


Abstract
Lapatinib, an oral tyrosine kinase inhibitor used for breast cancer, has been reported to cause idiosyncratic hepatotoxicity. Recently, it has been found that lapatinib forms a metabolite-inhibitor complex (MIC) with CYP3A4 via the formation of an alkylnitroso intermediate. Because CYP3A5 is highly polymorphic compared with CYP3A4 and also oxidizes lapatinib, we investigated the interactions of lapatinib with CYP3A5. Lapatinib inactivated CYP3A5 in a time-, concentration-, and NADPH-dependent manner using testosterone as a probe substrate with KI and kinact values of 0.0376 mM and 0.0226 min−1, respectively. However, similar results were not obtained when midazolam was used as the probe substrate, suggesting that inactivation of CYP3A5 by lapatinib is site-specific. Poor recovery of CYP3A5 activity postdialysis and the lack of a Soret peak confirmed that lapatinib does not form a MIC with CYP3A5. The reduced CO difference spectrum further suggested that a large fraction of the reactive metabolite of lapatinib is covalently adducted to the apoprotein of CYP3A5. GSH trapping of a reactive metabolite of lapatinib formed by CYP3A5 confirmed the formation of a quinoneimine-GSH adduct derived from the O-dealkylated metabolite of lapatinib. In silico docking studies supported the preferential formation of an O-dealkylated metabolite of lapatinib by CYP3A5 compared with an N-hydroxylation reaction that is predominantly catalyzed by CYP3A4. In conclusion, lapatinib appears to be a mechanism-based inactivator of CYP3A5 via adduction of a quinoneimine metabolite.
Introduction
Lapatinib (Fig. 1), an oral dual tyrosine kinase inhibitor was first approved by the U.S. Food and Drug Administration in 2007 for the targeted treatment of breast cancer overexpressing human epidermal growth factor receptor 2 (HER2/neu). In clinical use, lapatinib is administered with capecitabine to patients having advanced metastatic breast cancer that is resistant to anthracyclines, taxane, and trastuzumab (Kroep et al., 2010). In comparison with trastuzumab, a monoclonal antibody selective for HER2, lapatinib binds to epidermal growth factor receptor as well, and it can be given orally. Thus, lapatinib is able to exert an inhibitory effect on two distinct tyrosine kinase domains, both of which are responsible for the proliferation of cancer cells. Although general lapatinib-associated toxicities such as diarrhea and skin rash are clinically manageable, lapatinib has been known to cause potentially fatal liver damage in some patients, leading to a black box warning in July 2008 (Gomez et al., 2008). The mechanism of the hepatotoxicity arising from lapatinib is currently unknown.

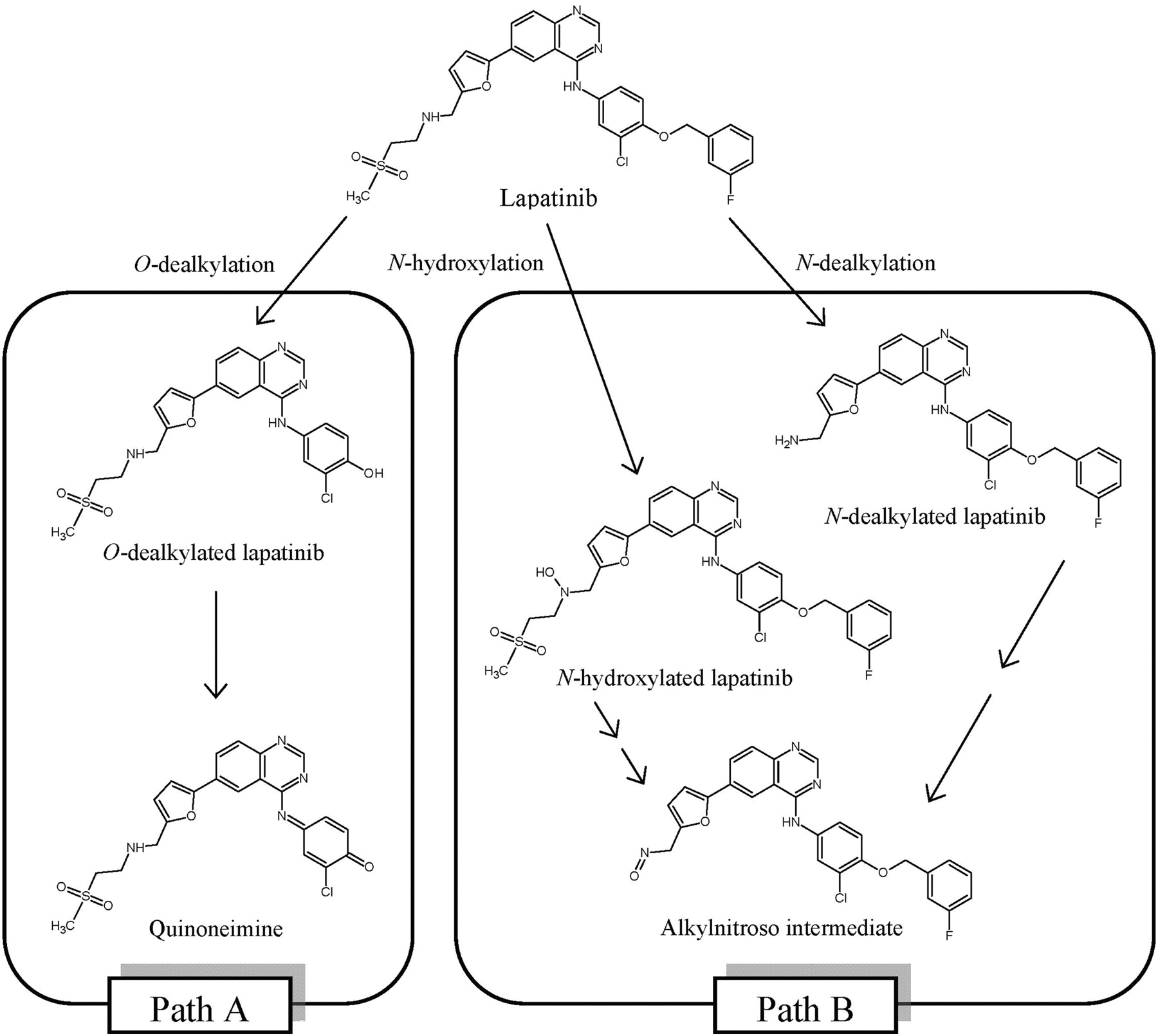
Chemical structures of lapatinib and its metabolites. Paths A and B denote the predominant human CYP3A5- and CYP3A4-mediated metabolic reactions of lapatinib.
It has been noted that lapatinib undergoes extensive metabolism in the liver, mediated mainly by CYP3A4/5 (European Medicines Agency, Assessment Report for Tykerb, 2008, http://www.emea.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000795/WC500044960.pdf). Our previous study established that lapatinib is metabolized by human liver P450s to form O- and N-dealkylated metabolites (Fig. 1) (Teng et al., 2010). These metabolites, together with others, have also been detected in the plasma, urine, and feces of human subjects (Castellino et al., 2011). Some of the products harbor structures that can be further metabolized to reactive metabolites capable of interacting with cellular proteins. For instance, the O-dealkylated metabolite can undergo further oxidation to form a quinoneimine, a highly reactive species that has the potential to interact with cellular proteins, initiating processes leading to toxic effects. This process parallels the metabolism of acetaminophen, in which one of its metabolites, N-acetyl-p-benzoquinone imine reacts with hepatocellular proteins, leading to liver injury. In a separate pathway, lapatinib is oxidized to a hydroxylamine that can be further oxidized to a nitroso intermediate, which can form a MIC with CYP3A4, thereby inhibiting its enzymatic activity (Takakusa et al., 2011). Thus, both O- and N-dealkylated metabolites of lapatinib have the potential of modifying cellular proteins, resulting in direct toxicities/drug interactions, and/or forming haptens that may trigger immune responses and subsequent toxicities (Kalgutkar et al., 2007).
Whereas CYP3A4 is typically assessed for drug interactions due to its relative abundance in the liver and intestines and its broad substrate selectivity, the role of CYP3A5 should not be neglected. A compelling reason is the differential expression levels of CYP3A5 among individuals. Unlike CYP3A4, the level of CYP3A5 is dependent on the allele that determines its endogenous expression (Kuehl et al., 2001). For instance, patients who have the 3A5 *3/*3 allele do not express CYP3A5. Conversely, patients with the 3A5 *1/*1 allele have a CYP3A5 level that can represent up to 50% of the total CYP3A content. In other words, CYP3A5 is highly polymorphic compared with CYP3A4, making it a potentially significant contributor to pharmacokinetic variability for some substrates. For instance, drugs that are metabolized mainly by CYP3A5 would have altered hepatic clearance, depending on the expression levels of CYP3A5. This could pose a problem if the affected drugs have narrow therapeutic indices or generate toxic reactive intermediates. For example, the therapeutic activities of some drugs (such as simvastatin and cyclosporine) can be affected by the differential expression levels of CYP3A5 (Hu et al., 2006; Kim et al., 2007).
Another impetus to investigate drug-CYP3A5 interactions is the substrate-dependent differences observed in the extent and mechanism of inhibition of CYP3A4 versus CYP3A5, even though these enzymes are 83% homologous and have similar substrate specificity (de Wildt et al., 1999). An example is verapamil, which forms a MIC with CYP3A4 but not with CYP3A5 (Wang et al., 2005). Whereas a recent study performed by our group demonstrated that lapatinib forms a MIC with CYP3A4 (Takakusa et al., 2011), the interaction between lapatinib and CYP3A5 was not fully explored. The overarching aim of this article was to establish the nature of interaction between lapatinib and CYP3A5. Considering the differential expression levels of CYP3A5 in the human population, our study could provide a basis for further research on the role of CYP3A5 in idiosyncratic hepatotoxicity caused by lapatinib.
Materials and Methods
Chemicals.
High-performance liquid chromatography-grade methanol and acetonitrile (ACN) were purchased from Tedia Company Inc. (Fairfield, OH). Lapatinib was purchased from LC Laboratories (Woburn, MA). GSH, Tergitol-type NP-40, prednisolone, carbamazepine, and safranin O were purchased from Sigma-Aldrich (St. Louis, MO). Testosterone was purchased from Merck (Darmstadt, Germany). Midazolam was purchased from CCM Duopharma Biotech Bhd (Selangor, Malaysia). Human recombinant P450 3A5 Supersomes (rCYP3A5) and the NADPH-regenerating system consisting of NADPH A (NADP+ and Glc-6-PO4) and B (glucose-6-phosphate dehydrogenase) were obtained from BD Gentest (Woburn, MA). 6β-Hydroxytestosterone and 1′-hydroxymidazolam were obtained from Cerilliant Corporation (Round Rock, TX). Water was obtained using a Milli-Q water purification system (Millipore Corporation, Bedford, MA). O-Dealkylated lapatinib was synthesized in-house (Teng et al., 2010). All other reagents used were analytical grades.
Time-, Concentration-, and NADPH-Dependent CYP3A5 Inactivation.
Two probe substrates, testosterone and midazolam, were tested in the experiments. Incubations (n = 3) were performed in 96-well plates. Primary incubations consisting of 20 pmol/ml rCYP3A5, 100 mM potassium phosphate buffer (pH 7.4), 1.6 μl of NADPH B and 1.6 μl of lapatinib were prewarmed at 37°C for 3 to 5 min. The concentrations of lapatinib used in the testosterone and midazolam assays were 2.5, 5, 10, 20, 30, 40, and 50 μM. To initiate the reactions, 8 μl of NADPH A was added to the mixture. The final volume in each well was 160 μl, and the final organic concentrations in the incubation mixture were 0.1% DMSO and 0.9% ACN (v/v). At 0, 3, 8, 15, 22, and 30 min after the addition of NADPH A, 8 μl of the primary incubation was transferred to 72 μl of secondary incubation containing either 200 μM testosterone or 25 μM midazolam (probe substrates), the NADPH-regenerating system, and 100 mM potassium phosphate buffer (pH 7.4). The secondary reaction mixture was incubated for another 10 min at 37°C before a 50-μl aliquot was removed and quenched with an equal volume of ice-cold ACN containing 0.5 μM prednisolone [internal standard (IS) for testosterone assay] or carbamazepine (IS for midazolam assay). The quenched samples were centrifuged at 16,000g at 4°C for 15 min, and the supernatants were removed for the respective determination of either testosterone 6β-hydroxytestosterone or 1′-hydroxymidazolam activity by LC/MS/MS. Negative control incubations were prepared by replacing 8 μl of NADPH A with 100 mM potassium phosphate buffer (pH 7.4). The inactivation assay was repeated for O-dealkylated lapatinib at 5, 25, and 50 μM, using testosterone as a probe substrate. In addition, to evaluate the effect of exogenous nucleophile in the inactivation of CYP3A5 by lapatinib, the experiment was repeated by including 2 mM GSH in the primary incubation mixture.
MIC Screening via Dialysis.
Incubations (n = 3) containing 200 pmol/ml rCYP3A5, NADPH B, 100 mM potassium phosphate buffer (pH 7.4), and 50 μM lapatinib were prewarmed at 37°C. After 3 to 5 min, NADPH A was added to initiate the reaction. The total volume of the primary incubation mixture was 200 μl, and final concentrations of organic solvents in the incubation were 0.1% DMSO and 0.9% ACN (v/v). At 30 min after the addition of NADPH A, 20 μl of the primary incubation was transferred to the secondary incubation containing 200 μM testosterone (probe substrate), the NADPH-regenerating system, and 100 mM potassium phosphate buffer (pH 7.4). The secondary incubation mixtures were further incubated for 10 min at 37°C before quenching with an equal volume of ice-cold ACN and 0.5 μM prednisolone (IS). After this, 170 μl of each primary incubation was transferred into dialysis membrane tubing (Dialysis Membrane-50, molecular mass cutoff 12,000–14,000 Da; HiMedia Laboratories, Mumbai, India) and placed in a beaker filled with 200 ml of 100 mM potassium phosphate buffer (pH 7.4). The buffer system was maintained at 4°C with constant stirring, and one fresh buffer change at the second hour. After another 4 h, 20 μl of the mixture was transferred to the secondary incubation and subjected to the same experimental conditions as before. Negative controls were prepared by excluding lapatinib in the incubation mixtures. All samples were centrifuged for 16,000g at 4°C for 15 min, and the supernatant was removed for determination of testosterone-6β-hydroxylase activity by LC/MS/MS. To account for the degradation of enzymatic activity during dialysis, the residual enzymatic activities of the incubation mixtures pre- and postdialysis were normalized to their respective negative control activities.
Measurement of Residual CYP3A5 Activity by LC/MS/MS.
The LC/MS/MS system consisted of a 1290 Infinity LC system interfaced with a 6430 triple quadrupole mass spectrometer equipped with an electrospray ionization (ESI) source. Both instruments were controlled using MassHunter Workstation software (version B.04.00; Agilent Technologies, Santa Clara, CA). Chromatographic separation was achieved using an ACQUITY UPLC BEH C18 column (1.7 μm, 50 × 2.1 mm i.d.; Waters, Milford, MA). The column and autosampler temperatures were 45 and 6°C, respectively. The mobile phases were Milli-Q water with 0.1% formic acid (solvent A) and ACN with 0.1% formic acid (solvent B) delivered at a flow rate of 0.6 ml/min. The elution conditions for 6β-hydroxytestosterone and prednisolone were as follows: linear gradient 20 to 70% B (0–1.40 min), isocratic 95% B (1.41–1.99 min), and isocratic at 20% B (2.00–2.50 min). For 1′-hydroxymidazolam and carbamazepine, the elution conditions were as follows: linear gradient 20 to 80% B (0–1.40 min), isocratic 95% B (1.41–1.99 min), and isocratic at 20% B (2.00–2.50 min). All analyses were performed in the ESI positive mode. The MS source conditions for 6β-hydroxytestosterone and prednisolone were the following: gas temperature, 350°C; gas flow, 12 l/min; nebulizer gas pressure, 40 psi; and capillary voltage 3500 V. For midazolam and carbamazepine, the MS source conditions were similar except that the capillary voltage was set to 4000 V. The multiple reaction monitoring (MRM) transitions and compound-dependent MS parameters are summarized in Table 1.
MRM transitions and compound-dependent MS parameters for the detection of 6β-hydroxytestosterone, prednisolone (IS for 6β-hydroxytestosterone), 1′-hydroxymidazolam, and carbamazepine (IS for 1′-hydroxymidazolam)
Spectral Difference Scanning.
A mixture of 100 pmol/ml rCYP3A5, 50 μM lapatinib, NADPH A, and 100 mM potassium phosphate buffer (pH 7.4) was prewarmed at 37°C for 3 to 5 min. NADPH B was added to initiate the reaction, and the reaction mixture (final volume of 500 μl) was immediately scanned from 430 to 495 nm at 1-min intervals over a 10-min duration at 37°C using a Hitachi U-2900 spectrophotometer (Hitachi High-Technologies Corporation, Tokyo, Japan). The negative control was prepared by replacing NADPH B with 100 mM potassium phosphate buffer (pH 7.4). The positive control was prepared by replacing rCYP3A5 with rCYP3A4. The spectral differences were obtained by comparing the UV absorbances between the sample and reference cuvettes. Spectra were generated by normalizing the UV absorbance peaks at various time points (1–10 min) against 0 min.
Reduced CO Difference Spectroscopy.
Reaction mixtures (n = 3) containing 50 μM lapatinib, NADPH B, and 640 pmol/ml rCYP3A5 in 100 mM potassium phosphate buffer (pH 7.4) were prepared and prewarmed at 37°C. After 3 to 5 min, NADPH A was added to initiate the reaction. The reaction mixture (final volume of 250 μl) was incubated for another 30 min at 37°C. The reaction was terminated by the addition of 850 μl of ice-cold quenching buffer containing 1 mM EDTA, 20% glycerol, 1% Tergitol-type NP-40, 2 μM safranin O, and 100 mM potassium phosphate buffer (pH 7.4). The quenched mixture was split into two 550-μl tubes (sample and reference tubes). CO was bubbled into one of the tubes (sample tube) and stopped after approximately 100 bubbles had been passed into the mixture. Then 1 mg of sodium dithionite was added to both tubes and vortex-mixed gently, and 500 μl was transferred from each tube into a cuvette. The reduced CO difference spectrum for the sample and reference was acquired by scanning from 400 to 500 nm using a U-2900 spectrophotometer (Hitachi High-Technologies Corporation). Negative controls were prepared by excluding NADPH from the incubation mixture. The extent of reduction in the peak at 450 nm was determined using a CYP450 concentration calculated as [(ΔA450 − ΔA490)/0.091 = nanomoles of P450 per milliliter] (Guengerich et al., 2009).
GSH Trapping of a Reactive Metabolite of Lapatinib.
Incubations containing 50 pmol/ml rCYP3A5, the NADPH-regenerating system, 100 mM potassium phosphate buffer (pH 7.4), and 5 mM GSH were prewarmed at 37°C for 3 to 5 min. Lapatinib or O-dealkylated lapatinib was added to initiate the reaction, and the final volume of the reaction mixture was 500 μl. The final concentrations of organic solvents were 0.1% DMSO and 0.9% ACN (v/v). After 60 min, 500 μl of ice-cold ACN was added to the incubation to quench the reaction. Then, the samples were centrifuged at 16,000g for 15 min at 4°C. The supernatant was transferred to a clean microtube and dried by a gentle flow of nitrogen gas (TurboVap LV; Caliper Life Science, Hopkinton, MA). The residue was reconstituted with 100 μl of ACN-water mixture (3:7, v/v), vortex-mixed, and centrifuged at 16,000g at 4°C for 15 min. The supernatant was removed for LC/MS/MS analysis. Negative controls were prepared by the exclusion of either lapatinib, O-dealkylated lapatinib, NADPH, or GSH in the incubation mixture.
Detection of GSH Adduct.
The formation of a GSH adduct with the reactive intermediate of lapatinib was analyzed on the same LC/MS/MS system used for the measurement of residual CYP3A5 activity. Chromatographic separation was achieved using an ACQUITY UPLC BEH C18 column (1.7 μm, 100 × 2.1 mm i.d. column; Waters). The column and sample temperatures were 45 and 6°C, respectively. The mobile phases were Milli-Q water with 0.1% formic acid (solvent A) and ACN with 0.1% formic acid (solvent B) and delivered at a flow rate of 0.45 ml/min. The elution conditions were as follows: linear gradient 5 to 60% B (0–6.25 min), isocratic at 95% B (6.26–7.09 min), and isocratic at 5% B (7.10–8.00 min). The adduct was analyzed in the positive ESI mode using MRM and product ion scanning (PIS). A MRM transition of m/z of 778 to m/z 665 was performed with a 20-ms dwell time. PIS at m/z 778 was performed in the scan range of m/z 150 to 800 with a 0.4-s scan time. The compound-dependent MS parameters for both scans were 100 and 20 V for fragmentor and collision energy voltages, respectively. The MS source conditions were 350°C, 12 l/min, 40 psi, and 4000 V for gas temperature, gas flow rate, nebulizer gas pressure, and capillary voltage, respectively.
Data Analysis.
All chromatographic peak integration was performed using MassHunter Workstation software (version B.04.00). The mean of triplicate analyses was used to calculate the natural log of percentage residual CYP3A5 enzyme activity normalized to 0 min against preincubation time for the time-, concentration-, and NADPH-dependent CYP3A5 inactivation experiments. The data were fitted to linear regression, and the observed first-order inactivation rate constant, kobs, was determined. Kinetic parameters inactivator concentration at half-maximum rate of inactivation (KI) and inactivation rate constant at infinite inactivator concentration (kinact) were determined using a Kitz-Wilson plot (Kitz and Wilson, 1962) to reflect the degree of inactivation of the specific P450 enzyme. These plots were acquired using GraphPad Prism 4.0 (GraphPad Software Inc., San Diego, CA).
In Silico Molecular Analysis.
An X-ray crystallographic structure of human CYP3A4 (Protein Data Bank code 3NXU) was obtained from the RCSB Protein Data Bank. This structure has a good resolution of 2.0 Å, R value of 0.232, and R free value of 0.262. The protein file contains two copies of CYP3A4. The difference between the two copies is small (root mean standard deviation 0.255 Å); thus, the second copy, chain B, was removed. The ligand associated with chain B and all water molecules associated with both chains were also removed. An oxygen atom was added to the heme group using the same procedure as that adopted by Rydberg et al. (2008). The protein chain and its associated ligand were processed using the default settings for the Protonate 3D feature in the software Molecular Operating Environment (MOE) to add hydrogen atoms and determine the ionization state of the residues. Residues 265 to 267 and residues 281 to 288 were missing from the 3NXU structure. Thus, an autohomology model of CYP3A4 was developed using MOE to reconstruct these missing residues.
At present, there are no three-dimensional structures of human CYP3A5 in the RCSB Protein Data Bank; hence, homology modeling was performed in MOE to create a three-dimensional structure for it. The FASTA sequences of the human CYP3A5 (identification number P20815) were obtained from UniProtKB (http://www.uniprot.org). The prepared structure of CYP3A4 was used as the template structure. The Protein Align module in MOE was used to align the CYP3A5 sequence with the template sequence. The BLOSUM62 matrix with a gap opening penalty of 7 and gap extension penalty of 1 was used for the alignment process. The Homology Model module was then used to create the homology models. AMBER99 force fields with calculation of implicit solvation energies in the Generalized Born model were used for the modeling process. The resultant CYP3A5 model was subsequently energy-minimized and evaluated using a Ramachandran plot.
Molecular docking of lapatinib to both CYP3A4 and CYP3A5 was performed using the Dock feature in MOE. The active sites in both enzymes were first identified using the Site Finder feature in MOE. A simple pharmacophore was then used to ensure that at least one of the atoms in lapatinib is within 4.0 Å from the oxygen atom that is bonded directly to heme (Julsing et al., 2008). A total of 1000 different conformations were generated for lapatinib using the pharmacophore. The top 100 conformations were retained and further refined by energy minimization. The MMFF94x force field of the Distance Model was used for energy minimization. Side chains of residues within 6 Å of the ligand were allowed to move during energy minimization. After energy minimization, all 100 conformations were manually inspected to retain those with known site of metabolism that were within 4.0 Å from the oxygen atom.
Results
Time-, Concentration-, and NADPH-Dependent CYP3A5 Inactivation.
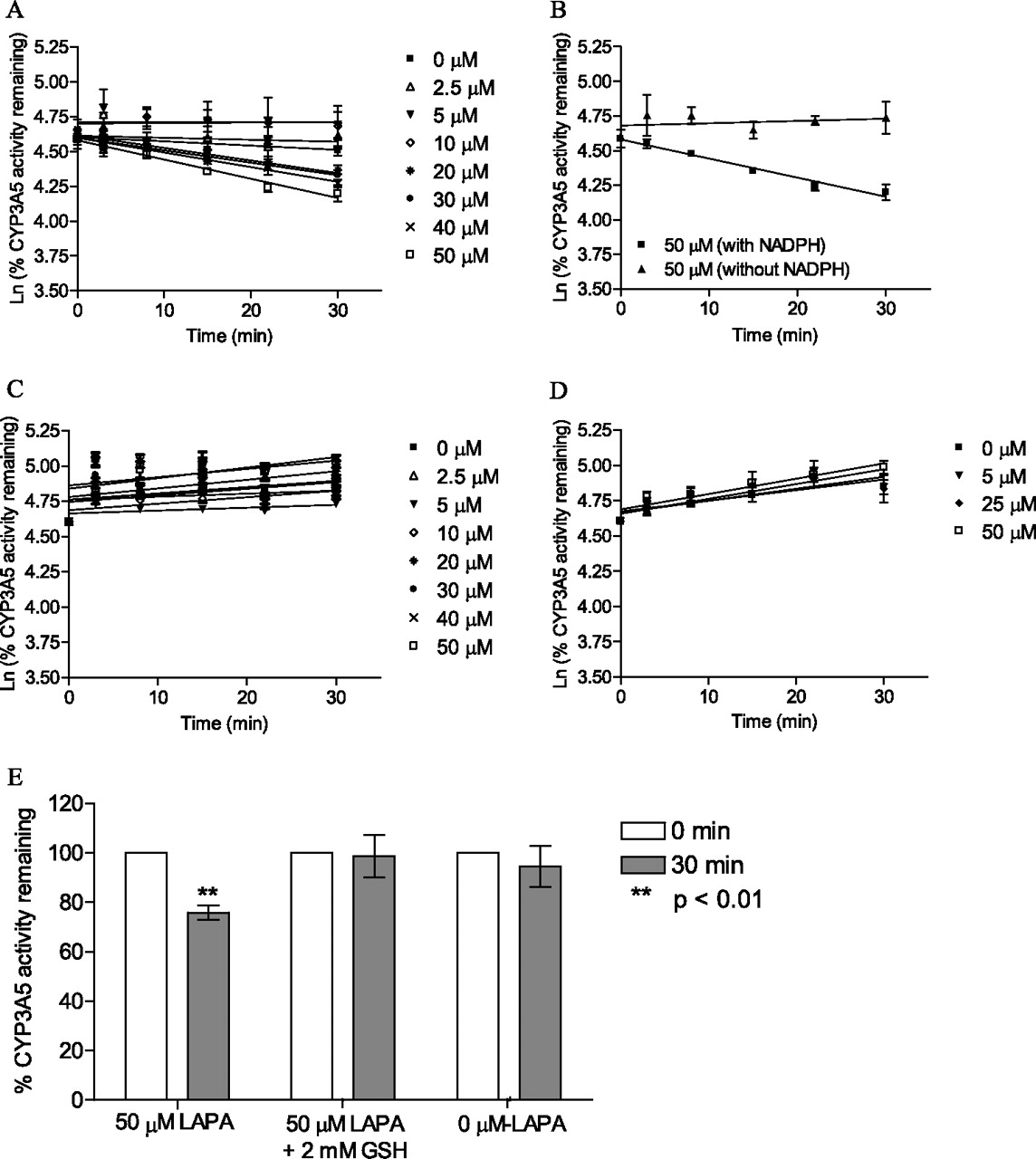
To determine the nature of the interaction between CYP3A5 and lapatinib, time-, concentration-, and NADPH-dependent metabolism of testosterone (6β-hydroxylation) and midazolam (1′-hydroxymidazolam) by lapatinib were examined. Figure 2A shows that testosterone-6β-hydroxylation activity declined in a time- and concentration-dependent manner when CYP3A5 was preincubated with lapatinib in the presence of NADPH for up to 30 min. Lapatinib caused a loss in testosterone hydroxylase activity that followed pseudo-first-order kinetics. The kinetic parameters were determined from a Kitz-Wilson plot. Figure 2B shows that lapatinib inhibited CYP3A5 in a NADPH-dependent manner. The absence of NAPDH in CYP3A5 incubations caused no significant decline in enzyme activity when incubated with lapatinib over the preincubation time of 30 min. On the other hand, the presence of NADPH showed a pronounced time-dependent decrease in CYP3A5 activity by lapatinib at 50 μM. The KI and kinact values for lapatinib were 0.0376 mM and 0.0226 min−1, respectively. The goodness of the fit of the Kitz-Wilson plot was 0.9981.

Inhibition assay of CYP3A5 by lapatinib using testosterone as probe substrate (A), using testosterone as probe substrate in the presence and absence of NADPH (50 μM lapatinib data are presented) (B), and using midazolam as probe substrate (C). D, inhibition assay of CYP3A5 by O-dealkylated lapatinib using testosterone as probe substrate. E, effect of GSH on residual CYP3A5 activity. rCYP3A5 was incubated with 50 μM lapatinib (LAPA) alone, with 50 μM lapatinib and 2 mM GSH, or without lapatinib. For all these plots (A–E), each point represents the mean of three replicates with less than 10% S.D.
To further assess inactivation of CYP3A5 by lapatinib, another well characterized CYP3A5 probe substrate, midazolam, was used. Figure 2C shows that midazolam 1′-hydroxylation activity was not affected in either a time- or concentration-dependent manner when CYP3A5 was preincubated with lapatinib in the presence of NADPH for up to 30 min. When CYP3A5 was preincubated with O-dealkylated lapatinib, no clear time-dependent inhibition of testosterone hydroxylase activity was observed (Fig. 2D). Increasing trends in the percentages of residual CYP3A5 activity were observed in Fig. 2, C and D, as the preincubation time was increased. We repeated the experiments and confirmed that the observed trends were reproducible. Because the increasing trend was also observed with 0 μM concentrations of each substrate (lapatinib for Fig. 2C and O-dealkylated lapatinib for Fig. 2D), the trend was possibly derived from systematic analytical variation (e.g., gradual increase in LC-MS ion enhancement effect) rather than the activation of CYP3A5 metabolism. Addition of GSH (exogenous nucleophile) blocked the extent of enzyme inactivation caused by lapatinib. As shown in Fig. 2E, the inactivation of CYP3A5 by lapatinib was not statistically significant in the presence of GSH compared with the incubation to which GSH was not added (p < 0.01).
MIC Screening via Dialysis.
To assess whether lapatinib was generating a MIC with CYP3A5 as it did with CYP3A4 (Takakusa et al., 2011), incubations were subjected to extensive dialysis in cold buffer over time to determine whether the inhibition of P450 activity was reversible (Nomeir et al., 2004). When dialysis was performed on incubations containing 50 μM lapatinib with CYP3A5, the difference in CYP3A5 enzymatic activities pre- and postdialysis was not significant (p > 0.05) (Fig. 3A).
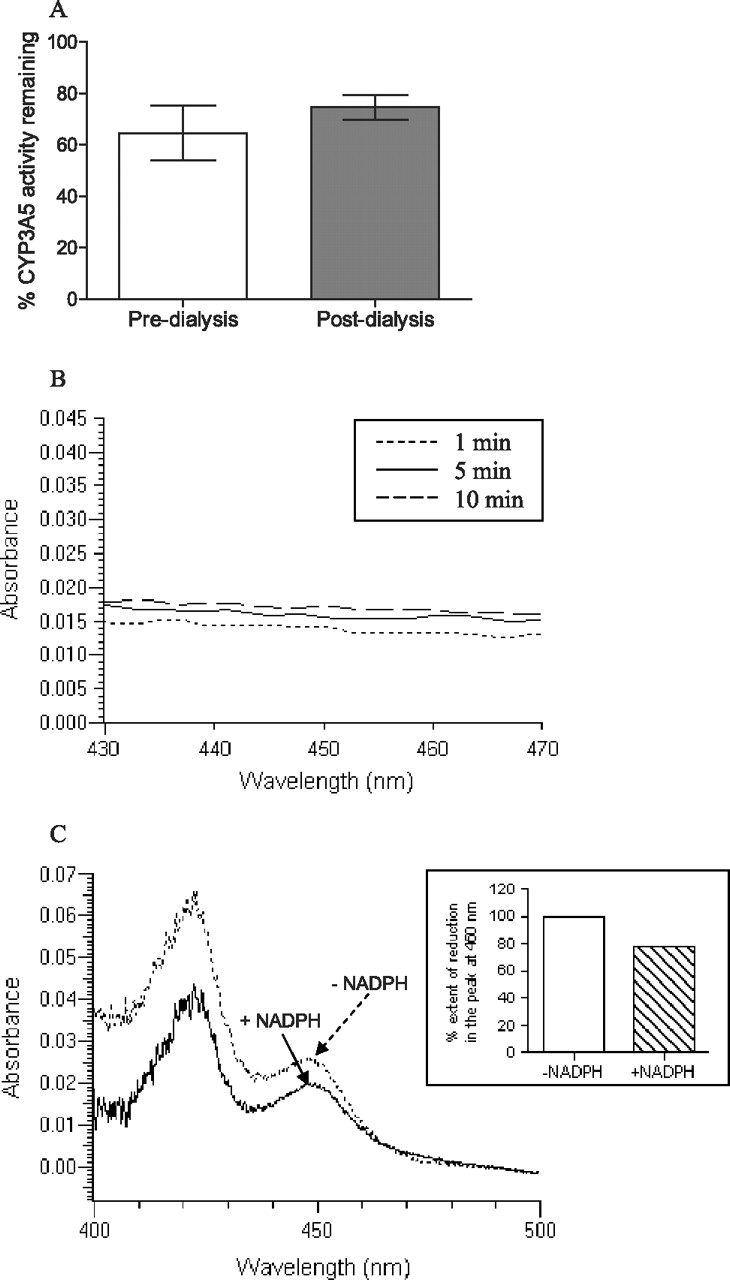
A, triplicate incubations containing 50 μM lapatinib were subjected to dialysis and the percentages of residual CYP3A5 activity were compared with the incubations with 50 μM lapatinib predialysis (control). The percentage activity of CYP3A5 remaining was not significantly different pre- or postdialysis. B, UV absorbance spectral differences obtained from the incubations of lapatinib with CYP3A5 over 10 min. There was no Soret peak observed in the absorbance range of 448 to 458 nm. C, spectral difference plot of reduced CO spectroscopy. Lapatinib (50 μM) was incubated with CYP3A5 with (+) and without (−) NADPH.
Spectral Difference Scanning.
MIC-forming compounds normally show a characteristic peak in the Soret region around 448 to 458 nm (Polasek and Miners, 2008). When spectral differences were obtained by scanning from 430 to 495 nm for 10 min, lapatinib showed no observable peak in the Soret region when incubated with CYP3A5 (Fig. 3B). However, there was an observable peak at 457.4 nm when lapatinib was incubated with rCYP3A4 (data not shown).
Reduced CO Difference Spectroscopy.
Ferrous (reduced) cytochrome P450 forms a complex with CO to give a spectrally detectable peak at 450 nm. When 50 μM lapatinib was incubated with CYP3A5 and NADPH for 30 min, there was a mean reduction of 22% in the peak at 450 nm compared with that in the negative control incubation in the absence of NADPH (Fig. 3C). The measured concentrations of CYP3A5 with and without NADPH were 154 ± 50 and 194 ± 70 pmol/ml, respectively.
GSH Trapping of a Reactive Metabolite of Lapatinib.
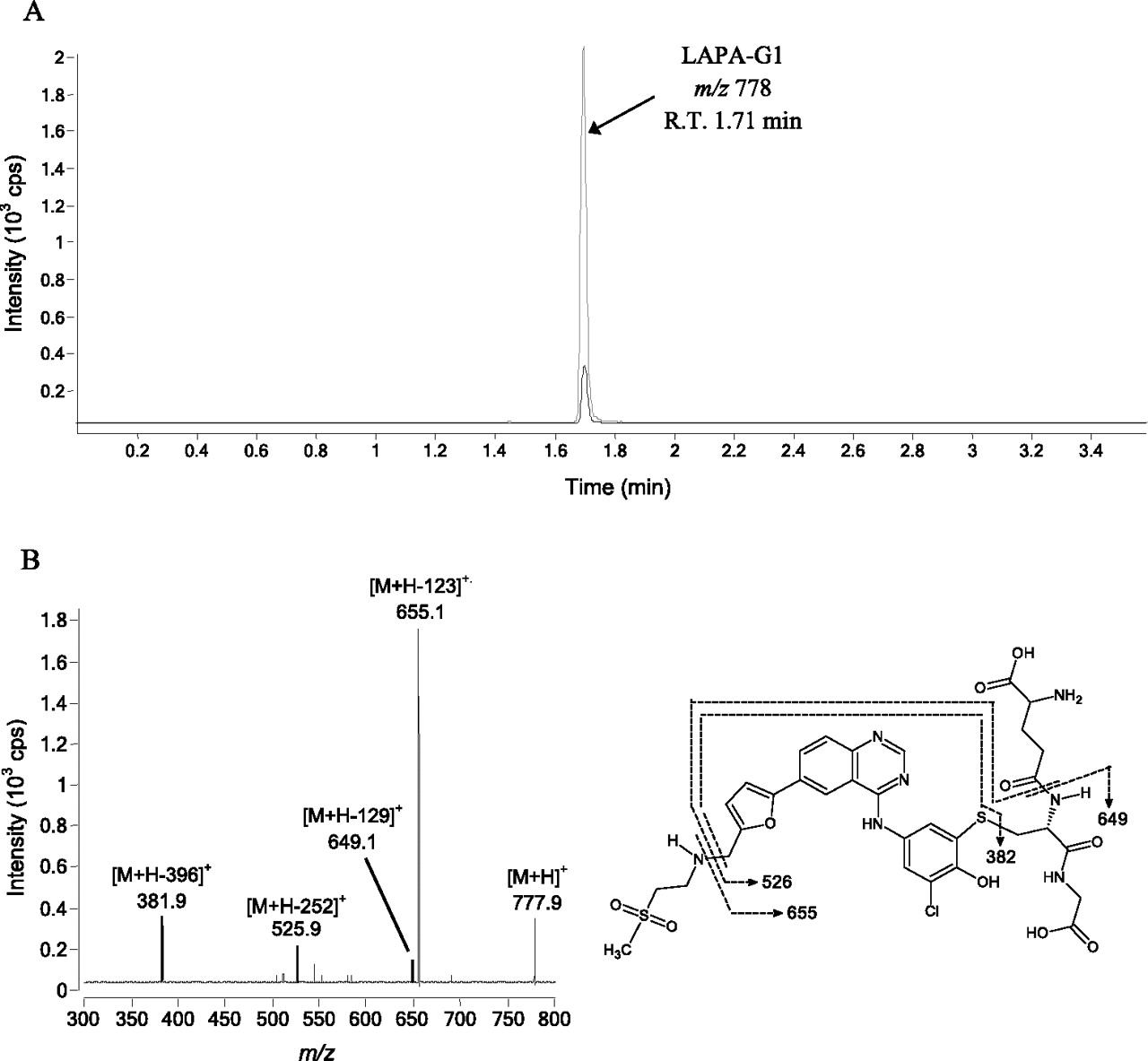
A GSH trapping assay was performed by incubating lapatinib with rCYP3A5 in the presence of GSH to confirm the generation of reactive metabolite. MRM scans of incubations containing lapatinib or its O-dealkylated metabolites (Fig. 1) were performed. In the incubation containing 50 pmol/ml rCYP3A5, 50 μM lapatinib, 5 mM GSH, and NAPDH, a peak eluting at 1.71 min was observed (Fig. 4A). The same peak was also observed in incubations of the O-dealkylated metabolite of lapatinib as the substrate. The parent ion of the peak was at m/z 778. In negative controls, no distinct peaks were observed. PIS at m/z 778 was performed to elucidate the structure of the metabolite eluting at 1.71 min. The PIS spectrum yielded product ions at m/z 655.1, 649.1, 525.9, and 381.9 (Fig. 4B). This fragmentation pattern is characteristic of an adduct formed between GSH and the quinoneimine metabolite of lapatinib as described previously (Teng et al., 2010). For instance, the neutral mass loss of 129 corresponded to the pyroglutamic acid moiety of GSH.

A, MRM chromatograms of rCYP3A5 incubations with GSH with 50 μM lapatinib (LAPA, black trace) and 50 μM O-dealkylated lapatinib (gray trace). B, PIS spectrum of quinoneimine metabolite-GSH adduct (m/z 778). R.T., retention time.
In Silico Molecular Analysis.
The conformations of lapatinib that are likely to lead to N-hydroxylation in CYP3A4 and CYP3A5 are shown in Fig. 5A. It can be seen that lapatinib adopts different conformations for N-hydroxylation in CYP3A4 and CYP3A5. In particular, the two phenyl rings of lapatinib have van der Waals interactions with Phe57, Phe108, and Phe213 in CYP3A4. Phe57 and Phe108 have previously been shown to be involved in van der Waals interactions with various substrates (Julsing et al., 2008; Mannu et al., 2011) to stabilize their binding. These interactions were not seen in CYP3A5, possibly due to the substitution of Phe57 and Phe108 with Leu57 and Leu108, respectively. This may contribute to the reduction of binding energy in CYP3A5.

Close-up views of the conformations of lapatinib in CYP3A4 and CYP3A5 for (A) N-hydroxylation, showing the involvement of Phe57, Phe108, and Phe213 in CYP3A4 and the lack of involvement of these residues in CYP3A5 and (B) O-dealkylation, showing the involvement of unfavorable steric interaction between lapatinib and Phe108 in CYP3A4 if it adopts that conformation. Conformations in CYP3A4 are drawn in brown, and conformations in CYP3A5 are drawn in dark green.
The conformations of lapatinib that are likely to lead to N-dealkylation in CYP3A4 and CYP3A5 are shown in Supplemental Fig. 1. The two conformations are similar, with a heavy atom root mean square deviation of 2.23 Å. In this conformation, lapatinib has a hydrogen bond interaction with Thr309.
The conformations of lapatinib that are likely to lead to O-dealkylation in CYP3A4 and CYP3A5 are shown in Fig. 5B. The two conformations are similar at the fluorophenyl end but differ at the sulfone end because the Phe108 in CYP3A4 will have unfavorable steric interaction with the sulfone group, thus causing it to adopt a different position from that in CYP3A5. The binding energies of the various conformations are shown in Table 2.
Binding energies of the various conformations of lapatinib in CYP3A4 and CYP3A5
Discussion
Because lapatinib was found to form a MIC with CYP3A4 (Takakusa et al., 2011), we aimed to investigate the chemical interaction between lapatinib and CYP3A5. Our findings confirmed time-, concentration-, and NADPH-dependent inhibition of CYP3A5 by lapatinib and suggested metabolism-based inactivation of the enzyme. It was further noted that the kinact/KI ratio of lapatinib in inactivating CYP3A5 (0.601 min−1 mM−1) was lower than that associated with CYP3A4 (11.8 min−1 mM−1) (Teng et al., 2010) when testosterone was used as the probe substrate. This suggested that lapatinib was less potent in inactivating CYP3A5 than CYP3A4. However, comparisons of kinact/KI ratios are insufficient for assessing the relative clinical significance of the inactivation of P450 enzymes (Fowler and Zhang, 2008), and further clinical studies are warranted.
Because the substrate specificity of CYP3A isoforms is low, inactivation of CYP3A5 by lapatinib using midazolam as a probe substrate was further investigated. Results of these experiments showed no significant inhibition by lapatinib of the 1′-hydroxylation of midazolam by CYP3A5. This contrasting but pertinent result suggests that the inactivation of CYP3A5 by lapatinib is site-specific and was consistent with the fact that there are multiple modes of binding of substrates on CYP3A5 because of its large active site (Ekins et al., 2003). From a clinical perspective, one would expect that any potential drug-drug interactions arising from the inactivation of CYP3A5 by lapatinib would be substrate-specific and not all drug substrates of CYP3A5 will be “victim” drugs even when they are eliminated predominantly by CYP3A5.
O-Dealkylated lapatinib failed to demonstrate time-dependent inhibition of CYP3A5 activity. This finding suggests that the rate-limiting step of the inactivation process is the conversion of lapatinib to O-dealkylated lapatinib and not the subsequent oxidation of the latter to a reactive species. Coincubation with GSH prevented the inactivation of CYP3A5 by lapatinib. For a classic MBI, we did not expect the rate of inactivation of CYP3A5 to be affected by exogenous scavenger nucleophiles such as GSH. Because our observation was not characteristic of a classic MBI, we repeated the experiments three times and confirmed that the results were reproducible. Our findings suggested that the quinoneimine metabolite of lapatinib could be released from the enzymatic active site and interact with enriched GSH in vitro. Under oxidative stress conditions in which the level of GSH is reduced in vivo, the quinoneimine metabolite might interact with other cellular proteins in addition to the inactivation of CYP3A5.
Although it was confirmed that lapatinib inactivates CYP3A5 in a metabolism-based reaction, it was not clear whether the inactivation was pseudo-irreversible (i.e., formation of a MIC) or irreversible (mechanism-based inhibition). To ascertain the exact nature of enzyme inhibition, a dialysis experiment was performed to investigate the recovery of enzymatic activity postdialysis. If lapatinib forms a MIC with CYP3A5, the tight but reversible coordination complex that the metabolite forms with the heme moiety of the enzyme should be exchanged with the solvent (buffer) during dialysis, leading to a recovery of enzymatic activity (Ma et al., 2000). If lapatinib is a MBI, there would be no recovery of enzymatic activity because the covalent bonds formed are irreversible. The pores of the dialysis membrane tubing (molecular mass cutoff 12,000–14,000 Da) are large enough for metabolites to diffuse out into the dialysis buffer without the loss of CYP3A5. Collectively, our results showed that there was insignificant recovery of CYP3A5 6β-testosterone hydrolase activity postdialysis. This result suggested that lapatinib is a MBI of CYP3A5.
It has been established that MIC forms a Soret peak at approximately 450 nm (Polasek and Miners, 2008). Spectral difference spectroscopy showed no Soret peak in the UV absorbance spectrum of inactivated CYP3A5, which further confirmed that lapatinib is predominantly a MBI of CYP3A5. However, a Soret peak was observed when lapatinib was incubated with CYP3A4, indicating that lapatinib forms a MIC with CYP3A4 as we had observed previously (Takakusa et al., 2011). Taken together, our data highlighted the fact that CYP3A4 and CYP3A5 have different susceptibilities to inhibition despite similar substrate specificity. Similar examples of such differential interactions with CYP3A4 and CYP3A5 include ketoconazole and erythromycin (Niwa et al., 2008).
Because the formation of a MIC was ruled out, there remained two other possible mechanisms of inactivation of CYP3A5 by lapatinib as a MBI: alkylation of the P450 heme component or modification of the P450 apoprotein (Polasek and Miners, 2007). Reduced CO spectroscopy demonstrated a reduction of 22% of the 450-nm peak when lapatinib was incubated with CYP3A5 in the presence of NADPH. These results suggested that a small fraction of the reactive intermediate may form an adduct with the heme component, whereas a larger percentage forms a covalent adduct with the P450 apoprotein of CYP3A5.
The addition of GSH to the CYP3A5 incubation containing lapatinib and NADPH led to the formation of a GSH conjugate of quinoneimine intermediate derived from the O-dealkylated metabolite. PIS of the GSH adduct (m/z 778) showed mass spectral fragments identical to those produced when CYP3A4 was incubated with either lapatinib or its O-dealkylated metabolite (Teng et al., 2010). This adduct was formed in both CYP3A4 and CYP3A5 via conjugate addition of GSH to the quinoneimine metabolite. Because CYP3A5 has a greater propensity to generate an O-dealkylated metabolite than CYP3A4 (Takakusa et al., 2011; current in silico data), CYP3A5 could be an important enzyme responsible for the generation of the reactive quinoneimine in those patients who have higher expression of CYP3A5 enzyme. Further attempts should be made to identify the quinoneimine-CYP3A5 protein adduct either using mass spectrometry technique.
We recently demonstrated that the inactivation of CYP3A4 by lapatinib is mostly due to quasi-irreversible MIC formation (Takakusa et al., 2011). The finding of a hydroxylamine and oxime metabolites of lapatinib suggested the formation of a nitroso intermediate, which forms an MIC with CYP3A4. Of interest, neither hydroxylamine nor oxime was detected in metabolites formed by CYP3A5, which explains the lack of MIC formation by lapatinib with CYP3A5 (Takakusa et al., 2011). Two pathways, namely oxidation of secondary amines to primary amines (N-dealkylation pathway) or hydroxylamines (N-hydroxylation pathway) (Lindeke et al., 1979; Cerny and Hanzlik, 2005), have been proposed for the reaction sequence from secondary amines to nitroso intermediates. In the case of lapatinib, it was suggested that N-hydroxylation of lapatinib is the initial step in the pathway to MIC formation because one of the metabolites formed by CYP3A4 was characterized as a hydroxylamine metabolite. In addition, CYP3A5 was shown to have a significantly lower ability to generate hydroxylamine even though it formed the primary amine metabolite (Takakusa et al., 2011). To further elucidate the mechanism responsible for the significant differences between MIC formation by CYP3A4 and mechanism-based inhibition by CYP3A5, in silico docking studies were performed in the current article to better understand the propensities for the enzymes to form the N-dealkylated, N-hydroxylated, and O-dealkylated metabolites. Comparison of the binding energies (Table 2) indicates that N-hydroxylation is favored over O-dealkylation and N-dealkylation in CYP3A4, whereas in CYP3A5 O-dealkylation is preferred over N-dealkylation and N-hydroxylation. From our modeling results, this difference in the metabolic pathway of lapatinib between CYP3A4 and 3A5 is probably due to Phe108 of CYP3A4, which is substituted with Leu 108 in CYP3A5. The Phe108 residue in CYP3A4 appears to be important in the π-stacking interaction with the 3-chloro-4-methoxy phenyl ring on the lapatinib structure that helps orient the lapatinib side chain alkylamino group toward the heme center (Fig. 5A). In contrast, substitution of Phe108 for leucine in CYP3A5 now allows for reorientation of the lapatinib structure to favor interaction of the distal fluorobenzyl methylene carbon with the heme group (Fig. 5B). Generation of site-directed mutants will be required to further investigate these interactions.
An indirect consequence of reactive metabolite formation is the covalent modification of endogenous proteins, which then trigger immunological responses. Indeed, immune-mediated toxicities and host genetic influences have also been suggested to contribute to hepatotoxicity caused by drugs. In a recent study, patients possessing specific HLA polymorphisms (HLA-DQA1*02:01) within the same major histocompatibility complex were found to be at greater risk of having lapatinib-induced hepatotoxicity (Spraggs et al., 2011). Because the formation of a metabolite-protein complex is one triggering factor leading to immune-mediated responses, the covalent adduction of lapatinib with CYP3A5 or other cellular proteins might possibly account for the immune-mediated hepatotoxicity. One possible follow-up study is to identify any lapatinib-enzyme/protein adducts from hepatic samples of affected patients.
In conclusion, our results demonstrate that lapatinib is a MBI of CYP3A5 primarily via the covalent modification of its apoprotein. Idiosyncratic hepatotoxicity caused by lapatinib may be attributed to a complex interaction of factors including polymorphisms of CYP3A5 and host immune and genetic status. Further clinical studies should be conducted to examine whether the polymorphism of CYP3A5 in a specific population plays a significant role in lapatinib-induced hepatotoxicity. The correlation between the covalent adduction of lapatinib with CYP3A5 and/or other cellular proteins and the immune-mediated toxicological endpoint should also be explored.
Authorship Contributions
Participated in research design: Chan, New, Ho, and Nelson.
Conducted experiments: Chan, New, Chua, and Yap.
Contributed new reagents or analytic tools: Chan and Nelson.
Performed data analysis: Chan, New, Chua, Yap, and Nelson.
Wrote or contributed to the writing of the manuscript: Chan, New, Chua, Yap, Ho, and Nelson.
Acknowledgments
This article is dedicated to the late Professor Sidney D. Nelson and his wife Joan Nelson.
Footnotes
This work was supported by the National University of Singapore, Academic Research Fund Tier 1 [Grants R-148-000-13-112, R-148-000-117-133, R-148-000-136-112]; and National University of Singapore Department of Pharmacy [Final Year Project Grant R-148-000-003-001].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- P450
- cytochrome P450
- MIC
- metabolite-intermediate complex
- ACN
- acetonitrile
- r
- recombinant
- DMSO
- dimethyl sulfoxide
- IS
- internal standard
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- ESI
- electrospray ionization
- MS
- mass spectrometry
- MRM
- multiple reaction monitoring
- PIS
- product ion scanning
- MOE
- Molecular Operating Environment
- MBI
- mechanism-based inactivator
- rCYP3A5
- recombinant P450 3A5 Supersomes.

- Received February 6, 2012.
- Accepted April 10, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}