration of OATP expression and activities can affect the plasma concentration of drugs, thereby significantly influencing drug toxicities, therapeutic efficacies, and drug-drug interactions (DDIs) (Kim, 2003; Hagenbuch and Gui, 2008).
To date, 11 human OATPs have been discovered and classified into six families (Hagenbuch and Meier, 2003), but only four OATPs (OATP1A2, 1B1, 1B3, and 2B1) have been extensively studied and better characterized (Hagenbuch and Meier, 2003). In the human liver, OATP1A2 was proposed to be expressed specifically in cholangiocytes (Lee et al., 2005). The other three OATPs are localized in the basolateral membrane of hepatocytes and mediate the uptake of a variety of compounds from blood into hepatocytes (Hsiang et al., 1999; König et al., 2000; Abe et al., 2001). Within the hepatocytes, the compounds can then be biotransformed by phase I and phase II enzymes and subsequently eliminated via bile or urine. Therefore, hepatic OATPs, in combination with metabolizing enzymes in hepatocytes and efflux transporters in the apical side of hepatocytes, are involved in the biliary excretion of many drugs and their metabolites. OATP1B1 and 1B3 have similarly broad substrate specificities and share 80% similarity in their amino acid sequences (Hagenbuch and Gui, 2008). In contrast, OATP2B1 has a rather narrow substrate spectrum but broader organ distribution. There is increasing evidence for clinically significant DDIs mediated by OATPs. For example, fibrate-statin interactions have been implicated in several severe adverse reactions including rhabdomyolysis and myopathy after the coadministration of these drugs (East et al., 1988; Bruno-Joyce et al., 2001; Neuvonen et al., 2006). Clinical studies showed that the coadministration of gemfibrozil and cerivastatin caused a significant increase in the AUC of cerivastatin in plasma, thereby resulting in severe side effects, such as rhabdomyolysis. Fifty-two deaths were reported in patients taking cerivastatin worldwide and 12 of 31 cerivastatin-related deaths in the United States took place in patients who were receiving a cerivastatin-gemfibrozil combination. Reports of these deaths resulted in the withdrawal of cerivastatin from the market in 2001. A variety of in vitro model systems, including transfected cell lines expressing single or multiple transporters (such as Madin-Darby canine kidney II cells, HeLa cells, and human embryonic kidney 293 cells), and OATP-transfected Xenopus laevis oocytes have been used to identify the contribution of hepatic OATPs to DDIs (Poirier et al., 2007; Xia et al., 2007). However, the absence of physiologically expressed transporters and drug-metabolizing enzymes in these model systems can lead to uncertain conclusions (Noé et al., 2007). Reliable in vitro model systems for transporter investigations, especially for their related DDI studies, are required to retain the expression of most, if not all, of the transporters, metabolizing enzymes, and regulatory factors controlling their expression. Hepatocytes contain all P450 isoforms, as well as phase II enzymes and transporters, and are organized in a physiological pattern. Hepatocytes are widely accepted as a valuable tool for characterizing transporter functions (Hewitt et al., 2007). Study of the inhibition of a particular transporter might assist in the investigation of transporter-mediated DDIs. Although there is an increasing need to prospectively define these transporter-mediated DDIs, the substantial overlaps in substrate and inhibitor specificities between transporters and drug-metabolizing enzymes as well as the limitations of the in vitro tools available have frustrated efforts to prospectively understand the extent of transporter-mediated DDIs.
RNAi is an invaluable tool for gene function characterization in mammalian cells and thus could be an excellent addition to the traditional methods for DDI studies in human hepatocytes. Since its first application in multidrug resistance protein 1 silencing in 2003, RNAi has been extensively used in transporter function characterization and drug resistance (Yu, 2007) but rarely applied in hepatocytes, with no published record in human hepatocytes (Tian et al., 2004; Yue et al., 2009). In the present study, a sandwich-cultured human hepatocyte model in which OATP expression and activity were efficiently knocked down by siRNA was developed and validated. Given the critical importance of DDIs in drug discovery and development, the contribution of hepatic OATP transporters to drug interaction were also evaluated in this model system. RNAi could be a new technology to predict transporter-related DDIs in human hepatocytes.
Materials and Methods
Materials.
Common cell culture media and supplements such as Dulbecco's modified Eagle's medium (DMEM), insulin-transferrin-selenium-G (100×), bovine serum albumin, penicillin/streptomycin, and l-glutamine were obtained from Invitrogen (Carlsbad, CA). Matrigel was obtained from BD Biosciences (San Jose, CA). Dexamethasone, estrone-3-sulfate (E3S), and gemfibrozil were purchased from Sigma-Aldrich (St. Louis, MO). Cerivastatin, desmethyl cerivastatin, and hydroxy cerivastatin were obtained from Toronto Research Chemicals (Toronto, ON, Canada). [3H]Estrone-3-sulfate and [3H]cerivastatin were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA) and American Radiolabeled Chemicals (St. Louis, MO), respectively. The Accell siRNA reagents, including 5× siRNA buffer, Accell siRNA delivery media, Accell SMARTpool siRNA oligos, Accell human (green) cyclophilin B siRNA (as positive control), and Accell nontargeting pool (as negative control; used as “control siRNA” throughout this study) were obtained from Dharmacon (Lafayette, CO). Rabbit anti-human OATP polyclonal antibody (H60) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
RNAi in Sandwich-Cultured Human Hepatocytes.
Freshly isolated and cryopreserved human hepatocytes were purchased from CellzDirect (Durham, NC) and Celsis IVT (Baltimore, MD). The demographics of the liver donors used in this study are shown in Table 1. The cryopreserved human hepatocytes were thawed at 37°C in a water bath and purified by Percoll before seeding. The viability of human hepatocytes was determined by trypan blue exclusion, and ≥80% viability was required. In brief, 0.5 × 106 hepatocytes in 0.5 ml of DMEM containing 0.1 mM nonessential amino acids, 2 mM l-glutamine, 4.5 g/l glucose, 10% fetal bovine serum, 1× insulin-transferrin-selenium, and 0.1 μM dexamethasone were seeded onto 24-well multiwell plates precoated with collagen IV (BD Biosciences). Hepatocytes were allowed to attach at 37°C for 1 h with 95% air and 5% CO2, and the media were replaced by fetal bovine serum-free DMEM containing 0.1 mM nonessential amino acids, 2 mM l-glutamine, 1× insulin-transferrin-selenium, and 0.1 μM dexamethasone. Cells were cultured for another 2 to 3 h before the initiation of RNAi treatment. In brief, 0.5 ml of Accell delivery media containing 1 μM Accell siRNA designed to silence a specific target gene or control siRNA was added to hepatocytes in 24-well multiwell plates. Cells were incubated at 37°C overnight and overlaid by 0.25 mg/ml Matrigel. Cells were incubated at 37°C for 72 h (mRNA and protein analysis) or 96 h (functional assay) before assessment of RNAi knockdown effect. All experiments were conducted in duplicate.
Demographics of human donors for sandwich-cultured human hepatocytes in this study
Quantitative Real-Time Polymerase Chain Reaction Analyses.
Sandwich-cultured human hepatocytes were washed three times with ice-cold phosphate-buffered saline. Total RNA and protein were isolated simultaneously from cultured cells with the AllPrep RNA/Protein Kit (QIAGEN, Valencia, CA) following the manufacturer's instructions. Antisense amplified RNA (aRNA) was synthesized using the MessageAmp II aRNA Amplification Kit (Applied Biosystems/Ambion, Austin, TX) according to the manufacturer's protocol. Cy5-labeled cDNA was generated using reverse transcription. In brief, 40 μl of a reaction mixture containing 1 to 2 μg of aRNA, 3 μl of 3 nmol/μl Cy5-labeled random nanomers, 4 μl of 100 mM DDT, 2 μl of 10 mM dNTPs, and 2 μl of SuperScript II reverse transcriptase (Invitrogen, Burlington, ON, Canada) in First Stand Buffer was incubated at 42°C for 16 h and at 70°C for 10 min. cDNA was purified using QiaQuick PCR Purification (QIAGEN) according to the manufacturer's protocol. SYBR Green protocols were used to amplify the cDNA of interest by real-time quantitative PCR using an Mx3000P system (Stratagene, La Jolla, CA). The Quantitect primers used for amplification were purchased from QIAGEN (Mississauga, ON, Canada), and the reaction buffer used was PerfeCTa SYBR Green SuperMix, Low ROX (Quanta BioSciences, Gaithersburg, MD). Ten-microliter PCR amplifications consisted of 1.0 μl of cDNA templates, 5.0 μl of SuperMix, and 1.0 μl of 10× primer mix. Reactions were initially denatured to 95°C for 3 min, followed by 40 cycles consisting of denaturation at 94°C for 15 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s. After amplification, samples were denatured at 95°C for 1 min, annealed at 55°C for 30 s, and gradually heated to 95°C to obtain a dissociation curve. The expression levels of each gene of interest were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and calibrated against untreated control samples using comparative quantitation in MxPro software. The results are presented as fold changes in gene expression (GAPDH normalized gene expression levels in the treated samples relative to untreated control samples).
Microarray Analysis.
Cy5-labeled cDNA samples were analyzed by a DTEx microarray (NoAb BioDiscoveries, Mississauga, ON, Canada). Before hybridization, slides were denatured and prehybridized in Nexterion Block E (Schott North America Inc., Louisville, KY) for 15 min at 55°C. Then 500 ng of labeled cDNA and 1 μg of human Cot-1 DNA (Invitrogen) was added to Nexterion Hybridization Buffer (Schott North America Inc.) and heated to 95°C for 5 min. Hybridization was performed in custom-made slide chambers at 60°C for 14 to 16 h. Slides were washed at room temperature sequentially in 2× SSC, 0.2% SDS for 15 min, 2× SSC for 10 min, and 0.2× SSC for 10 min. Slides were scanned and microarray images were acquired using ScanArray software and the ProScanArray HT Microarray Analysis System (PerkinElmer Life and Analytical Sciences, Woodbridge, ON, Canada). After image acquisition, fluorescent signals from the DTEx microarray were assessed using QuantArray (PerkinElmer Life and Analytical Sciences). Results were exported to GeneLinker Gold (Improved Outcomes Software, Kingston, ON, Canada) for normalization, data analysis, and the generation of hierarchical cluster dendrograms and matrix plots. Normalization of the DTEx microarray gene expression levels was performed in two steps: 1) global normalization of all the gene expression levels on the DTEx microarray was performed to determine which of the 12 control genes displayed a constant level of gene expression across all the treated and untreated samples and 2) gene-specific normalization of all the gene expression levels on the DTEx microarray to the control gene(s) from step 1. In this set of experiments, GAPDH gene expression levels were used to normalize the gene expression levels on the DTEx microarrays. Fold change data were generated by comparing normalized gene expression levels between treated and untreated samples. Histograms were generated from normalized gene expression data using Microsoft Excel.
Immunoblotting Analyses.
When needed, protein content was determined using the BCA protein assay (Pierce Endogen, Rockford, IL). Proteins were separated on a 4 to 12% gradient polyacrylamide gel and subsequently transferred electrophoretically to a polyvinylidene difluoride membrane. The membrane was blocked in Tween-Tris-Buffered Saline containing 1.0% Tween 20 with 4% nonfat milk and incubated with rabbit polyclonal anti-human OATP IgGs (1:100, v/v) overnight. The membrane was then incubated with a secondary goat anti-rabbit horseradish peroxidase-coupled antibody (1:1000, v/v) for 1 h. The immunolabeled proteins were further visualized by enhanced chemiluminescence (GE Healthcare, Chalfont St. Giles, UK). Parallel immunoblotting analyses of β-actin served as the internal control for normalization. The protein bands were semiquantified by National Institutes of Health Image Software.
Uptake Assays in Sandwich-Cultured Human Hepatocytes.
Before the studies, the sandwich-cultured human hepatocytes were washed twice with prewarmed Hanks' balanced salt solution (HBSS). The uptake assay was initiated by addition of HBSS-containing [3H]E3S or [3H]cerivastatin (2 μM, substrates for OATP) or in the presence of 100 μM rifampin (inhibitor of OATP). After incubation for 3 min at 37°C, hepatocytes were washed three times with ice-cold HBSS and lysed with 0.5% Triton X-100 for 30 min at room temperature. The radioactivity in the cell lysates was measured by a liquid scintillation counter, and the protein amounts were determined by the BCA protein assay.
P450 Enzyme Activity Assays in Cultured Hepatocytes.
The activity assays were measured in the hepatocytes cultured for 1 h after plating, and the sandwich-cultured hepatocytes were treated with an OATP siRNA mixture targeting OATP1B1, 1B3, and 2B1 for 96 h. Cultured human hepatocytes were washed twice with prewarmed Krebs-Henseleit bicarbonate (KHB) buffer (pH 7.4) and preincubated in KHB buffer containing 3 mM salicylamide at 37°C for 10 min. KHB buffer containing P450 probe substrates (25 μM phenacetin, 25 μM tolbutamide, 100 μM S-mephenytoin, 10 μM midazolam, 20 μM dextromethorphan, and 20 μM amodiaquine) and 3 mM salicylamide were added directly to the hepatocytes to initiate the reaction, and the cells were incubated for 60 min. The incubation buffer was collected, and the cells were subsequently washed with ice-cold KHB buffer three times and lysed with 0.5% Triton X-100 to determine protein concentration. Two volumes of acetonitrile containing 0.1% formic acid and 500 μM carbutamide (internal standard) were added to the incubation buffer and centrifuged at 3000g for 10 min. The concentrations of the metabolite from each P450 substrate in the supernatant were analyzed by LC-MS/MS.
In Vitro Metabolism Studies of Cerivastatin in Sandwich-Cultured Human Hepatocytes.
Sandwich-cultured human hepatocytes were washed twice with prewarmed KHB buffer (pH 7.4). Solutions containing cerivastatin (2 μM), in the absence or presence of the test compounds at different concentrations, were added to the hepatocytes to start the reaction. After incubation for 60 min at 37°C, the incubation buffer was collected for further analysis. The hepatocytes were washed with ice-cold KHB buffer three times and lysed with 0.5% Triton X-100 for 30 min at room temperature, and the cell lysates were collected. The amounts of the two major metabolites of cerivastatin, desmethyl cerivastatin (M1) and hydroxy cerivastatin (M23), in both the incubation buffer and cell lysates were analyzed by LC-MS/MS.
LC-MS/MS for the Metabolites of Cerivastatin.
Quantitation of the two major metabolites of cerivastatin, M1 and M23, was performed using the LC-MS/MS method. The analytes were separated by an HPLC system with a Synergi Hydro RP C18 column (50 × 2.1 mm, 4 μm; Phenomenex, Torrance, CA), an Agilent 1100 system (Agilent Technologies, Palo Alto, CA), and a Valco Valve Diverter (Valco Instruments Co. Inc., Houston, TX) using a gradient of solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). The initial mobile phase was 90:10 (A:B), followed by linear gradient transitioned to 5:95 (A:B) over 5 min. For the first 1.5 min, the samples were diverted to the waste, and from 1.5 to 5 min, the samples were switched into the mass spectrometer for sample collection. The flow rate was 0.5 ml/min. The precursor/product pair ions of M1 (446.1/342), M23 (476.1/280.1), and carbutamide (272.1/156, as internal standard) were monitored under the multiple reaction monitoring mode using a TurboIonSpray source in the positive ionization mode on an API 4000 triple quadrupole mass spectrometer (Applied Biosystems, Foster City, CA).
Results
Effect of siRNA-Mediated OATP Gene Silencing on the Morphology of Human Hepatocytes.
siRNA oligos targeting the OATP gene(s) in Accell siRNA delivery media were induced into sandwich-cultured human hepatocytes. During the culture, the morphological changes of sandwich-cultured human hepatocytes were examined with a phase-contrast microscope. Because Williams' E medium has been widely used and recognized to maintain the morphology and function of hepatocytes during the culture (Jasmund et al., 2007), the morphology of human hepatocytes cultivated with Williams' E medium was used as a normal morphological control. There were no substantial effects observed on the morphology of sandwich-cultured human hepatocytes by either individual siRNA oligos against each OATP (1B1, 1B2, or 2B1) or mixed siRNA oligos against these three OATPs (Fig. 1). In addition, in the optimization experiments, the fluorescent control, cyclophilin B siRNA, showed 60 to 80% silencing efficiency without disturbing the morphology of human sandwich-cultured hepatocytes (data not shown). These findings suggested that the siRNA reagents in this study maintained the normal morphology of sandwich-cultured human hepatocytes.
The morphology of sandwich-cultured human hepatocytes in Accell delivery media containing control siRNA oligos (A), sandwich siRNA oligos against OATP1B1 (B), siRNA oligos against OATP1B3 (C), siRNA oligos against OATP2B1 (D), mixed siRNA oligos against OATP1B1, 1B3, and 2B1 (E), and sandwich-cultured human hepatocytes in Williams' E medium (F). The hepatocytes were obtained from human donor Hu0837.
Effect of OATP Knockdown on OATP mRNA and Protein Expression.
The knockdown effects of siRNA against OATP(s) in sandwich-cultured human hepatocytes obtained from three human donors on mRNA and protein expression levels were assessed by qPCR and immunoblotting analyses (Fig. 2A). After individual OATP knockdown by siRNA, qPCR analyses revealed that in three lots of human hepatocytes, the mRNA expression levels of OATP1B1 and OATP1B3 were reduced to 2.4 to 5.8% compared with the control, whereas OATP2B1 mRNA expressions were only decreased to 14.4 to 27.7%. The mRNA expression levels of OATPs (1B1, 1B3, and 2B1) in hepatocytes treated with the mixed siRNA are illustrated in Fig. 2B. The mRNA levels of hepatic OATPs in hepatocytes from three donors were reduced to 1.5 to 5.0% for OATP1B1, 3.0 to 8.5% for OATP1B3, and 6.0 to 7.5% for OATP2B1 compared with the control (Fig. 2B).
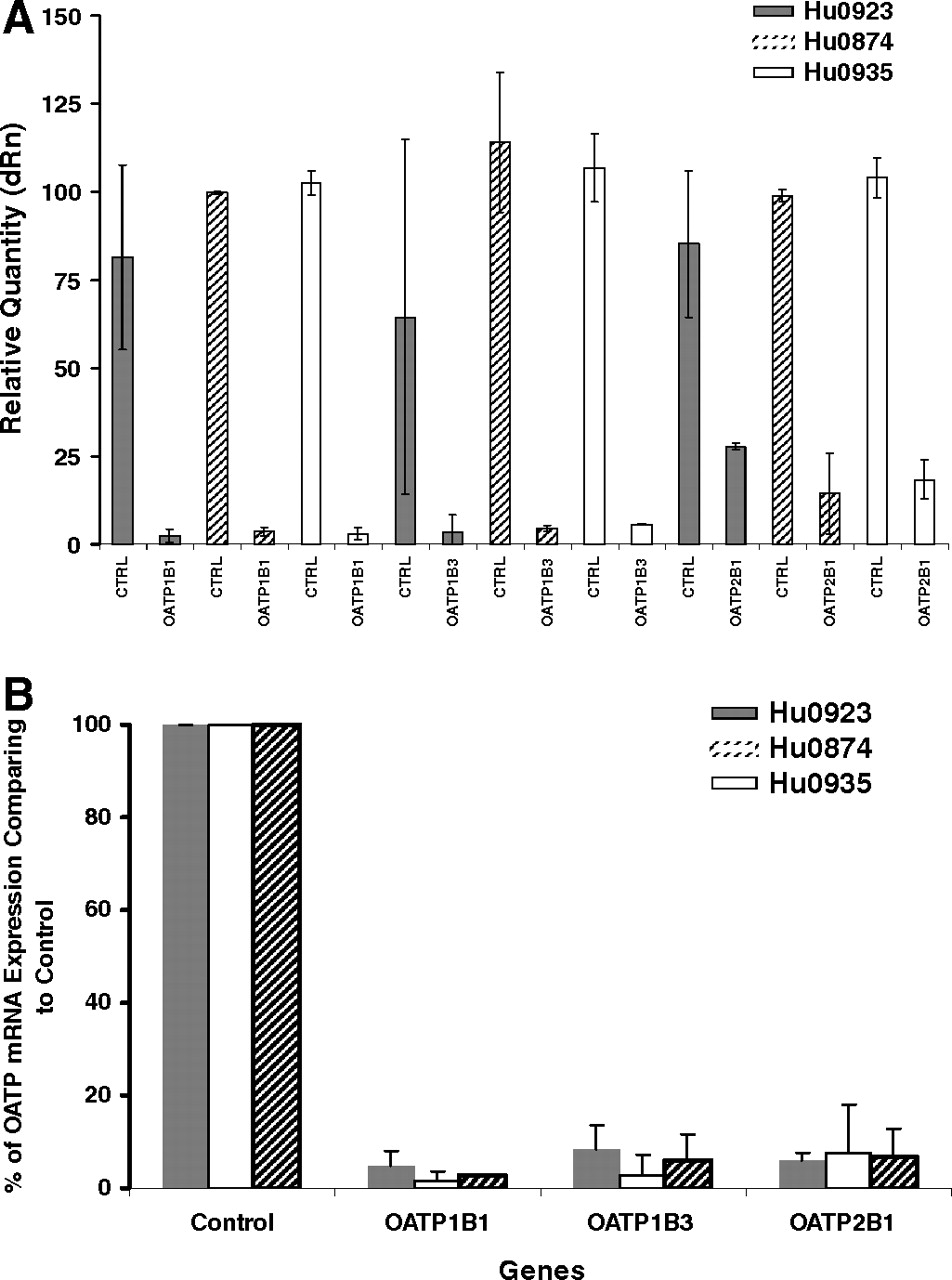
Hepatic OATP mRNA expression in sandwich-cultured hepatocytes treated with OATP siRNA oligos. OATP mRNA values are expressed relative to control (CTRL, control siRNA-treated cells) values. Each bar shows the mean ± S.D. of duplicate experiments. Hepatocytes obtained from three human donors, Hu0923 ( ), Hu0874 (▨), and Hu0935 (□), were used in this study. A, relative quantities of each OATP mRNA expression in sandwich-cultured human hepatocytes treated with the corresponding individual OATP siRNA. dRn, normalized fluorescence intensity. B, relative OATP mRNA expression in sandwich-cultured human hepatocytes treated with OATP siRNA mixture against OATP1B1, 1B3, and 2B1.
), Hu0874 (▨), and Hu0935 (□), were used in this study. A, relative quantities of each OATP mRNA expression in sandwich-cultured human hepatocytes treated with the corresponding individual OATP siRNA. dRn, normalized fluorescence intensity. B, relative OATP mRNA expression in sandwich-cultured human hepatocytes treated with OATP siRNA mixture against OATP1B1, 1B3, and 2B1.
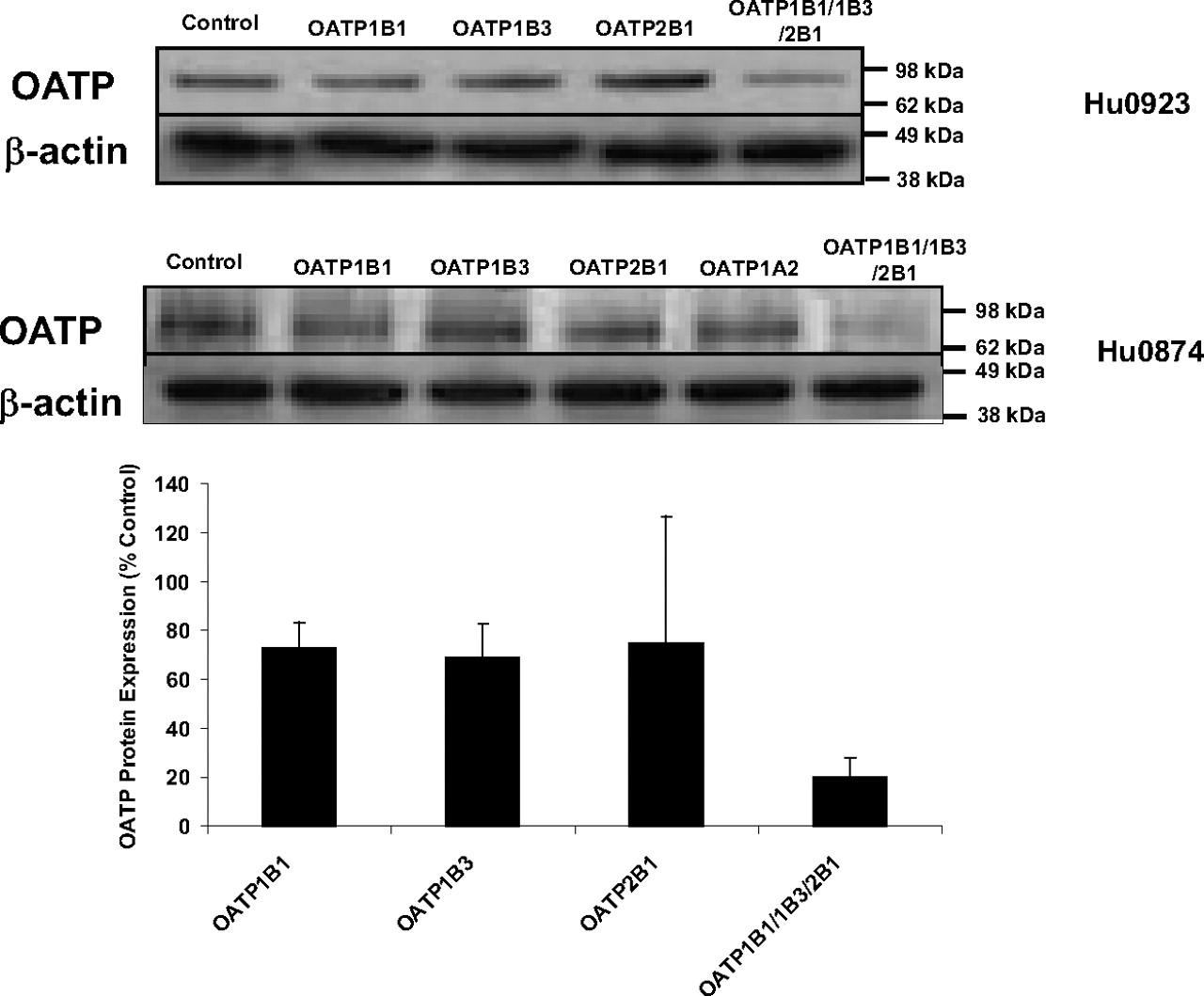
The parallel immunoblotting analyses of hepatocyte lysates with anti-OATP-C polyclonal IgGs indicated that the OATP protein expression was substantially reduced in lysates of hepatocytes treated with the OATP siRNA mixture targeting OATP1B1, 1B3, and 2B1. However, there was no significant difference in effect on OATP protein expression between individual OATP siRNA-treated and control siRNA-treated hepatocytes from three human donors (Fig. 3). This finding could be due to the lesser selectivity of OATP antibody. The commercial available anti-OATP-C polyclonal IgG is recommended by the manufacturer to detect OATP1B1 and, to a lesser extent, OATP1B3 of human origin. Figure 3 shows representative Western blot results of hepatic OATP protein expression from two lots of human hepatocytes. Together, these findings suggested that the siRNA against OATP(s) substantially silenced the OATP(s) mRNA and protein expression in sandwich-cultured human hepatocytes.

Representative OATP immunoblotting in sandwich-cultured hepatocytes treated with OATP siRNA. OATP expression in two lots of human hepatocytes, Hu0923 (above) and Hu0874 (below), are shown. Parallel actin immunoblots were included as internal controls. The densitomeric results were also shown.
Role of OATP Knockdown on the mRNA Expression of ADME-Associated Genes.
To determine whether the introduction of OATP siRNA oligos induced the alteration in the gene expression of other, nontarget, proteins, qPCR or microarray analysis was used to identify the mRNA expression of nontarget OATP members and ADME-associated genes in sandwich-cultured human hepatocytes. Gene expression changes greater than 1.5-fold or less than 0.5-fold were regarded as significant gene expression induction or reduction. qPCR analysis illustrated that OATP1B1 siRNA not only significantly inhibited OATP1B1 mRNA expression in the three lots of human hepatocytes but also reduced OATP1B3 mRNA expression in the lots Hu0935 and Hu0874, whereas OATP1B1 siRNA induced the mRNA expression of OATP2B1 in Hu0923 and Hu0935 (Fig. 4). Likewise, OATP1B3 siRNA significantly decreased the mRNA expression of OATP1B3 and OATP1B1 without any appreciable effect on the expression of OATP2B1 in three lots of human hepatocytes. OATP2B1 siRNA inhibited OATP2B1 mRNA expression significantly but had no notable influence on the mRNA expression of OATP1B1 in hepatocytes from three human donors. In Hu0935, OATP2B1 siRNA decreased OATP1B3 mRNA expression to 0.45-fold.

mRNA expression of OATP1B1, 1B3, and 2B1 in sandwich-cultured human hepatocytes treated with individual OATP siRNAs. mRNA values of OATP genes in three lots of human hepatocytes, (A) Hu0923, (B) Hu0874, and (C) Hu0935, are expressed relative to control (control siRNA-treated cells) values. Each bar shows the mean ± S.D. of duplicate experiments.
The expression of 145 ADME-associated genes involved in drug metabolism and conjugation or transport in human hepatocytes was evaluated by microarray analysis. The mRNA expression of five nontarget genes was significantly affected by the OATP siRNA mixture targeting OATP1B1, 1B3, and 2B1 in Hu0923, namely, ABCA10 (2.0-fold), PEPT1 (1.8-fold), CYP2B6 (1.7-fold), SULT1E1 (1.9-fold), and SULT2A1 (1.6-fold) (Fig. 5). Such nonspecific mRNA changes were donor-dependent. Except for CYP2B6, the mRNA expression of which was induced by 1.7-fold, the other four nontarget genes were not significantly affected by the OATP siRNA mixture in Hu0874. There were no significant changes (within ±50% range) observed for the mRNA expressions of the other ADME-associated genes treated with an OATP siRNA mixture in either human hepatocyte lot (data not shown).

mRNA expression of ADME-associated genes in sandwich-cultured human hepatocytes treated with OATP siRNA mixture. mRNA values of five genes whose expression was nonspecifically affected are depicted relative to control (control siRNA-treated cells) values. Each bar shows the mean ± S.D. of duplicate experiments.
Inhibitory Effects of OATP Gene Silencing on E3S Uptake in Sandwich-Cultured Human Hepatocytes.
The uptake of E3S was analyzed to characterize the function of OATP in sandwich-cultured human hepatocytes. E3S was used as a prototypical hepatic OATP substrate. Because rifampin at 100 μM was believed to totally inhibit OATP-mediated uptake of E3S in sandwich-cultured human hepatocytes (with an IC50 value of 25.9 μM) (data not shown) (Vavricka et al., 2002; Lau et al., 2004), the uptake of E3S in the presence of 100 μM rifampin was considered to be due to passive diffusion and was used as inhibitory control in this study. In hepatocytes from human donor Hu0879, the individual siRNA against OATP1B1, 1B3, or 2B1 reduced the E3S uptake to 35.2, 43.0, and 68.0%, respectively, and the OATP siRNA mixture decreased the uptake of E3S even more to 23.2% of control, indicating that siRNA-mediated OATP silencing suppressed the OATP function effectively in sandwich-cultured human hepatocytes (Fig. 6A). To determine the interindividual variability in the expression of drug transporters in human hepatocytes, such uptake assays were also conducted in the hepatocytes from another three human donors, Hu0935, Hu0923, and Hu0874. The knockdown efficiencies for the OATP siRNA mixture were identical, and approximately 70 to 80% of OATP-mediated E3S uptake was reduced in knockdown hepatocytes (Fig. 6B). Given that hepatocytes from Hu0874 were cryopreserved, these findings demonstrated that the knockdown efficiencies of OATP activity mediated by siRNA were comparable in hepatocyte cultures irrespective of whether fresh or cryopreserved human hepatocytes were used.
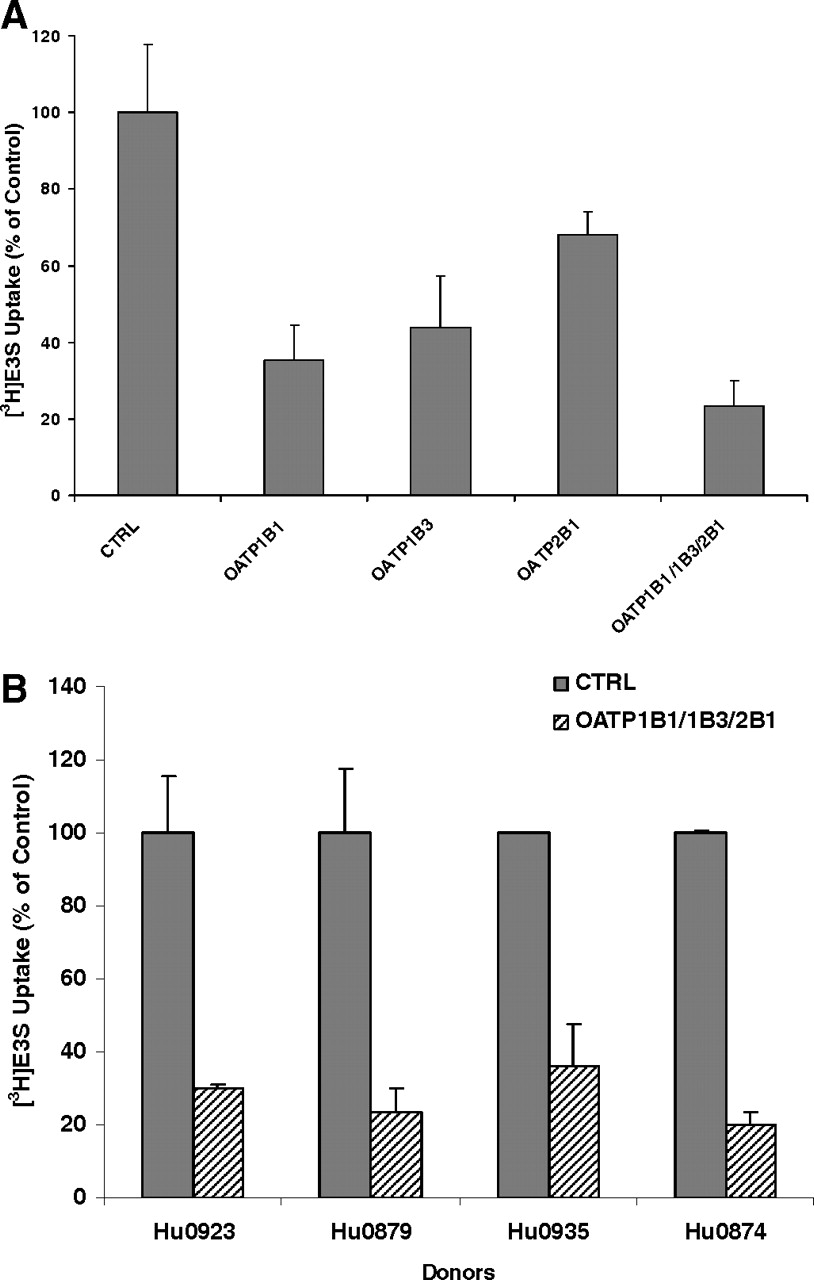
Uptake transport of E3S in sandwich-cultured human hepatocytes treated with OATP siRNA. [3H]E3S uptake values are expressed as a percentage of the values in hepatocytes treated with control siRNA (CTRL), adjusted for the contribution of passive diffusion. Each bar shows the mean ± S.D. of duplicate experiments. A, [3H]E3S uptake in sandwich-cultured human hepatocytes from donor Hu0879 treated with each OATP siRNA or the OATP siRNA mixture. B, comparison of [3H]E3S uptake knockdown efficiencies in OATP siRNA mixture-treated sandwich-cultured human hepatocytes obtained from four human donors.
Effect of siRNA Reagents on P450 Enzyme Activity in Sandwich-Cultured Human Hepatocytes.
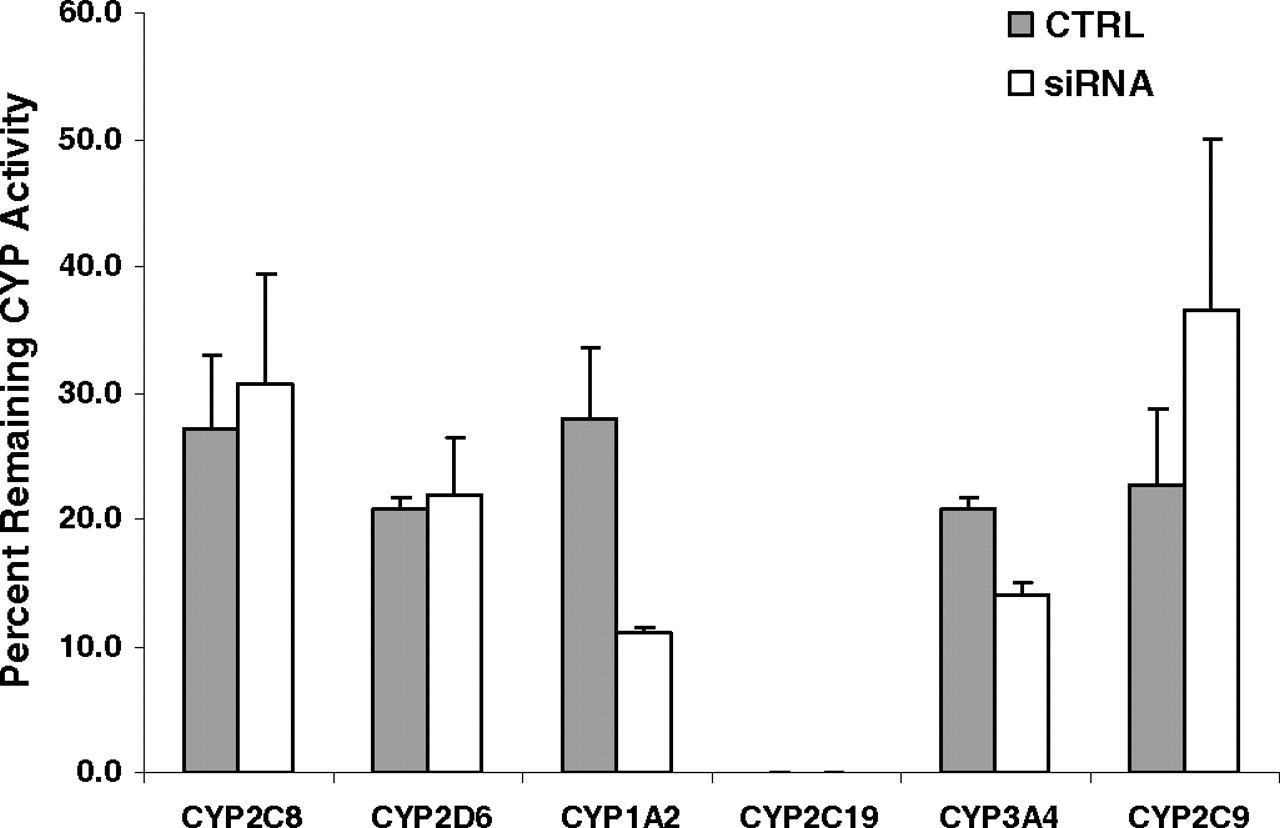
P450 activity in sandwich-cultured human hepatocytes treated with an siRNA mixture against OATP1B1, 1B3, and 2B1 for 96 h was assessed to evaluate the effect of siRNA reagents on the activities of CYP3A4, 2C9, 2C19, 2D6, 2C8, and 2E1. The sandwich-cultured hepatocytes without siRNA reagent treatment were used as a noninhibitory control, and human hepatocytes cultured for 1 h after plating were used as a control with 100% P450 activity remaining. As shown in Fig. 7, the activities of all P450 enzymes declined substantially in both treated and nontreated human hepatocytes cultured for 5 days. siRNA reagents did not significantly affect the activities of CYP2C8 and CYP2D6. However, the OATP siRNA mixture treatment showed a weak inhibitory effect on CYP3A4, ∼50% inhibition on CYP1A2, and a slight induction on CYP2C9 in sandwich-cultured human hepatocytes compared with their respective control cells.

Effects of siRNA reagents on P450 enzyme (CYP) activity in sandwich-cultured human hepatocytes. P450 enzyme activities in siRNA-treated and nontreated sandwich-cultured hepatocytes cultured for 5 days are expressed as a percentage of the values in hepatocytes cultured for 1 h after plating. Each bar shows the mean ± S.D. of duplicate experiments.
Effect of OATP Gene Knockdown on Cerivastatin Clearance in Sandwich-Cultured Human Hepatocytes.
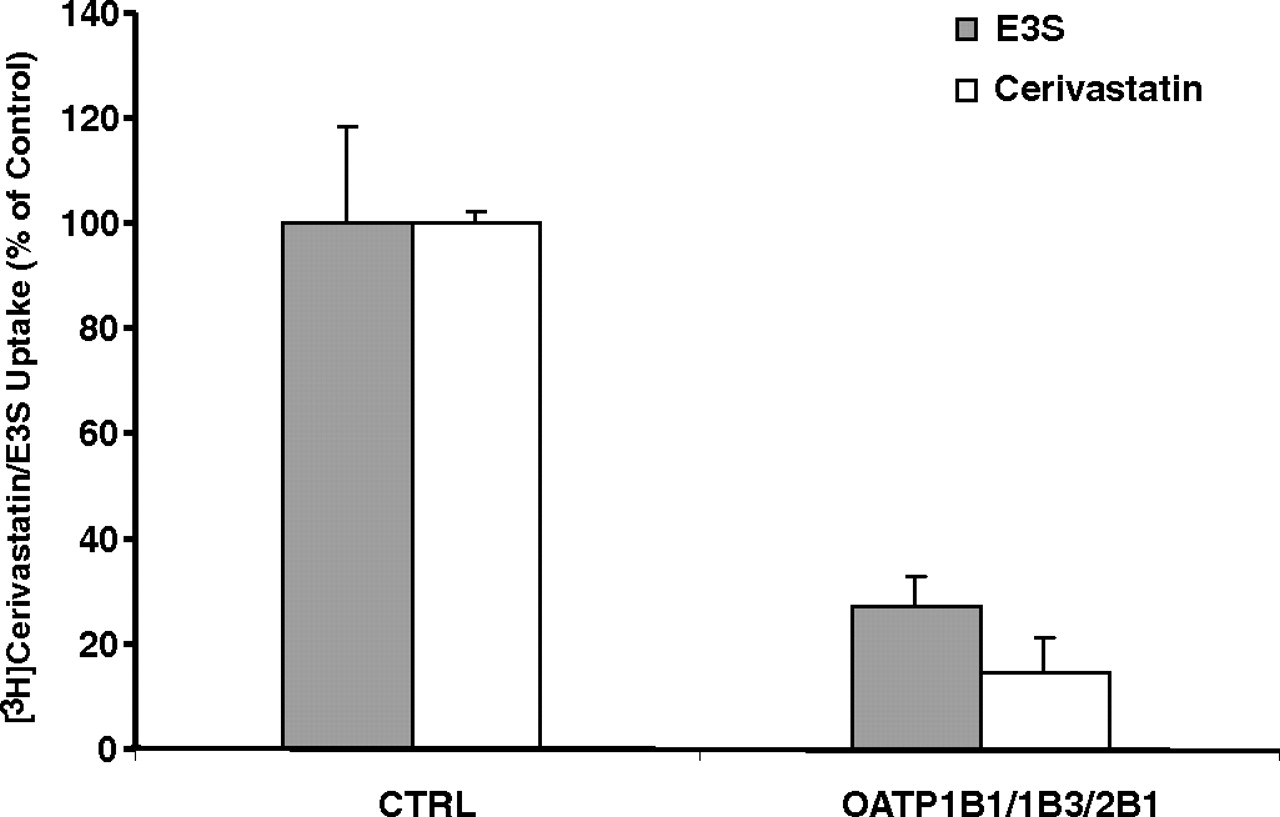
The inhibition of OATP-mediated cerivastatin uptake into sandwich-cultured human hepatocytes by the OATP siRNA mixture was monitored after a 3-min incubation of hepatocytes with 2 μM cerivastatin. Rifampin at a concentration of 100 μM was used as the inhibitory control to completely inhibit OATP-mediated cerivastatin uptake. The OATP siRNA mixture reduced cerivastatin uptake to <20% of the control, adjusted for the contribution of passive diffusion, indicating that OATP-mediated cerivastatin uptake was efficiently inhibited by siRNA in sandwich-cultured human hepatocytes (Fig. 8). The assays conducted in both OATP knockdown hepatocytes and control hepatocytes indicated that neither of the two known major metabolites of cerivastatin, M1 or M23 (Prueksaritanont et al., 2002; Shitara et al., 2004), was detectable in the incubation buffer or cells after a 3-min incubation.

Uptake transport of cerivastatin in sandwich-cultured human hepatocytes treated with an siRNA mixture targeting OATP1B1, 1B3, and 2B1. [3H]Cerivastatin uptake values are expressed as a percentage of the values in control cells (CTRL), adjusted for the contribution of passive diffusion. The knockdown efficiencies were monitored by the measurement of the uptake of 2 μM [3H]E3S in parallel. Each bar shows the mean ± S.D. of duplicate experiments.
To evaluate the effect of OATP knockdown on cerivastatin clearance, sandwich-cultured human hepatocytes treated with the OATP siRNA mixture were incubated with 2 μM cerivastatin as substrate for 1 h. The amounts of the two major metabolites of cerivastatin, M1 and M23, in the incubation buffer and hepatocytes were quantified using LC-MS/MS. The corresponding hepatocytes treated with nontargeting siRNA were used as control cells. OATP knockdown decreased the amount of M1 by 50 and 40% in the buffer and hepatocytes, respectively. However, there was no effect on M23 formation by OATP knockdown observed in the incubation buffer and hepatocytes (Fig. 9).

Effects of OATP knockdown on the metabolism of cerivastatin in sandwich-cultured human hepatocytes. The content values of two major metabolites of cerivastatin, M1 and M23, are expressed as a percentage of the values in control cells (CTRL). Each bar shows the mean ± S.D. of duplicate experiments.
Effects of Gemfibrozil on Cerivastatin Metabolism in Sandwich-Cultured Human Hepatocytes.
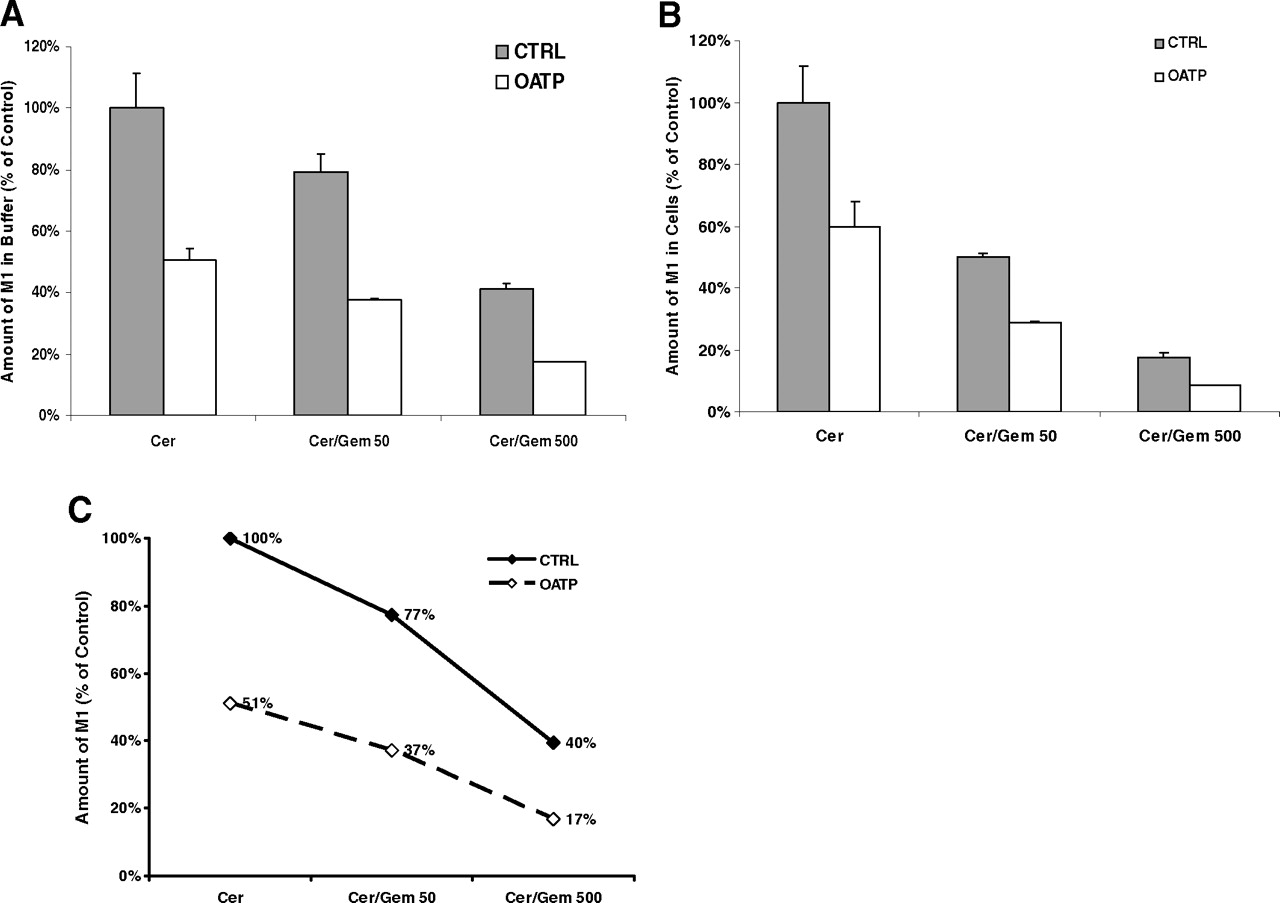
To understand the enzyme- and OATP-mediated DDIs in the sandwich-cultured human hepatocyte model, the effects of gemfibrozil on cerivastatin metabolism in sandwich-cultured human hepatocytes were assessed by monitoring the production of the metabolites of cerivastatin, M1 and M23. Gemfibrozil at a concentration of 50 or 500 μM with 2 μM cerivastatin was used to treat OATP knockdown hepatocytes and control hepatocytes, and the incubation buffer and cell lysates were collected for the bioanalytical quantification of M1 and M23. A concentration-dependent inhibitory effect of gemfibrozil (50 or 500 μM) on M1 formation in incubation buffer and hepatocytes was detected in OATP-knockdown hepatocytes and control cells (Fig. 10). Likewise, at concentrations of 50 and 500 μM, gemfibrozil drastically reduced the M23 content in a concentration-dependent manner in incubation buffer, whereas M23 was not detectable in hepatocytes (Fig. 11). These findings demonstrated that gemfibrozil substantially inhibited cerivastatin metabolism.

Effects of gemfibrozil (Gem) on the formation of demethyl cerivastatin (Cer, M1) in the control (CTRL) and OATP knockdown sandwich-cultured human hepatocytes. The amount of M1 in the incubation buffer (A) and cell lysates (B) was monitored. Metabolite content values are expressed as a percentage of the values in control (control siRNA and no gemfibrozil treatment) cells. Each bar shows the mean ± S.D. of duplicate experiments. C, effect of gemfibrozil on the total M1 formation in the incubation buffer and hepatocytes.

Effects of gemfibrozil (Gem) on the formation of hydroxyl cerivastatin (Cer, M23) in control (CTRL) and OATP knockdown sandwich-cultured human hepatocytes. The amount of M23 in the incubation buffer (A) and cell lysates (B) were monitored. Metabolite content values are expressed as a percentage of the values in control (control siRNA and no gemfibrozil treatment) cells. Each bar shows the mean ± S.D. of duplicate experiments. C, effect of gemfibrozil on the total M23 formation in the incubation buffer and hepatocytes.
Discussion
Human hepatic OATP1B1, 1B3, and 2B1 function can have an impact on drug disposition, hepatic clearance, and drug-drug interactions (Kim, 2003; Hagenbuch and Gui, 2008). However, the specific contribution of hepatic OATP to clinically significant drug-drug interactions remains to be defined. The lack of selective inhibitors and substrates as well as effective and reliable in vitro tools has compromised mechanistic evaluation on OATP-mediated DDIs. Although selective probe substrates, inhibitors, antibodies, recombinant enzymes, and in vitro methods have been applied to quantify the involvement of specific P450s to better understand their role in drug disposition, the deficiency of similar tools has frustrated the identification of the roles played by transporters in DDIs. In this study, a novel in vitro sandwich-cultured human hepatocyte model was developed, wherein the expression and activity of hepatic OATP were specifically and efficiently knocked down by siRNA. The interplay between P450 enzymes and OATP transporters in DDI can also be evaluated using this model system.
RNAi technology has been extensively used to study the function of genes in vitro in mammalian cell culture and in vivo in model organisms (Hannon, 2002; Lee and Sinko, 2006). The knockdown efficiency of target genes by siRNA is dependent on effective siRNA duplexes, efficient delivery methods, and off-target effects. Being primary cells, human hepatocytes are typically difficult to transfect. The low delivery efficiency of traditional transfection methods has restricted the application of siRNA to hepatocytes. So far, only two studies on the role of efflux transporters on drug biliary elimination using RNAi technology in rat hepatocytes have been reported (Tian et al., 2004; Yue et al., 2009). Although adenovirus-mediated shRNA generally achieves high delivery efficiency, it is time consuming and tedious. In the current study, a novel siRNA system was applied in sandwich-cultured human hepatocytes. As demonstrated in Fig. 2, up to 99% of mRNA expression of OATP1B1 and OATP1B3 was reduced by their individual siRNAs and the OATP siRNA mixture. The knockdown efficiencies of OATP2B1 mRNA expression were 80 and >90% by OATP2B1 siRNA and an OATP siRNA mixture, respectively. Furthermore, the knockdown of the three hepatic OATPs by the OATP siRNA mixture led to the reduction of OATP protein expression by approximately 80%. However, when an individual OATP was knocked down by its specific siRNA, OATP protein expression had no notable change, which may be due to the lack of specificity of the commercial anti-OATP-C polyclonal antibodies. These antibodies are against OATP1B1, but they also recognize OATP1B3 in human cells (Santa Cruz Biotechnology, Inc.). Functional assays are more accurate and unequivocal in assessing siRNA knockdown effects on target genes than are assays that quantify the changes in mRNA and protein expression levels. OATP-mediated E3S uptake in human hepatocytes was substantially decreased by individual OATP siRNAs and the OATP siRNA mixture (Fig. 6). Such E3S uptake knockdown was detected in cryopreserved or fresh hepatocytes obtained from four tested human donors. In both fresh and cryopreserved human hepatocytes, siRNA knocked down the mRNA and protein expressions and activities of hepatic OATPs efficiently, validating this model for the characterizing of OATP functions in vitro.
The off-target effects of siRNA have always been a concern to the application of this technique. The off-target effects by OATP siRNA on the expression of the major ADME-associated genes in cultured human hepatocytes were evaluated by qPCR and microarray analysis. As illustrated in Fig. 4, each individual OATP siRNA also changed the gene expression of other OATP members, which could be partly explained by the high homology that exists in DNA sequences of the three hepatic OATPs. Five nontarget genes of 145 ADME-related genes were significantly affected by the OATP siRNA mixture in human hepatocytes from donor Hu0923. However, such a nonspecific effect was donor-dependent. CYP2B6 is the only gene whose mRNA expression level was considerably elevated by the OATP siRNA mixture in both human donors. The current study is an attempt to reveal the combined contribution of the three hepatic OATPs on DDIs with cerivastatin. There are no published reports showing the involvement of CYP2B6 in the metabolism of cerivastatin. Therefore, the nonspecific effects on the expression of nontarget OATP members by individual OATP siRNA and ADME-associated genes by the OATP siRNA mixture were not considered relevant in this particular study.
In humans, cerivastatin is exclusively eliminated by biotransformation, undergoing two metabolic pathways mediated by CYP2C8 and CYP3A4. The two primary metabolites of cerivastatin, M1 and M23, are produced via demethylation and hydroxylation pathways, respectively (Mück, 2000; Prueksaritanont et al., 2002). In human liver, cerivastatin was actively taken up into hepatocytes by OATP transporters (Ishikawa et al., 2004). Several clinical and in vitro studies have been performed to investigate the mechanisms of cerivastatin DDIs between cerivastatin and gemfibrozil (Backman et al., 2002; Shitara et al., 2004). However, few of the in vitro systems physiologically express both transporters or P450s and, consequently, cannot model the interplay between these two systems on the disposition of drugs.
In the present study, the cerivastatin uptake assays in an in vitro sandwich-cultured human hepatocyte model revealed that 20 to 30% of cerivastatin was actively taken up into hepatocytes via OATP (Table 2). As a result, passive diffusion represents 70 to 80% of cerivastatin uptake in sandwich-cultured human hepatocytes compared with 20 to 30% of that reported in suspended human hepatocytes (Shitara et al., 2003). The siRNA efficiently inhibited 80 to 90% of OATP-mediated cerivastatin uptake into sandwich-cultured human hepatocytes.
The uptake of [14C] cerivastatin in sandwich-cultured human hepatocytes
Considering that the total uptake of cerivastatin mediated by passive diffusion is greater than that mediated by OATPs, it is intriguing that the 20 to 30% reduction in cerivastatin uptake into cultured human hepatocytes is responsible for the 50% decrease in M1 formation in the incubation buffer and hepatocytes (Fig. 9). In contrast, the formation of M23 in the buffer and hepatocytes was not affected by siRNA-mediated cerivastatin uptake reduction. Previous reports showed that cerivastatin demethylation (M1) is catalyzed by CYP2C8 and CYP3A4 equally, whereas its hydroxylation (M23) is mainly mediated by CYP2C8 rather than by CYP3A4 (Mück, 2000; Prueksaritanont et al., 2002). Further investigation has shown that the Km value for cerivastatin demethylation in the recombinant CYP2C8 was 0.23 μM, lower than that in the recombinant CYP3A4, which had a Km value of 1.37 μM (data not shown). Moreover, equivalent Km values of ∼0.2 μM were observed for cerivastatin hydroxylation by both CYP2C8 and CYP3A4. Consequently, one possible explanation for the greater inhibition of the formation of M1 than M23 by the siRNA is that CYP2C8 is more easily saturated than CYP3A4 by cerivastatin for demethylation and hydroxylation reactions. A slight decrease in cerivastatin contents in hepatocytes caused by the siRNAs would have a more pronounced affect on CYP3A4-mediated metabolism (M1) than on CYP2C8-mediated cerivastatin metabolism (M23).
The production of M23 was substantially lower than that of M1; M23 only accounts for 10% of total metabolites formed in cultured hepatocytes (data not shown). Because the intrinsic clearance change of cerivastatin can be expressed as the change of the formation of major cerivastatin metabolites (M1 and M23), the 20 to 30% reduction of cerivastatin uptake caused by OATP knockdown can decrease cerivastatin clearance by approximately 50%, suggesting that coadministration of a drug that is an OATP inhibitor may significantly increase cerivastatin blood concentration in vivo. This speculation is consistent with a previous clinical investigation, in which the inhibition of transporter-mediated cerivastatin uptake rather than P450-mediated cerivastatin metabolism was suggested as the cause for a 3- to 5-fold increase in the plasma AUC of cerivastatin in kidney transplant patients receiving cyclosporine compared with the AUC of cerivastatin in healthy volunteers (Mück et al., 1999).
Several clinically significant DDIs between gemfibrozil and statins have been reported. Gemfibrozil is a potent inhibitor of CYP2C8 and CYP2C9 (Wen et al., 2001; Fujino et al., 2003; Ogilvie et al., 2006). One clinical study in healthy volunteers showed that administration of the usual therapeutic doses of gemfibrozil markedly increased the AUC of cerivastatin by 5- to 6-fold and the AUC of metabolite M1 by 3-fold. In contrast, the level of metabolite M23 was reduced to 22% (Backman et al., 2002). Gemfibrozil is an inhibitor of OATP1B1 with an IC50 value of 72 μM in in vitro OATP1B1-expressing Madin-Darby canine kidney cells. In addition, in vitro metabolism studies in CYP2C8 and CYP3A4 expression systems elucidated that gemfibrozil inhibited CYP2C8-mediated cerivastatin metabolism (IC50 = 28 μM) more potently than CYP3A4-mediated metabolism (IC50 = 372 μM) (Shitara et al., 2004). To understand the enzyme- and OATP-mediated DDIs in this sandwich-cultured human hepatocyte model, the combination of 50 or 500 μM gemfibrozil and 2 μM cerivastatin was used to treat OATP-knockdown and control hepatocytes. A concentration-dependent inhibitory effect of gemfibrozil on M1 formation in the buffer and hepatocytes was observed in both OATP knockdown and control hepatocytes (Fig. 10, A and B). In addition, gemfibrozil decreased M1 formation more substantially in OATP-knockdown hepatocytes (from 51 to 17% treated with 500 μM gemfibrozil) than in control cells (from 100 to 40% treated with 500 μM gemfibrozil) (Fig. 10C), thus suggesting that the inhibition of CYP2C8 by gemfibrozil is not the sole mechanism for the decreased M1 formation, and that the inhibition of OATP by gemfibrozil at a higher concentration (500 μM) would contribute to the decreased M1 formation in vivo. In addition, gemfibrozil reduced M23 formation more drastically than M1 at the concentrations of 50 and 500 μM (Fig. 11). These results suggested that gemfibrozil inhibited cerivastatin hydroxylation more extensively than cerivastatin demethylation, which is consistent with the previous report in human hepatocytes (Prueksaritanont et al., 2002).
The predicted fold change of the AUC in the presence or absence of inhibitor (AUCinh/AUC) can be described as the ratio of the metabolic intrinsic clearance (CL), calculated as the rate of the total metabolite formation (ΔM). The fold change of AUC can thus be expressed by the following equation:
 In consideration of the assumption discussed previously, OATP knockdown contributed approximately a 2-fold change in the buffer AUC ratio in sandwich-cultured human hepatocytes, whereas 50 μM gemfibrozil increased the AUC ratio by 1.3-fold. However, the combination of OATP knockdown and 50 μM gemfibrozil treatment increased the ratio to 2.7-fold, which is slightly greater than the sum of the individual ratio change of 2.3-fold, suggesting that OATP and P450 have additional or synergistic effects on cerivastatin DDIs in sandwich-cultured human hepatocytes. A synergistic effect was observed when cultured human hepatocytes were treated with 500 μM gemfibrozil. OATP knockdown caused a 6.0-fold change in the AUC ratio in hepatocytes treated with 500 μM gemfibrozil, compared with a 2.5-fold change in nontreated human hepatocytes (Fig. 12). These results derived from the new in vitro model are similar to previous clinical observations and results in other in vitro studies (Backman et al., 2002; Shitara et al., 2004).
In consideration of the assumption discussed previously, OATP knockdown contributed approximately a 2-fold change in the buffer AUC ratio in sandwich-cultured human hepatocytes, whereas 50 μM gemfibrozil increased the AUC ratio by 1.3-fold. However, the combination of OATP knockdown and 50 μM gemfibrozil treatment increased the ratio to 2.7-fold, which is slightly greater than the sum of the individual ratio change of 2.3-fold, suggesting that OATP and P450 have additional or synergistic effects on cerivastatin DDIs in sandwich-cultured human hepatocytes. A synergistic effect was observed when cultured human hepatocytes were treated with 500 μM gemfibrozil. OATP knockdown caused a 6.0-fold change in the AUC ratio in hepatocytes treated with 500 μM gemfibrozil, compared with a 2.5-fold change in nontreated human hepatocytes (Fig. 12). These results derived from the new in vitro model are similar to previous clinical observations and results in other in vitro studies (Backman et al., 2002; Shitara et al., 2004).

The predicted AUC fold change of cerivastatin (Cer) in the presence or absence of gemfibrozil (Gem) (AUCinh/AUC) in control (CTRL) and OATP knockdown sandwich-cultured human hepatocytes. AUCinh/AUC was calculated as the ratio of the total metabolite formation of cerivastatin (M1 and M23).
In summary, in these studies an in vitro siRNA OATP sandwich-cultured human hepatocyte model against hepatic OATP1B1, 1B3, and 2B1 has been developed and characterized. In this model the siRNAs inhibited their mRNA and protein expression and knocked down OATP-mediated uptake of drug and endogenous compound efficiently and specifically. Furthermore, this model system was applied to the study of P450- and OATP-mediated DDIs. The clinically significant cerivastatin-gemfibrozil interaction investigated in this model revealed synergistic effects of OATP and P450 on this DDI when a high concentration of gemfibrozil was used, thereby suggesting that this RNAi knockdown model may reveal more OATP and P450 synergisms than can be elucidated with other in vitro systems. One limitation of this in vitro model is that the P450 activity drops significantly during the culture period (Fig. 7), which would lead to underestimation of the role of P450s in the interaction and thus of the interplay between OATP and P450. Therefore, caution needs to be exercised when this model is used to extrapolate the DDI to an in vivo situation.
Acknowledgments.
We thank Dr. Eric Lightcap of Oncology Biochemistry for valuable discussions on siRNA methodology. We also thank Dr. Gerald Miwa for his scientific comments on this manuscript. We sincerely thank Larry Cohen of Drug Metabolism and Pharmacokinetics (DMPK) for the great help with the quantitative bioanalytical methods validation, and we are grateful to Dr. Sandeepraj Pusalkar and Shanshan Chen of DMPK for excellent assistance with the metabolite identification of cerivastatin.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.032995.
-
ABBREVIATIONS:
- OATP
- organic anion-transporting polypeptide
- DDI
- drug-drug interaction
- AUC
- area under the concentration-time curve
- P450
- cytochrome P450
- DMEM
- Dulbecco's modified Eagle's medium
- E3S
- estrone-3-sulfate
- siRNA
- small interfering RNA
- PCR
- polymerase chain reaction
- aRNA
- amplified RNA
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- SSC
- standard saline citrate
- HBSS
- Hanks' balanced salt solution
- KHB
- Krebs-Henseleit buffer
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- ADME
- absorption, distribution, metabolism, and elimination.
- Received February 26, 2010.
- Accepted June 1, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}