


Abstract
The existence of a glucuronide conjugate of the major circulating clopidogrel metabolites, called clopidogrel acyl glucuronide (CAG), is already known. However, information regarding its pharmacokinetics (PK), metabolism, and clearance are modest. We investigated in vivo the potential CAG trans-esterification to clopidogrel (reaction occurring in vitro in particular conditions) by administering the metabolite to mice. Experiments were then carried out on men, clopidogrel administered alone or followed by activated charcoal intake (intestinal reabsorption blockade). Study objectives included: PK comparison of CAG, clopidogrel carboxylic acid (CCA), and clopidogrel in plasma, determination of their elimination patterns in urine and feces, and tracking of charcoal-induced changes in PK and/or urinary excretion that would indicate relevant enterohepatic recycling of CAG. In mice, CAG was rapidly hydrolyzed to CCA after oral administration, whereas by intravenous route metabolic conversion to CCA was delayed. No levels of clopidogrel were detected in mice plasma, excluding any potential trans-esterification or other form of back-conversion in vivo. PK experiments in man showed that CAG is hydrolyzed in the gastrointestinal tract (very low concentrations in feces), but there is no evidence of enterohepatic recirculation. Quantitation of the three moieties in stool samples accounted for only 1.2% of an administered dose, suggesting that other yet unknown metabolites/degradation products formed through metabolic processes and/or the activity of local microflora are mainly excreted by this route. In man CAG was confirmed as one of the major terminal metabolites of clopidogrel, with a PK behavior similar to CCA.
Introduction
Glucuronide conjugates represent one of the major types of phase II metabolites of xenobiotics. Since generally the biologic function of the aglycone is abolished by glucuronidation, conjugates are often considered as metabolites of modest interest; however, a few compelling cases in which glucuronides maintain/increase the biologic function of their parent compound (Barua and Sidell, 2004; Ohno et al., 2008) suggest that further inquiry into their metabolic fate is warranted.
In the particular case of clopidogrel, although the oxidative metabolism is quite well known, the conjugative metabolism has not been studied in detail. In terms of phase I metabolism, it is known that two oxidative steps, mediated by multiple P450 cytochromes, are required for the conversion of clopidogrel to its active metabolite (Savi et al., 2000; Kazui et al., 2010). Interestingly, activation by the P450 system is rate-limited and ultimately a quantitatively minor metabolic pathway. In parallel, about 85% of the drug released from dosage form is converted to clopidogrel carboxylic acid (CCA) (von Beckerath et al., 2005; Ksycinska et al., 2006), which is subsequently conjugated to clopidogrel acyl glucuronide (CAG) (Silvestro et al., 2010)—a quantitatively important metabolite that has not been studied in detail until now (see Fig. 1, schematic representation of clopidogrel metabolism).
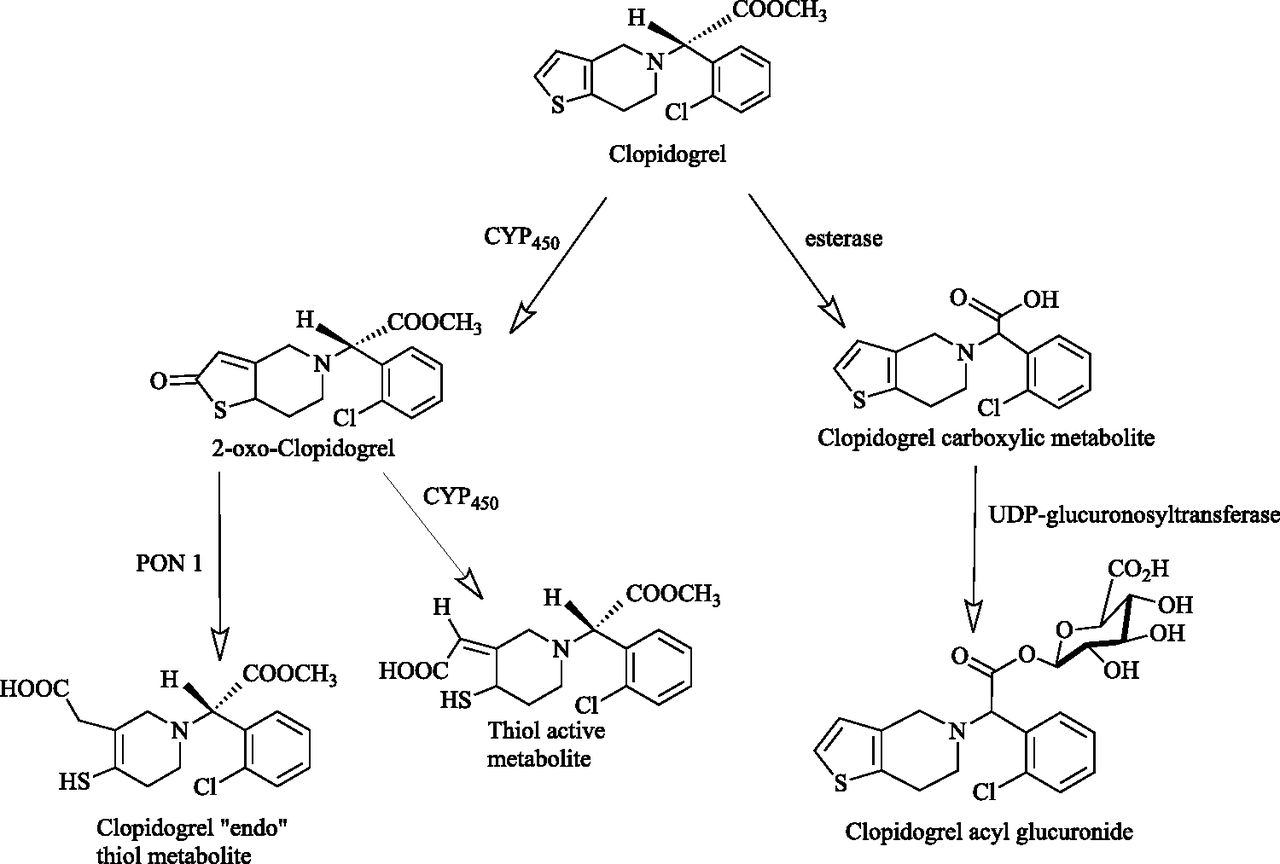
Representation of clopidogrel metabolism.
Although in vivo reactivity of CAG in particular remains to be clarified, it should be noted that acyl glucuronides of carboxylic acids are a class of conjugates generally prone to hydrolysis, molecular rearrangements, and interactions with cellular target molecules by covalent bindings (Ritter, 2000). So far, only binding to CYP2C8 has been demonstrated for CAG (Tornio et al., 2014), and it is unknown if the metabolite undergoes any type of metabolic conversion before being excreted from the human body.
In vitro, reactivity of CAG has already been demonstrated. It was shown that in specific conditions it converts to parent clopidogrel by trans-esterification (Silvestro et al., 2011), a reaction sometimes occurring also during metabolic processes (Boyer and Petersen, 1992; Knights et al., 2000; Celli et al., 2007; Fujino et al., 2014).
Should CAG contribute in vivo to any process resulting in back-conversion to clopidogrel, the amount reconstituted could be considerable, because the exposure to CAG in man [on the basis of area under the curve from time zero to infinity (AUC0–inf)] is 500 times higher than that of clopidogrel (Silvestro et al., 2013)]; furthermore, the newly formed clopidogrel would be again available for metabolism by P450s and thus partly converted to the active metabolite. Although it is clear that the confirmation of such a pathway could only provide mechanistic insight (quantitative data on clopidogrel and its active metabolite is already available in literature), the disposition of CAG was considered important knowledge to be gained, as any yet unknown intermediate reaction could prove useful in understanding the large pharmacokinetic (PK) variability of clopidogrel and its active moiety.
Rationale and Study Objectives.
The present studies represent a follow-up to previous work from which we reported the existence of CAG and described its in vitro back-conversion to clopidogrel by trans-esterification (Silvestro et al., 2011). The main questions to clarify now are, “Can this happen by any means in vivo also?” and “What is the metabolic fate of this conjugate?”.
First, in the absence of a CAG standard suitable for administration to humans, we conducted a study in mice to determine if this metabolite may back-convert to clopidogrel parent by trans-esterification or another reaction of the conjugated metabolite; the study was conducted on mice (C57BL) that have a similar glucuronidase tissue distribution to that of man (Gad et al., 2007).
Another important aspect to clarify was whether CAG undergoes enterohepatic recycling, since mass balance studies conducted with radiolabeled clopidogrel in man (Lins et al., 1999) showed that recycling occurs, although the moieties involved were not identified. Plasma levels of clopidogrel and its two main metabolites were compared in healthy volunteers treated with clopidogrel alone or in combination with activated charcoal; this bile-binding agent was administered according to a regimen designed to disrupt enterohepatic recycling, as already described in literature (Elomaa et al., 2001; Wang et al., 2014) and have minimal impact on clopidogrel absorption.
In view of a more comprehensive understanding of their metabolism, the determination of the main excretion route (urine and/or feces) for clopidogrel, CCA (as precursors) and CAG was also a set objective of the single -dose charcoal-interaction study in man.
It is noteworthy that a human study was preferred owing to the complex nature of the physiologic processes studied through PK determinations and the consideration that data gathered in any other model would be extremely difficult to extrapolate, raising concerns of relevance to a real clinical setting.
Materials and Methods
Standards, Reagents, and Medication
For the preparation of solutions for oral and intravenous administration in mice, clopidogrel acyl-β-d-glucuronide standards of adequate purity were purchased from Toronto Research Chemicals (Toronto, Canada).
The internal standards used for high-performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) analytical determinations were: d3-clopidogrel hydrogen sulfate (SynFine Research, Richmond Hill, ON, Canada), clopidogrel acyl-β- d-glucuronide (Toronto Research Chemicals Inc.), and 13C6-clopidogrel carboxylic acid (Alsachim, Illkirch-Graffenstaden, France). Commercially available reagents of analytical grade purity were used for sample processing.
Plavix 75 mg tablets (Sanofi, Paris, France) from a commercial batch (AY171) were used. Medical-grade activated charcoal was also procured from the market (from Silcarbon Aktivkohle GmbH, Kirchhundem, Germany).
Intravenous and Oral Pharmacokinetics Study in Mice
Study Design and Sample Collection.
All procedures used were in accordance with the standards set forth in the eighth edition of Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, The National Academies Press, Washington D.C.). Laboratory animals (C57BL/6 male mice, weighing 20 ± 4g, 25 ± 1 day of age) were bred, raised, and cared for at the Cantacuzino National Institute of Research-Development for Microbiology and Immunology (NIRDMIC) located in Bucharest, Romania. The experimental part was carried out in the Pharmacology Department of the National Institute for Chemical Pharmaceutical Research and Development (ICCF) located in Bucharest, Romania. The study was conducted according to a parallel design on an overall sample size of 71 laboratory animals (five per sampling point after each mode of administration plus six animals treated with normal saline only in view of obtaining blank plasma for preparation of analytical quality control samples). Animals randomized to the treatment arms, received in sterile conditions a dose of 200 μl freshly prepared solution of 1.25 mg/ml clopidogrel acyl glucuronide in normal saline, either per os (through gavage) or intravenously, via tail-vein injection. Blood samples (150 μl) were collected in prechilled tubes containing di-potassium ethylenediaminetetraacetic acid (K2EDTA) at 0.5, 1, 2, 4, 6, and 8 hours after oral dosing or at 0.25, 0.5, 1, 2, 4, 6, and 8 hours after intravenous administration. The samples were immediately immersed in water and ice bath until centrifugation (performed at a nominal temperature of 4°C, 1500g for a duration of 10 minutes). The separated plasma was frozen at −70°C and maintained at this temperature until analyzed. For sample processing and analysis we used a slight modification of a method already published (Silvestro et al., 2011), as described below.
Extraction of Clopidogrel, Clopidogrel Carboxylic Acid, and Clopidogrel Acyl Glucuronide from Mice Plasma Samples.
Plasma thawing was done on wet ice. Aliquots of 100 μl from postdose mice plasma samples were diluted with 200 μl of ice-cold acetonitrile, spiked with 20 μl of internal standard mix in acetonitrile (d3-clopidogrel hydrogen sulfate, clopidogrel acyl-β-d-glucuronide, and 13C6-clopidogrel carboxylic acid, 200 ng/ml), vortexed for 3 minutes, and then centrifuged for 5 minutes at 4000 rpm and 8°C. Supernatants (100 μl) were diluted with 100 μl ice-cold water containing 2% acetonitrile and 0.1% formic acid. The extracts were analyzed as described in the next paragraph.
Clopidogrel, Clopidogrel Carboxylic Acid, and Clopidogrel Acyl Glucuronide Quantification.
Six-point calibration curves were prepared in blank mice plasma (K2EDTA as anticoagulant) with concentrations ranging from 0.01 to 100.00 ng/ml for clopidogrel and from 1.00 to 10,000.00 ng/ml for clopidogrel acyl glucuronide and clopidogrel carboxylic acid. The quality control and calibration curve samples were also spiked with internal standard mix in acetonitrile (d3-clopidogrel hydrogen sulfate, clopidogrel acyl-β-d-glucuronide, and 13C6-clopidogrel carboxylic acid, 200 ng/ml) and subsequently extracted in the same manner described previously for study samples. Calibration curves and quality control samples (three concentration levels and in triplicate) were analyzed during each analytical sequence. Decisions regarding the acceptance of sequences were taken according to well-established bioanalytical rules (FDA, 2013; EMA, 2011). No sequences had to be rejected owing to quality control or calibration failure.
Human Oral Pharmacokinetics and Elimination Study
Study Design and Sample Collection.
Six subjects were enrolled and completed the human PK and elimination study. Study population comprised three male and three nonpregnant, nonlactating female volunteers, 18 to 51 years old (mean age 32.17 ± 14.48). The study was conducted at the Clinical Hospital of the Ministry of Health of the Moldavian Republic located in Chisinau. The study protocol was reviewed and approved by an Institutional Ethics Committee and all 6 subjects enrolled were informed about the study medication and procedures and gave consent for the participation in the study. Clinical investigations were conducted according to the Declaration of Helsinki principles and the medication administered consisted of a single oral dose of reference-listed drug (Plavix 75 mg, procured from the market) per study period. The design was two-way crossover: in one study period the subjects received just clopidogrel and in the other they received clopidogrel plus a regimen consisting of 20 g activated charcoal suspended in 240 ml of water, given at 6.0, 12.0, 24.0, 36.0, 48.0 and 60.0 hours after dosing. Blood samples (4 ml) for the quantification of parent clopidogrel, clopidogrel acyl glucuronide, and clopidogrel carboxylic acid in plasma were collected in prechilled tubes containing K2EDTA as anticoagulant, at 1.0, 2.0, 6.0, 9.0, 24.0, 36.0, 48.0, and 72.0 hours after dosing.
In the same study, urine was collected in both study periods up to 72 hours postdose, and fecal matter was collected over the same interval but only when clopidogrel was given without activated charcoal (as previous experience suggested that the presence of charcoal in stool samples would lead to ambiguous results).
Extraction of Metabolites from Biologic Samples.
Before analysis, plasma samples were thawed on wet ice, and 100-μl aliquots were spiked with 20 μl of solution of internal standard, which contained 200 ng/ml d3-clopidogrel hydrogen sulfate, 200 ng/ml clopidogrel acyl-β-d-glucuronide, and 200 ng/ml 13C6-clopidogrel carboxylic acid in acetonitrile, and then diluted with 200 μl ice-cold acetonitrile. Afterward they were vortex for 3 minutes and centrifuged at 4000 rpm and 8°C for 5 minutes. Supernatants (100 μl) were diluted with 100 μl ice-cold water containing 2% acetonitrile and 0.1% formic acid.
Urine samples were collected during the time intervals 0–12 , 12–24 , 24–36, 36–48, 48–60, and 60–72 hours postdose. The volume of each fresh urine sample was measured and 50-ml aliquots were mixed with 100 μl of acetic acid 99.8%, vortexed for 2 minutes, and frozen at –20°C. To obtain a single representative urinary excretion sample for each time interval, aliquots from individual samples were mixed in appropriate proportions according to initial sample volume. Before analysis, samples (100 μl) were thawed on wet ice, spiked with 20 μl of internal standard mix in acetonitrile (d3-clopidogrel hydrogen sulfate, clopidogrel acyl-β-d-glucuronide, and 13C6-clopidogrel carboxylic acid 200 ng/ml), and then diluted with 200 μl ice-cold acetonitrile. Afterward they were vortexed for 3 minutes and then centrifuged for 5 minutes at a nominal temperature of 8°C, with a speed of 4000 rpm. A volume of 100 μl supernatant was separated and diluted with 100 μl ice-cold water containing 2% acetonitrile and 0.1% formic acid.
Fresh fecal matter samples were frozen for storage at –20°C. Before analysis, samples were thawed on wet ice, weighed, and then diluted 1:10 (w/v) with an ice-cold solution containing 50% acetonitrile and 1% formic acid, as follows: Samples were first vortexed for 2 minutes with one-fifth of the calculated volume of the above solution for dilution and 250 mg of glass beads per gram of sample. The remaining volume of the solution was then added and the samples were vortexed again for 3 minutes and centrifuged at 4000 rpm and 8°C for 10 minutes. A volume of 100 μl of supernatant was recovered and processed in the same manner as previously described for thawed urine samples.
Clopidogrel, Clopidogrel Carboxylic Acid, and Clopidogrel Acyl Glucuronide Quantification.
Six-point calibration curves were prepared in appropriate matrix (in blank plasma, blank urine, or blank fecal matter samples that were spiked with internal standard, processed, and diluted according to the same protocol previously described for study samples). The concentration ranges of the calibration curves were 0.01–100.00 ng/ml for clopidogrel and 1.00–10,000.00 ng/ml for clopidogrel acyl glucuronide and clopidogrel carboxylic acid. Calibration curves and quality control samples (three concentration levels in triplicate) were analyzed during each analytical sequence. Decisions regarding the acceptance of sequences were made according to well-established bioanalytical rules (EMA, 2011; FDA, 2013). No sequences had to be rejected owing to quality control or calibration failure.
HPLC–MS/MS Analysis.
For the analytical determinations we used an HPLC binary gradient (LC-20 AD chromatographic pumps) by Shimadzu-Japan with a CTC-PAL autosampler (model HTS) manufactured by CTC Analytics (Zwingen, Switzerland). The HPLC system was coupled with a triple quadrupole mass-spectrometer model API 5000 (mice PK samples) or API 6500 (human PK and elimination samples) with an atmospheric pressure electrospray ionization source (model TurboIonSpray), all manufactured by Applied Biosystems/MDS SCIEX (Concord, ON, Canada). Separations were performed on Ascentis Express RP-Amide columns (100 × 2.1 mm, 2.7 μm) produced by Supelco (Bellefonte, PA). The mobile phase used was a gradient of 0.1% formic acid and acetonitrile at a flow rate of 0.2 ml/min. The injection volume was 10 μl, the temperature of the autosampler 3°C, and the temperature of the chromatographic column 55°C. Quantitative data were acquired in multiple reaction monitoring (MRM)–positive electrospray ionization mode. The MRM transitions considered were 322.2/184.0 for clopidogrel, 327.2/189.2 for clopidogrel-d3, 484.3/198.1 for clopidogrel acyl glucuronide, 308.2/95.0 for clopidogrel carboxylic acid, and 314.1/158.1 for 13C6-clopidogrel carboxylic acid.
Software for Pharmacokinetic Evaluations and Statistics.
Pharmacokinetic parameters pertaining to the human PK study were determined and statistically analyzed using SAS software (version 9.4; SAS Institute Inc., Cary, NC). For the determination of pharmacokinetic parameters from mean plasma concentration-versus-time curves constructed on mice data and for designing charts and graphs, Excel software was used (Microsoft Corporation, Redmond, WA).
Results
Mice PK and Metabolism Study.
No concentration of clopidogrel parent above the lower limit of quantification (LLOQ) of the bioanalytical method was identified in any of the mice plasma samples, permitting the conclusion that either the concentrations were below 0.01 ng/ml or, most probably, clopidogrel was not formed at all.
As the only detected analytes (out of the three moieties screened), the mean plasma concentration-versus-time profiles obtained for clopidogrel acyl glucuronide and clopidogrel carboxylic acid after intravenous and oral administration of clopidogrel acyl glucuronide in mice are presented in Fig. 2, A and B.

Metabolites determined in plasma after administration of clopidogrel acyl glucuronide by intravenous (N = 35, parallel, 5 animals per sampling point) and oral route (N = 30 parallel, 5 animals per sampling point).
In Fig. 2C we present in overlay mode and on ln-linear scale the plasma concentration-versus-time curves of both metabolites after intravenous and oral dosing.
Pharmacokinetic parameters estimated for the two quantifiable metabolites are presented in Table 1.
PK parameters estimated for clopidogrel acyl glucuronide and clopidogrel carboxylic acid after intravenous and oral administration of 200 μl of solution 1.25 mg/ml clopidogrel acyl glucuronide in mice
N = 35 parallel, 5 animals per sampling point.
The percentage ratio of oral versus intravenous AUCs within the sampling interval (0–8 hours) was estimated at 29.73%, suggesting that clopidogrel acyl glucuronide undergoes extensive presystemic hydrolysis resulting in the formation of the carboxylic acid derivative, not clopidogrel parent.
Pharmacokinetic Data Gathered in the PK and Elimination Study in Man.
The concentration-versus-time profiles for parent clopidogrel, clopidogrel acyl glucuronide, and clopidogrel carboxylic acid obtained in human subjects following administration of clopidogrel with and without charcoal are presented in Fig. 3.
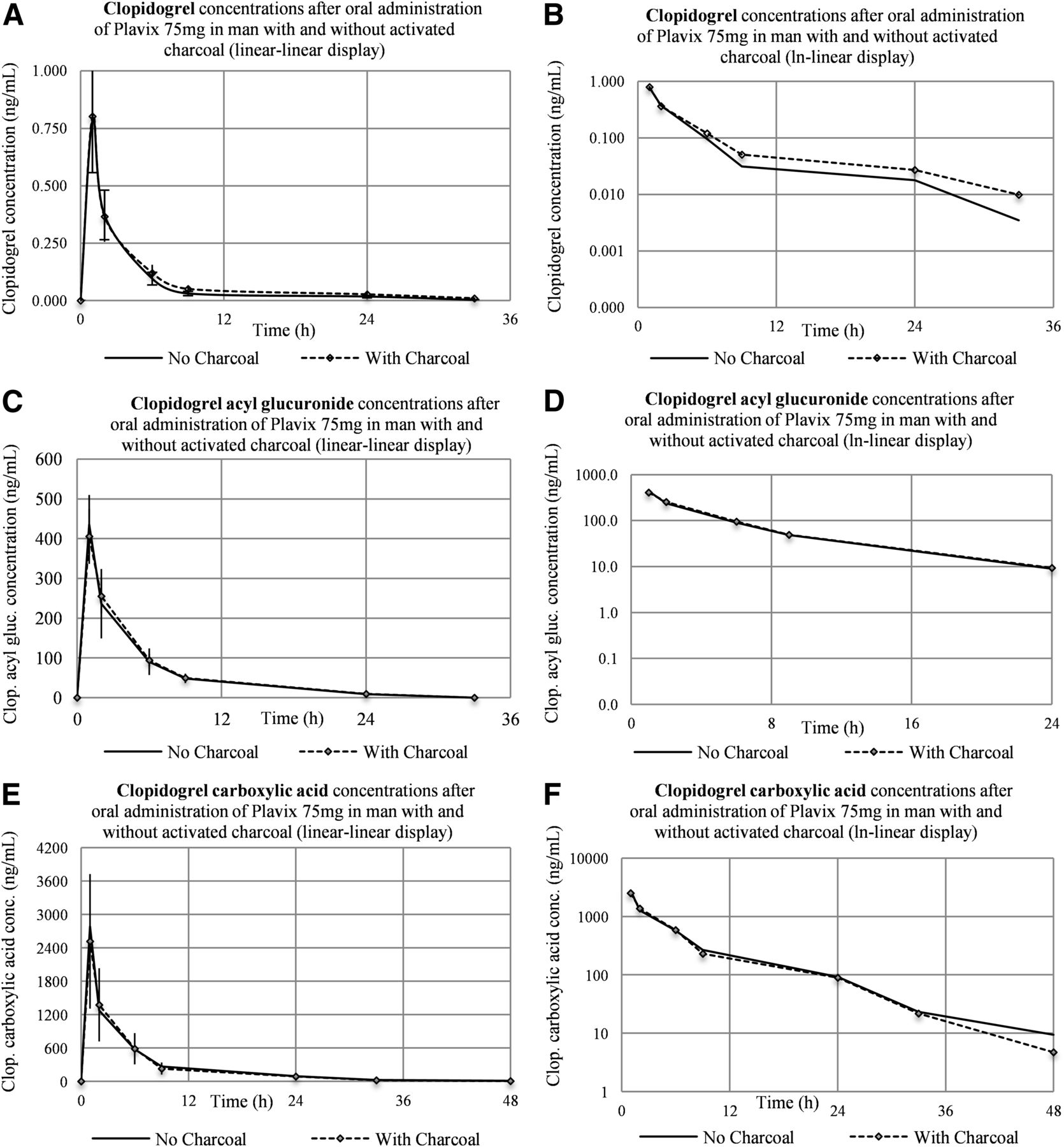
Plasma concentration-versus-time curves for the three analytes after administration of clopidogrel in human subjects (N = 6) with and without charcoal (linear-linear display on A, C, E and ln-linear display on B, D, F).
For the two metabolites the profiles are practically superimposable, irrespective of charcoal intake, whereas for clopidogrel the circulating levels registered during the elimination phase were slightly increased when charcoal was coadministered. Analysis of AUC data revealed that the increase was not statistically significant [P value returned by the analysis-of-variance (ANOVA) test for treatment effect was 0.055, above the 0.05 significance level].
The main pharmacokinetic parameters estimated for clopidogrel and its two metabolites are presented in Table 2.
Main PK parameters determined in human volunteers (N = 6) for clopidogrel, clopidogrel carboxylic acid, and clopidogrel acyl glucuronide after oral dosing with Plavix 75 mg with and without subsequent administration of activated charcoal (in a randomized, two-way crossover design study)
For clopidogrel parent the elimination half-life (t1/2) was 8.1 hours in standard dosing conditions and 10.6 hours when charcoal was coadministered; this difference was found to be not statistically significant (paired t test returned a value of 0.082, above the 0.05 significance level). For clopidogrel carboxylic acid t1/2 was 7.8 hours for clopidogrel alone and 6.8 hours when charcoal was coadministered, whereas for clopidogrel acyl glucuronide the same t1/2 of 5.6 hours was estimated for both administration regimens.
Elimination Data Gathered in the PK and Elimination Study in Man.
We found that about 15% of an administered clopidogrel dose (calculated as micromolar ratios) is recovered in urine in the form of the quantified analytes (see Fig. 4). The longest recovery times were found for clopidogrel carboxylic acid (urinary excretion still ongoing in the 60–72 hours postdose collection interval) and for clopidogrel acyl glucuronide (recovered in urine up to 60 hours postdose). For clopidogrel, only trace amounts were identified in urine (total recovery well below 0.001 μM) up to 36 hours postdose, whereas, as expected, unchanged clopidogrel not absorbed from the intestine was mainly recovered in feces. Quantitation of the analytes in stool samples accounted for only 1.2% of an administered dose.

Total recovery of clopidogrel, clopidogrel acyl-glucuronide, and clopidogrel carboxylic acid in urine and stool samples over 72 hours postdose after administration of clopidogrel in human subjects (N = 6).
The one-tailed paired t test was used to compare urinary excretion data over the time intervals 0–12, 12–24, 24–36, 36–48, 48–60, and 60–72 hours for the three analytes, after dosing with clopidogrel with or without subsequent administration of activated charcoal (see Fig. 5). It was found that the difference in amount recovered over the array of specified intervals was not statistically significant (P values were 0.231 for clopidogrel, 0.488 for clopidogrel carboxylic acid, and 0.181 for clopidogrel acyl glucuronide).

Total recovery of clopidogrel, clopidogrel acyl-glucuronide, and clopidogrel carboxylic acid in urine samples after administration of clopidogrel in human subjects (N = 6) with or without activated charcoal.
Urinary recovery by collection intervals for clopidogrel acyl glucuronide is presented in Fig. 6A, and the amount of urine excreted within the intervals is depicted in Fig. 6B.
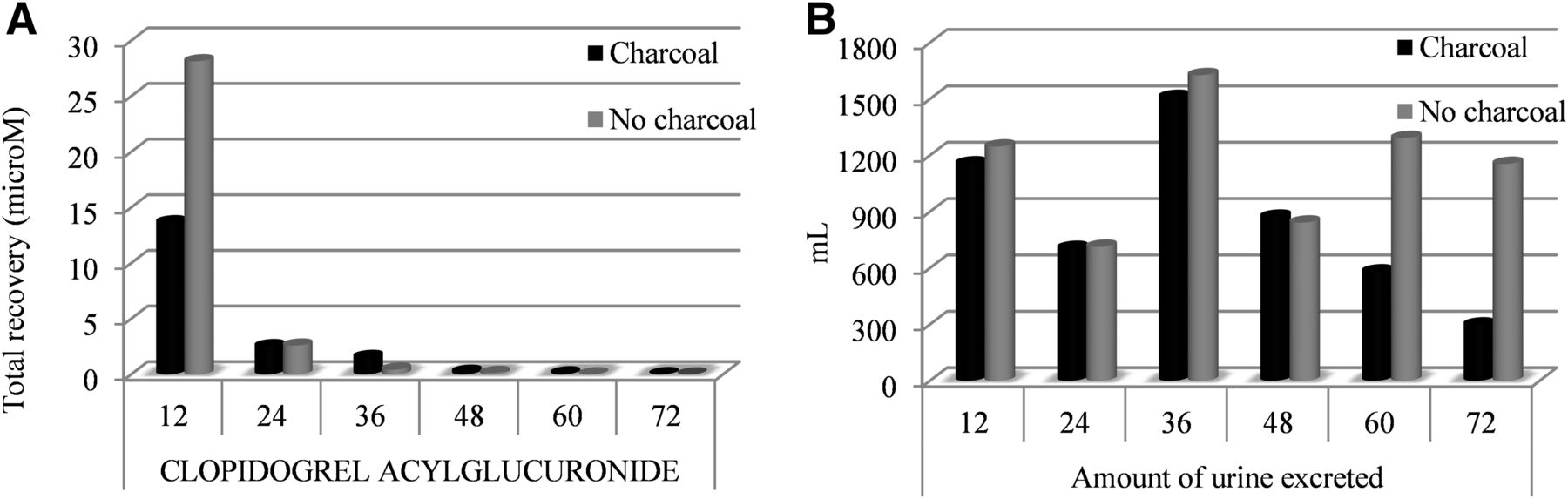
Recovery of clopidogrel acyl-glucuronide in urine (N = 6) displayed by collection intervals (A) and amount of urine excreted by collection intervals (B).
No statistically significant difference in urinary recovery of clopidogrel acyl glucuronide was identified in any of the collection intervals.
Discussion
The purpose of the present studies was to evaluate the pharmacokinetics, metabolic fate, and elimination pattern of clopidogrel acyl glucuronide, the main conjugated metabolite of clopidogrel. Since previous in vitro data have demonstrated that CAG can undergo trans-esterification resulting in the formation of parent clopidogrel, emphasis was placed on ascertaining if such a reaction could occur also in the in vivo setting. For each type of potential mechanistic conversion studied (trans-esterification/hydrolysis, deconjugation during enterohepatic recycling), a relevant physiologic model was chosen. For gaining insight into the biodisposition of the metabolite (as such) and for identifying the reaction products derived from the activity of β-glucuronidase and other hydrolases, a study was conducted in C57BL/6 mice of proper age to ensure peak enzymatic activity (Peng et al., 2013). For acquisition of quantitative data regarding the systemic availability and balance between urinary and fecal recovery of CAG after oral dosing with clopidogrel and for determining the likelihood of its involvement in enterohepatic recycling, the only clinically relevant option, given the complex metabolic processes involved, was to perform a study in man (Sörgel et al., 1989).
Mice PK and Metabolism Study.
After direct administration of clopidogrel acyl glucuronide to mice by oral (gavage) and intravenous (tail vein) routes, HPLC–MS/MS analysis of postdose PK samples has shown no generation of parent clopidogrel. While trans-esterification to clopidogrel did not take place in vivo, hydrolysis leading to the formation of the acidic derivative was the most important metabolic process observed for clopidogrel acyl glucuronide.
Oral data have revealed a very fast metabolism of clopidogrel acyl glucuronide within the first 2 hours from administration, probably occurring in the gastrointestinal tract by chemical degradation and/or enzymatic hydrolysis. The percentage ratio of oral versus intravenous AUCs estimated for the administered conjugated metabolite within the sampling interval (0–8 hours) was 29.73%.
By intravenous route, as metabolism was restricted only to systemic degradation of CAG, the rate of conversion to the carboxylic acid form was lower; specifically, while oral data showed that both the administered clopidogrel acyl glucuronide and the formed clopidogrel carboxylic acid reached peak levels simultaneously at 1 hour, following tail injection the time lag until maximal plasma levels of clopidogrel carboxylic acid was 6 hours and the peak concentrations reached were 2.5 times lower than after oral dosing. Nevertheless, total exposure to clopidogrel carboxylic acid was almost identical irrespective of the administration route of CAG (mean AUC ratio intravenous/per os was 1.05), thus showing that systemic conversion is also very extensive. This was to be expected considering that lysosomal and microsomal fractions expressing β-glucuronidase and esterases are widely expressed also in serum and organs other than the liver in the organism of C57BL/6 mice (Peng et al., 2013; Tegelström and Ryttman, 1981; Lusis and Paigen, 1977).
Human PK Data.
The use of activated charcoal as bile-binding agent for the purpose of impeding enterohepatic recycling of xenobiotics is already well established (Chyka et al., 2005; Stass et al., 2005; Taft, 2009). Also, PK studies of interaction between drugs and activated charcoal have been used previously for determining if the active ingredient or related molecules undergo extensive recycling; reduced exposure coupled with accelerated elimination of the investigated molecule in the charcoal study arm are classic indicators of discontinuing/minimizing the recycling process (Sörgel et al., 1989; Elomaa et al., 2001; Wang et al., 2014). For unbiased results, the administration schedule for activated charcoal must be individualized according to the biopharmaceutical properties of the studied drug to ensure that administration of the bile-binding agent does not also alter drug absorption. For clopidogrel in particular, whereas the early time of the peak analyte concentration (Tmax) can be misleading, it is important to note that absorption is slow and mainly occurs in the lower compartments of the gastrointestinal tract. With slow absorption and fast subsequent elimination of the absorbed fraction (mainly through extensive metabolism and to a lesser extent the result of actual excretion), the equilibrium between the two constants occurs much earlier than complete absorption of the prodrug. In fact, an in silico gastrointestinal simulation of regional absorption distribution of clopidogrel, recently published by our group, has shown that absorption only starts in the duodenum (33% of dose absorbed) and is completed through significant contribution (30%) from cecum and ascending colon (Savu et al., 2016). This behavior is quite typical, since clopidogrel is a weak base characterized by a dissociation constant (pKa) of 5.3 (NLM, 2012); therefore, it freely crosses cell membranes in gastrointestinal compartments where the pH is greater than 5.3. Considering these properties, administration of activated charcoal was started at 6.0 hours after clopidogrel dosing so that any decreased exposure possibly noted for the parent drug or the studied metabolites in the charcoal arm could only be attributed to recycling impairment and not decreased drug absorption.
The fact that clopidogrel concentrations remained practically unchanged irrespective of charcoal intake indicated that the administration schedule for the bile-binding agent was correctly designed for the intended purpose and that clopidogrel (as such) is not involved in any enterohepatic cycle.
Considering the pharmacokinetic data obtained for clopidogrel acyl glucuronide, with particular emphasis on elimination half-life (determined to be 5.6 hours irrespective of charcoal administration) and results of the comparison carried out between plasma profiles of the metabolite generated in the presence and absence of activated charcoal (charcoal/no charcoal ratios of 0.98 for Cmax and 1.10 for AUC), it can be concluded that any enterohepatic recycling of CAG that may occur is not significant. The conclusion is supported also by the statistic tests applied for comparison of the primary PK parameters of CAG in the two administration conditions (the ANOVA test checking for treatment as fixed-effect returned P values above the 0.05 significance level for both Cmax and AUC0–t data).
Human Elimination Data.
On the basis of the knowledge acquired it can be said that clopidogrel acyl glucuronide may be regarded as a quantitatively important yet terminal metabolite of the parent drug, not being capable of contributing to the regeneration of known moieties linked to active metabolite formation. However, the potential of acyl glucuronide to play other roles of significant importance in terms of clopidogrel activity cannot be yet excluded.
Quantitation of the analytes in stool samples accounted for only 1.2% of an administered dose, quite far from the mass balance study results previously reported in literature (Lins et al., 1999) that showed a cumulative fecal recovery of radioactivity ranging from 35 to 57% after single dosing with 75 mg of 14C-labeled clopidogrel. This fact strongly suggests that other metabolites and/or degradation products not yet characterized are involved in this elimination process. The finding is consistent with the report that twenty distinct metabolites of clopidogrel can be identified in biological matrices (EMA, 2004).
Urinary data confirm what we hypothesized on the basis of the previously presented plasma PK results of same subjects, namely that the acyl glucuronide derivative does not undergo significant enterohepatic recycling, if any. Should that have been the case, administration of charcoal would have accelerated elimination of the metabolite and not the opposite. There is also no evidence that any of the three quantified moieties is involved in enterohepatic recycling.
To conclude, despite the high tendency observed for it in vitro, no evidence was found to suggest that clopidogrel acyl glucuronide could reconvert to parent clopidogrel in vivo by trans-esterification. By comparing of PK profiles for clopidogrel and the conjugated metabolite alone and in the presence of activated charcoal, it can also be stated that it is unlikely that clopidogrel acyl glucuronide would be capable of reforming clopidogrel (as such) through participation in an enterohepatic cycle. So far it seems that the amount of clopidogrel converted by carboxylesterase 1 to the inactive carboxylic acid (about 85% of an administered dose) is not made again available for metabolization by P450s so that it might be oxidized and form the active thiol metabolite.
Acknowledgments
The authors would like to extend their gratitude to Angela Casarica, Department of Pharmaceutical Biotechnologies of ICCF, for her help in mice plasma processing and to Constanta Dulea and Adrian Ghita from Pharma Serv International for the help granted concerning the HPLC–MS/MS analysis of pharmacokinetic samples and respectively for aiding in the statistical analysis of PK data.
Authorship Contributions
Participated in research design: Savu, Silvestro, Rizea Savu, Mircioiu.
Conducted experiments: Savu, Silvestro, Remis, Yuksel.
Performed data analysis: Savu, Silvestro, Mircioiu.
Wrote or contributed to the writing of the manuscript: Savu, Silvestro, Surmeian, Mircioiu.
Footnotes
- Received April 28, 2016.
- Accepted July 8, 2016.
This work received financial support through the project entitled “CERO—Career profile: Romanian Researcher”, cofinanced by the European Social Fund for Sectoral Operational Programme Human Resources Development 2007–2013 [POSDRU/159/1.5/S/135760].
Abbreviations
- AUC
- area under the curve
- CAG
- clopidogrel acyl glucuronide
- P450
- cytochrome P450
- HPLC–MS/MS
- high-performance liquid chromatography–tandem mass spectrometry
- K2EDTA
- di-potassium ethylenediaminetetraacetic acid
- PK
- pharmacokinetics
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}