


Abstract
Current studies explored the effect of structural changes on the aldehyde oxidase (AO)-mediated metabolism of zoniporide (1). Zoniporide analogs with modifications of the acylguanidine moiety, the cyclopropyl group on the pyrazole ring, and the quinoline ring were studied for their AO-catalyzed metabolism using the human S9 fraction. Analysis of the half-lives suggested that subtle changes in the structure of 1 influenced its metabolism and that the guanidine and the quinoline moieties were prerequisites for AO-catalyzed oxidation to 2-oxozoniporide (M1). In contrast, replacement of the cyclopropyl group with other alkyl groups was tolerated. The effect of structural variation on AO properties was rationalized by docking 1 and its analogs into the human AO homology model. These studies indicated the importance of electrostatic, π-π stacking and hydrophobic interactions of the three motifs with residues in the active site. Differences in substrate properties were also rationalized by comparing their half-lives with cLogD, electrophilicity parameters [electrostatic potential (ESP) charges and energy of lowest unoccupied molecular orbitals (ELUMO)], and the energies of formation of tetrahedral intermediates (J Med Chem 50:4642–4647, 2007). Whereas the success of energetics in predicting the AO substrate properties of analogs was 87%, the predictive ability of other descriptors was none (cLogD) to 60% (ESP charges and ELUMO). Overall, the structure-metabolism relationship could be rationalized using a combination of both the energy calculations and docking studies. This combination method can be incorporated into a strategy for mitigating AO liabilities observed in the lead candidate or studying structure-metabolism relationships of other AO substrates.
Introduction
Aldehyde oxidase (AO) is a cytosolic enzyme that catalyzes the metabolism of a wide range of substrates with different chemical structures and functionalities (Beedham, 1985; Kitamura et al., 2006; Pryde et al., 2010; Garattini and Terao, 2011, 2012). Even though its role in drug metabolism has been known for decades, its importance in the drug metabolism field has emerged in the past decade (Pryde et al., 2010). Of consequence is the AO-catalyzed oxidation of heteroaromatic rings because these rings are commonly incorporated into new drug entities and form the backbone of several marketed drugs and drug candidates. Some drugs that are AO substrates are famciclovir (Rashidi et al., 1997), zaleplon (Kawashima et al., 1999), brimonidine (Acheampong et al., 1996), and N-[(2-diethylamino)ethyl] acridine-4-carboximide (Schofield et al., 2000) (Fig. 1).

Structures of drugs that undergo AO-mediated metabolism.
Although major clinical drug-drug interactions are unknown, the importance of understanding AO-mediated metabolism stems from the high clearance and poor exposure of AO substrates in the clinic. These in turn affect their progression through clinical evaluation (Pryde et al., 2010). Indeed, poor pharmacokinetics of AO substrates has resulted in discontinued development of several good candidates (Fig. 2). Failure to predict human pharmacokinetics is ascribed to lack of in vitro methods that can predict in vivo clearances or determine the contribution of the fraction of total clearance that is mediated by AO (Zientek et al., 2010). Furthermore, species differences among rat, mouse, dog, and monkey (the commonly used preclinical/toxicology species) and high and variable AO activity in humans make prediction a challenge (Beedham, 1987; Al-Salmy, 2001; Kitamura et al., 2006). Attempts have been made to recognize this liability early in a lead optimization stage and mitigate the potential of AO catalyzed metabolism at this stage. Several reports describing medicinal chemistry strategies to mitigate AO liabilities have been published recently (Magee et al., 2009; Jones et al., 2011; Linton et al., 2011; Tran et al., 2011; Pryde et al., 2012).

Structures of drug candidates discontinued from development because of AO-mediated metabolism.
An understanding of the structure-metabolism relationship between AO and its substrates can be useful in designing new drug candidates with minimal AO liability. Some efforts in understanding the relationship between the structure of azaheterocycles and their metabolism by AO have been reported. Ghafourian and Rashidi (2001) have reported on structure-activity relationship studies of phthalazine and quinazoline derivatives and the influence of substituents on AO-mediated oxidation. The findings from these studies are consistent with a nucleophilic attack on an electron-deficient sp2-hybridized carbon atom by AO (Hille, 2005; Torres et al., 2007) with electron-deficient groups favoring the oxidation reaction. Likewise, the role of hydrophobicity as well as electronic properties in determining their AO-mediated catalysis has also been demonstrated (Krenitsky et al., 1972; Hall and Krenitsky, 1986; Beedham et al., 1990). Even though these studies provide insights into the electronics and lipophilicity requirements of AO substrates, additional structure-activity relationships studies can help in accurately predicting substrate-enzyme interactions.
The aim of the current studies was to investigate AO-catalyzed oxidation of a series of zoniporide analogs to probe the structure-metabolism relationship of zoniporide and AO. Zoniporide [1-(quinolin-5-yl)-5-cyclopropyl-1H-pyrazole-4-carbonyl guanidine] (1, Fig. 3) was designed and synthesized as a highly selective inhibitor of the sodium/hydrogen exchanger (Guzman-Perez et al., 2001; Knight et al., 2001; Tracey et al., 2003). The AO-mediated metabolism of 1 to 2-oxozoniporide (M1) (Fig. 3) appears to play a major role in its elimination in humans and rodents. The role of AO in the oxidation of 1 to M1 has been confirmed in vitro by incubating zoniporide with the human S9 fraction in the presence of the AO inhibitor, raloxifene (Obach, 2004; Dalvie et al., 2010). Enzyme kinetic studies revealed that the Km and Vmax values for the formation of M1 were 3.4 μM and 74 pmol · min−1 · mg protein−1, respectively (Dalvie et al., 2010).

Conversion of zoniporide (1) to 2-oxozoniporide (M1) by AO.
To this end, the metabolic stability of zoniporide and its analogs was determined by estimating their half-lives in the human S9 fraction and the effect of variation in zoniporide structure on its AO-mediated biotransformation to M1 was investigated. The differences were rationalized by comparing their half-lives with cLogD and electrophilicity parameters (partial atomic charges and ELUMO), and the energies of formation of tetrahedral intermediates as reported by Torres et al. (2007). In addition, zoniporide and its selected analogs were docked into the AO homology model in an attempt to explore the pattern of interactions between zoniporide and the active site residues in AO.
Materials and Methods
Materials.
Zoniporide and its analogs were synthesized at Pfizer Global Research and Development (Groton, CT) using previously reported synthesis procedures (Guzman-Perez et al., 2001). The pooled hepatic human S9 fractions were purchased from In Vitro Technologies (Baltimore, MD). All other chemicals were of the highest purity commercially available.
Metabolite Identification Studies.
Incubations for metabolite identification were performed at 37°C for 60 min in 100 mM potassium phosphate buffer (pH 7.4) containing magnesium chloride (10 mM) and liver S9 fractions (protein concentration, 2.0 mg/ml) (total volume 1.0 ml). Reactions were initiated by addition of zoniporide or its analogs (final concentration, 10 μM), dissolved in a mixture of acetonitrile and dimethyl sulfoxide (DMSO) (9:1). The final concentration of organic solvent in the incubation mixture was <1%, and the final concentration of DMSO was <0.1%. Reactions were terminated by addition of acetonitrile (2 ml) followed by centrifugation at 3000g for 10 min. The supernatant was decanted into a new tube and evaporated to dryness under a stream of nitrogen at 35°C. The resulting residue was reconstituted in 200 μl of a mixture of 0.1% formic acid solution and acetonitrile (7:3) and analyzed by HPLC-MS. The components in the incubation mixtures were separated on a Luna C4 100A column (3.0 μm, 150 × 2.0 mm; Phenomenex, Torrance, CA) at ambient temperature. The mobile phase consisted of 0.1% formic acid (solvent A) and acetonitrile (solvent B) and was delivered at 200 μl/min for 50 min. The initial composition of solvent B was maintained at 1% for 5 min and then increased in a linear manner as follows: 30% at 25 min; 50% at 35 min; and 90% at 40 min. Solvent B was then maintained at 90% for up to 45 min and decreased to 1% in the next 2 min. The column was allowed to equilibrate at 1% solvent B for 5 min before the next injection. The HPLC effluent going to the mass spectrometer was directed to waste through a divert valve for the initial 5 min after sample injection. Mass spectrometric analyses were performed on a Thermo Finnigan LCQ Deca XP ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA), which was interfaced to an Agilent HP-1100 HPLC system (Agilent Technologies, Palo Alto, CA) and equipped with an electrospray ionization source. The parameters for the electrospray ionization source were as follows: capillary temperature, 270°C; spray voltage, 4.0 kV; and capillary voltage, 4.0 V. Both mass spectrometers were operated in a data-dependent scanning mode to MS3. The mass spectrometer was operated in a positive ion mode with data-dependent scanning. The ions were monitored over a full mass range of m/z 100 to 1000. For a full scan, the automatic gain control was set at 5.0 × 108, maximum ion time was 10 ms, and the number of microscans was set at 1. For MSn scanning, the automatic gain control was set at 1.0 × 108, maximum ion time was 100 ms, and the number of microscans was set at 1. For data-dependent scanning, the default charge state was 1, default isolation width was 3.0, and the normalized collision energy was 35.0. The data obtained were analyzed using Xcalibur software (version 1.4; Thermo Fisher Scientific).
Metabolic Stability.
Metabolic stability was assessed for zoniporide and its analogs that showed an AO-catalyzed oxidative metabolite in the metabolite identification studies (described above). Metabolic stability was expressed as in vitro half-life (t1/2) and was determined from the disappearance of the parent during incubation with human S9 fractions. Half-life experiments were performed using an automated procedure on a Beckman Biomek FX system (Beckman Coulter, Inc., Fullerton, CA), and incubations were conducted at 37°C for 120 min in 100 mM potassium phosphate buffer (pH 7.4) containing human liver S9 fractions (protein concentration, 2.0 mg/ml). Reactions were initiated by addition of zoniporide or its analogs (final concentration, 1 μM). The final reaction volume of the incubation mixture was 250 μl. Samples (25-μl aliquots) were removed at 0, 10, 20, 30, 60, and in some cases 120 min and then were quenched with 75 μl of acetonitrile containing 0.1 μM buspirone as an internal standard and centrifuged at 3000 rpm (GH3.8A rotor; Beckman Coulter) for 15 min at room temperature. A 50-μl aliquot of supernatant was transferred to a 96-well plate, mixed with 50 μl of Milli-Q water (Millipore Corporation, Billerica, MA), and analyzed by liquid chromatography-tandem mass spectrometry. All incubations were performed in duplicate.
The peak areas of the parent compound in aliquots terminated at 0 min and were defined as 100%, and the percentage of the parent compound remaining at other times was calculated by comparing peak areas at these times with those at 0 min. The rate constant for the disappearance of the parent compound, k, estimated from the plot of the percentage of the parent compound remaining at each time and the time from which a t1/2 was calculated using eq. 1:

Enzyme Kinetics.
Zoniporide analogs 4 (1–130 μM), 8 (1–25 μM), 9 (1–130 μM), 10 (0.01–13 μM), 14 (1–25 μM), and 15 (1–25 μM) were incubated with pooled human liver S9 (0.1 mg/ml) and EDTA (0.1 mM) in 100 μl of 100 mM phosphate buffer and 10 mM MgCl2 at 37°C for 10 min. Under the incubation conditions chosen, metabolite formations were linear with respect to time of incubation and protein concentration, and substrate depletion was less than 10%. The incubations were commenced with addition of a mixture of liver S9 and buffer that had been prewarmed to 37°C, and the reactions were terminated with the addition of 200 μl of acetonitrile containing 100 nM buspirone (which was used as an internal standard). All compounds were dissolved in a mixture of acetonitrile and DMSO (9:1). The final concentration of organic solvent in the incubation mixture was <1% and the final concentration of DMSO was <0.1%. DMSO had no effect on metabolite formation at a final concentration of less than 0.1% (v/v). The samples were vortexed and centrifuged for 10 min at 3000g and further diluted with water (4-fold dilution). The diluted mixtures were filtered through a Millipore MultiScreen-HA filter plate (0.45 μm), and filtrates were analyzed by liquid chromatography-tandem mass spectrometry to assess the formation of AO-catalyzed oxidative metabolite.
Filtered incubation mixtures were injected (40 μl) onto a 2.0-mm i.d. × 50 mm Zorbax XDB C18 column (5-μm particle size) pre-equilibrated in 10 mM ammonium acetate solution containing 5% acetonitrile at a flow rate of 0.4 ml/min. The HPLC system consisted of two Shimadzu LC-10AD pumps (Shimadzu, Columbia, MD), a CTC PAL autoinjector (CTC Analytics, Carrboro, NC), and a API 4000 triple quadrupole mass spectrometer (MDS Sciex, Concord, ON, Canada) operated in the multiple reaction monitoring mode. The analytes were eluted using a 4.3-min gradient elution program. The initial solvent composition of 5% acetonitrile was changed linearly to 80% in 2.8 min, followed by a gradient increase to 95% in 0.2 min. It was held at 95% for an additional 0.4 min, and then the solvent composition was returned in a single step to the initial conditions for reequilibration over an additional 0.8 min. Oxidative metabolites was detected using a MDS Sciex API 4000 triple quadrupole mass spectrometer equipped with a turbo ion source. The multiple reaction monitoring transitions used were m/z 351 → 278 for 4, m/z 339 → 280 for 8, m/z 341 → 282 for 9, m/z 337 → 278 for 10, 14, and 15, and m/z 386 → 122 for buspirone (internal standard).
The formation rate of the oxidative metabolite at each concentration was expressed as the ratio of the peak area of the oxidative metabolite to the peak area of the internal standard, and this was used with their corresponding substrate concentrations to calculate the kinetic parameters Km (Michaelis-Menten constant). GraphPad Prism (version 5; GraphPad Software, Inc., San Diego, CA) was used to fit the data. The single binding site model (eq. 2) gave the best fit for Km (v, rate of oxidation; S, substrate concentration). Experiments were performed in triplicate for each compound to assess the reproducibility of the Km values.

In Silico Methods.
Computation of cLogD7.4.
The molecular structures of zoniporide and its analogs (2–16) were prepared using ChemSketch software (version 11; ACD Labs, Toronto, ON, Canada). The cLogD7.4 values were then estimated using cLogD calculation module in the ACD software.
Estimation of partial atomic charges and ELUMO.
The molecular structures of 1 and its analogs (2–16) were prepared using ChemDraw molecular editor (suite 12.0; CambridgeSoft Corporation, Cambridge, MA).
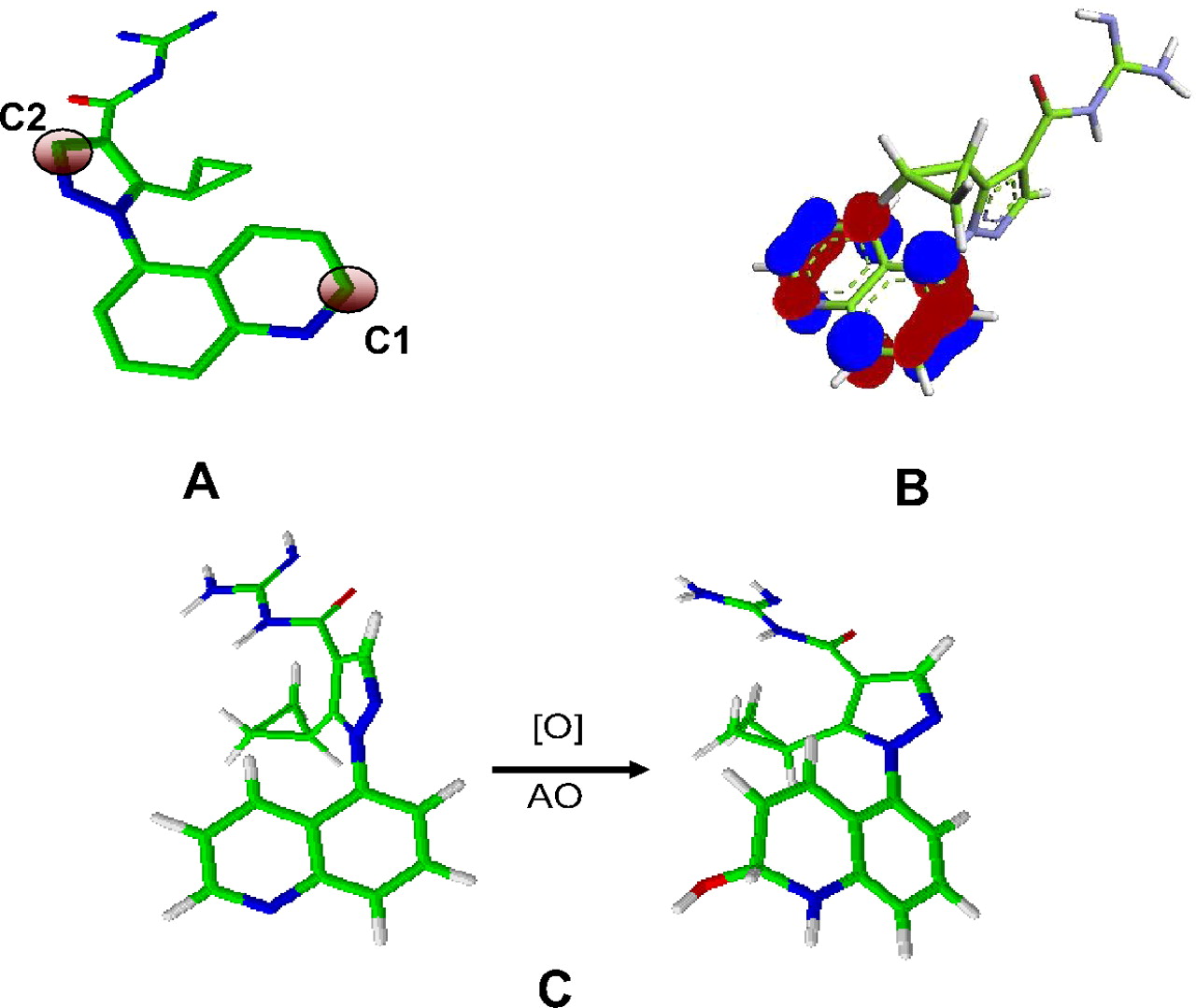
Electrostatic potential charge (ESP) charges on 1) carbon atoms adjacent to the nitrogen atoms in the quinoline ring of 1 to 15 and benzimidazole 16 and 2) carbon atom adjacent to the nitrogen atom in the pyrazole ring (C2) (Fig. 4A) were computed using the Merz-Kollman electrostatic potential surface approach (Torres et al., 2007). The electrostatic potential surfaces were constructed by sampling the points external to the van der Waals radii of atoms as defined in Gaussian 09 (Gaussian, Inc., Wallingford, CT). An increasing positive value of ESP charge signified an increase in electrophilicity at the site of attack and therefore its increased reactivity.

A, electron-deficient carbons in the zoniporide molecule. C1 and C2 are carbon atoms adjacent to nitrogen atoms in the quinoline and pyrazole ring, respectively. B, LUMOs of zoniporide delocalized on the bicyclic heteroaromatic ring. The ELUMO values were computed for these LUMOs to correlate with half-lives. C, formation of a tetrahedral intermediate resulting from AO-mediated oxygenation reaction. The ΔG values computed from these intermediates were correlated with half-lives.
For ELUMO, the LUMOs were first generated for all compounds. Only the ELUMO estimates of LUMOs that were delocalized on the heteroaromatic ring (Fig. 4B) were used for subsequent comparison with t1/2 and to assess its predictive ability. ELUMO values for zoniporide and analogs were generated using the quantum mechanics-based Becke three-parameter Lee-Yang-Parr density functional theory on 6-31G** basis sets and visualized with GaussView 5 (Gaussian, Inc.). An increasing negative value of ELUMO indicated increased electrophilicity and therefore increased reactivity.
Estimation of energies of tetrahedral intermediate formation after AO-mediated oxygenation of heteroaromatic rings.
Tetrahedral intermediate structures were generated by addition of the hydroxyl anion (simulation of MoCo) to the carbon atom adjacent to the nitrogen atom of the quinoline ring in 1 to 15 and the carbon atom between the two nitrogens of the benzimidazole ring of 16 (Fig. 4C). A hydrogen atom was added on the nitrogen atom, resulting in a net addition of water as described by Torres et al. (2007). All structures were optimized by density functional theory at a Becke three-parameter Lee-Yang-Parr level with 6-31G* basis on all constituent atoms as defined in Gaussian 09 (Supplemental Fig. 1) (Becke, 1988). The differences between the energy of the products and the substrates plus the hydroxyl anion yielded relative reaction energies for formation of tetrahedral intermediates (ΔG, kilocalories per mole). A lower ΔG value suggested a favorable formation of the tetrahedral intermediate and hence increased rate of reaction. No transition state search was performed for the reactive intermediate of the modeled transformation, but it was approximated from product stability as suggested by Hammond's postulate. Differences in the zero-point energies between zoniporide and its analogs and its corresponding tetrahedral intermediates were also computed for all analogs to verify whether they are influenced by the rotation or vibration of these intermediates. The differences in the values were <0.2 kcal/mol for most compounds (12 of 16) and <0.5 kcal/mol for all compounds, suggesting their minimal influence on the final energies. The final energies reported include zero-point energy corrections.
Assessment of predictive ability of partial charge, ELUMO, and ΔG.
To assess whether the three parameters could predict the substrate properties of zoniporide analogs, the compounds were first divided into two categories: the compounds that had a half-life within 2-fold of t1/2 of 1 were considered to be similar or better AO substrates (<52 min) and the ones with longer half-life (>52 min) were considered to be weak AO substrates. Thus, eight compounds exhibited better or similar substrate properties, whereas seven compounds were poor substrates when their half-lives were compared with that of 1. A descriptor was considered to be predictive if a compound displaying a higher ESP charge and lower ELUMO or ΔG value (relative to those estimated for 1) had a shorter half-life than zoniporide. The overall success of a descriptor was determined from the ratio of the number of compounds whose t1/2 values were in agreement with values of that descriptor to the total number of compounds that were used in the study (16 compounds).
Docking Studies.
The substrate binding orientations were predicted by molecular mechanics-based docking approaches as described previously (Sun et al., 2009, 2012; Sun and Scott, 2010, 2011). To prepare for docking input files, the density functional theory energetically minimized structures of zoniporide, and its analogs were modified by AutoDockTools (The Scripps Research Institute, La Jolla, CA) with Gasteiger atomic charges assigned and flexible torsions defined. The three-dimensional coordinates of AO homology model published by Dastamalchi and Hamzeh-Mivehrod (2005), which was prepared with the protein preparation wizard within the Maestro program (Maestro User Manual, version 9.0; Schrödinger LLC, Camberley, UK) was used in the docking study. The template structure was further modified with polar hydrogens, Kollman partial charges, and solvation parameters added using AutoDockTools. AutoGrid 4.0 (The Scripps Research Institute) was applied to define the active site space of aldehyde oxidase, which precalculates the grids of van der Waals, hydrogen bonding, and electrostatic, torsional, and solvation interactions between the protein template and zoniporide analogs. Because of the unavailability of the molybdenum ion on its atom type list in AutoGrid, the corresponding parameters such as the van der Waals radii and atomic solvation volume were estimated using those numbers for iron. However, the partial charge calculation was based on molybdenum, as reported previously (Pryde et al., 2010; Linton et al., 2011). Docking of zoniporide and its selected analogs 4, 9, 10, 12, 14, 15, and 16 was performed in an unrestricted manner. The docking process was accomplished on Pfizer's high-performance computing Linux clusters using AutoDock 4.0 (The Scripps Research Institute), which searched globally optimized conformations and orientations for all compounds using a Lamarckian genetic algorithm, a hybrid of genetic algorithms and adaptive local search method, with 100 million evaluations performed for each output binding pose of a total 100 per compound. Docking pose clusters with the lowest binding energies in which the site of metabolism was in close proximity of the hydroxyl (<5.0 Å) group of molybdenum pyranopterin catalytic center were chosen for analysis and visualized using PyMOL (Schrödinger, LLC). The docking scores are provided in the Supplemental Table 1.
Results
Metabolite Identification.
Table 1 depicts the chemical structures of zoniporide analogs (2–18) that were used in the study. Structural variants of zoniporide with modifications of the acylguanidine moiety (Fig. 5A), the cyclopropyl group (Fig. 5B) on the pyrazole ring, and the quinoline moiety (Fig. 5C) were selected from the Pfizer database to probe the structure-metabolism relationship. Because the metabolic AO liability of selected zoniporide analogs was unknown, all compounds were first incubated at 10 μM concentrations with the human S9 fraction in the absence of cofactors (such as NADPH) to assess their potential to turnover by AO. Compounds that were metabolized displayed an additional peak in the total ion chromatograms upon mass spectrometric analysis of the incubation mixture. The peak had a molecular ion (MH+) that was 16 Da greater than the molecular ion of 1, suggesting addition of an oxygen atom (Table 1). The relationship of the observed metabolites with the parent compound was confirmed from their respective mass spectral fragment ions. Because the fragmentation pattern of the metabolites was similar to that of M1, it was assumed that the position adjacent to the nitrogen atom in the quinoline ring was the site of oxidation for all compounds. In addition, previous reports on AO metabolism have indicated that oxidation takes place at carbon atoms adjacent to nitrogen atoms in the heteroaromatic rings (Torres et al., 2007). No further attempts were made to confirm the structures of the oxidative metabolites. The site of metabolism and inhibition of formation of the metabolite upon addition of raloxifene (Obach, 2004) to the incubation mixture further confirmed that the metabolite was catalyzed by AO.
Structures of zoniporide analogs and their respective oxidative metabolites and estimated half-lives after incubation with human S9 fractions
Incubations to identify the metabolite of zoniporide analog were performed at substrate concentrations of 10 μM, and the rates of AO-catalyzed metabolism were determined at concentrations of 1 μM. Half-lives were determined with substrate concentrations of 1 μM.

Modifications probed at three sites in the zoniporide molecule: acylguanidine moiety (A), cyclopropyl group (B), and quinoline ring (C).
Metabolic Stability Determination.
Substrate depletion experiments were conducted, and half-lives were estimated to determine the metabolic stability of the compounds against AO for those that showed a 2-oxo metabolite in the metabolite identification studies. To get a reliable estimate, these studies were conducted at a substrate concentration of 1 μM. Replacement of the acylguanidine moiety with either a carboxylic acid (2) or an unsubstituted amide (3) resulted in analogs that were more stable toward metabolism by AO (t1/2 = 110 min or greater) compared with zoniporide (1), which had a t1/2 of 26 min (Table 1). In contrast, the diethylamide analog 4 was rapidly metabolized by AO (t1/2 = 37 min) in the substrate depletion assay. The minimal change in the half-life of 4 (compared with 1) suggested that the diethylamide moiety was also tolerated by the active site of AO.
Analogs, in which the cyclopropyl group in 1 was substituted with a methyl (5), n-propyl (6) or n-butyl (7), isopropyl (8), or methoxymethyl (9) group, were selected from the sample bank to assess the effect of the cyclopropyl group on AO-mediated oxidation of zoniporide. Whereas the half-life of 5 was similar to that of 1 (t1/2 of 33 for 5 versus 26 for 1), replacement of the cyclopropyl group with n-propyl or n-butyl resulted in a modest increase in metabolic stability (41 min for 6 and 48 min for 7) relative to that for 1 (Table 1). In contrast to n-alkyl analogs, which showed a trend of decreasing AO activity with increasing chain length, incorporation of an isopropyl group (8) resulted in an analog that was a better substrate for AO (t1/2 of 8 = 15 min), especially compared with the n-propyl congener 6 (t1/2 = 41 min). Of interest, the methoxymethyl group (9) in which the methylene group of 6 was replaced by an oxygen atom resulted in a significant increase in metabolic stability with a t1/2 value of 122 min (Table 1). To understand the importance of the position of the cyclopropyl group in 1, the metabolic stability of 10, a zoniporide congener with the cyclopropyl group at the 3-position was also evaluated. In contrast to 1, 10 was metabolized ∼3 times faster by AO (t1/2 of 10 = 3.1 min) signifying that substitution at the 3-position enhanced the AO substrate property of the molecule (Table 1).
The effect of modification of the quinoline ring on AO-mediated catalysis of 1 was evaluated using three types of zoniporide analogs. As shown in Table 1, incorporation of a substituent at the 6-position of the quinoline ring (11–13) produced compounds that showed greater resistance to AO-mediated oxidative metabolism as judged by an increase in their half-lives relative to that of 1. Likewise, replacement of the quinoline ring with benzimidazole, indole, and indazole ring (16–18) produced compounds that were either devoid of AO activity completely (no turnover was observed in the preliminary analysis using 10 μM substrate concentrations) or were poor substrates for AO-mediated oxidation (16). In contrast, a change in the position of the pyrazolyl-acylguanidine moiety to the 4- or 6-position (14 and 15) resulted in an enhanced rate of AO-mediated oxidation to the corresponding 2-oxo metabolites (t1/2 of 14 and 15 were 14 and 5 min, respectively).
Substrate Saturation Experiments.
Substrate saturation studies were performed with selected compounds (8, 9, 10, 14, and 15) to get a better understanding of their binding properties. These compounds were selected because they showed rapid AO-mediated clearance compared with zoniporide in the substrate depletion assay. Although the Km for formation of the 2-oxo metabolite of each compound could be computed using enzyme kinetics experiments, estimation of the Vmax was difficult because of the lack of synthetic standards of metabolites.
Analysis of the reaction velocity for 2-oxo metabolite formation versus the substrate concentration curve revealed classic Michaelis-Menten kinetics for all compounds (Supplemental Fig. 2), with apparent Km estimates of 2.2, 0.4, 1.8, and 1.2 μM, for 8, 10, 14, and 15, respectively (Table 2). Whereas compounds 10, 14, and 15 showed 2- to 8-fold higher binding affinity, the affinity of 8 was similar (Km 1.4-fold lower) to that of 1 [(Km 3.4 μM; (Reproduced from the original article Drug Metab Dispos 38:641–564)]. Substrate saturation kinetic experiments were also conducted to evaluate the Km estimate of N,N-diethylamide analog 4 because replacement of the positively charged acylguanidine moiety with a neutral N,N-diethylamide analog exhibited minimal changes in the half-lives of 1 versus 4. Like the other analogs, oxidation of 4 by AO also displayed normal Michaelis-Menten kinetics (Supplemental Fig. 2); however, the Km for the formation of the 2-oxo-metabolite of 4 was ∼5-fold higher (16 μM) than that of 1 (Table 2). Even though the methoxymethyl analog 9 was a weak substrate for AO (t1/2 = 120 min), the compound was subjected to Michaelis-Menten kinetics to assess its affinity. Analysis of the rate of formation of the 2-oxo metabolite versus the concentration curve yielded an estimated Km value of 9.0 μM, which was three times higher than that for 1 (Table 2) (Supplemental Fig. 2).
Km values of selected zoniporide analogs
The Km values are means (S.D.) of three different experiments. All compounds exhibited classic Michaelis-Menten kinetics (Supplemental Fig. 2). The Vmax values could not be estimated because of the lack of synthetic standards of metabolites. The velocity at various substrate concentrations was expressed as a peak area ratio of 2-oxo metabolite to the internal standard, buspirone, per minute per milligram of protein.
Molecular Modeling Studies.
In an attempt to rationalize the differences in the AO-mediated metabolism of zoniporide and its congeners, 1 was docked into the human AO homology model originally constructed by Dastmalchi and Hamzeh-Mivehrod (2005). Molecular docking approaches have been commonly used to elucidate important interactions between substrates and active site residues of drug-metabolizing enzymes (Sun and Scott, 2010). This invaluable information can be used for chemical design and redesign to enhance pharmacokinetic profiles and reduce toxicities. To perform structure-based drug metabolism prediction, experimentally determined (such as use of X-ray crystallography) structures of drug-metabolizing enzymes are required. In the absence of the crystal structures, homology models of the enzymes can be used to evaluate the binding modes and predict interactions of the functional groups in the molecule with the key amino acid residues (de Groot, 2006; Afzelius et al., 2007; Sun and Scott, 2010). Because the crystal structure of human AO was not available, the three-dimensional homology model of human AO, which was constructed using the crystal structure of bovine xanthine dehydrogenase (XDH) as a template and guided by multiple alignments using bovine, rabbit, and rat sequences of AO as well as the sequence of chicken XDH, was used. The structure was further refined by molecular dynamics simulation and energy minimization (Pryde et al., 2010; Linton et al., 2011).
After zoniporide modeling, the pose cluster with the lowest binding energy in which the site of metabolism was in close proximity to the hydroxyl group (<5.0 Å) of the molybdenum pyranopterin catalytic center was chosen for analysis. For AO-mediated oxidative metabolism to take place, the site of metabolism should be in proximity to the cofactor. It was assumed that the electron transfer occurred only when the substrates bind closely to the MoCo cofactor, which was thus used as one of the criteria to analyze the clustered docking poses. It is important to note that the energy (docking score) of this pose was quite similar to that of other poses as seen from the docking scores (Supplemental Table 1). The assumption was aligned with docking of various substrates reported previously that applied docking constraints of 2.5 to 3.5 Å between atoms from substrate known to be involved in the oxidation (atom that will be oxidized) and the molybdenum atom from the protein (Dastmalchi and Hamzeh-Mivehrod, 2005; Pryde et al., 2010; Linton et al., 2011). Zoniporide appeared to adopt an orientation in which the site of metabolism in the molecule was 2.8 Å away from proximity to the hydroxyl group of the molybdenum cofactor (2.8 Å is a reasonable distance to form a tetrahedral intermediate) (Fig. 6). The distance was consistent with that observed after docking of other substrates into the homology model. These docking poses predicted the sites of metabolism to be between 2.1 and 3.1 Å and have been in good agreement with the experimental results (Pryde et al., 2010; Linton et al., 2011). Several other key molecular interactions between zoniporide and AO active site residues were also identified in the docking studies. These included the quinoline ring π-π stacking with Phe923, the electrostatic interaction between the guanidine moiety and Glu882, and some hydrophobic interactions of the cyclopropyl moiety with the active site residues Val811, Leu1018, and Ile1085. These interactions together juxtaposed the α-carbon atom for oxygenation by MoCo. The primary interactions with Glu882 and Phe923 were also consistent with those observed by Dastamalchi and Hamzeh-Mivehrod (2005) when the phthalazine or the quinazoline derivatives were docked in the active site.

Docking of zoniporide in the homology model of human AO. Human AO was modeled using the crystal structure of bovine XDH as a template. The construction of the homology model was guided by multiple alignments using bovine, rabbit, and rat sequences of AO as well as the sequence of chicken XDH, and the structure then refined by molecular dynamics simulation and energy minimization (Dastmalchi and Hamzeh-Mivehrod, 2005).
Relationship between Half-Lives and Structural Properties of Zoniporide and Its Analogs.
In an effort to discern the differences in metabolism of 1 and its structural analogs, the relationship of their half-lives with their molecular (cLogD7.4) and other structural properties was analyzed. Only the compounds that showed turnover in the human S9 incubation were used in this analysis. Hence, the molecular and electronic properties of zoniporide (1) and analogs 2 to 16 were computed (Table 3), whereas the indole (17) and indazole (18) analogs were omitted from this analysis because of their lack of AO activity (Table 1).
Half-lives and physicochemical (cLogD7.4) and structural properties (Merz-Kollman partial atomic charge, ESP; ELUMO, and ΔG) of zoniporide and its analogs
The procedure used in the computation of these parameters is found under Materials and Methods.
Lipophilicity of compounds is an important determinant for binding to enzymes and their metabolic clearance. In general, there exists good correlation between P450-mediated turnover and high lipophilicity as measured by cLogP or cLogD (van de Waterbeemd et al., 2001; Lewis et al., 2004; Smith, 2011). Therefore, one can predict the susceptibility of a compound to metabolism by estimating either LogP or LogD of compounds. Superficially, there seemed to be a relationship between the lipophilicity and half-life when the cLogD values of all zoniporide analogs and their AO metabolic stability were compared (Table 3). The compounds with a negative (or lower) cLogD (2 and 9) were stable whereas the most lipophilic compound (4) showed susceptibility to AO-catalyzed metabolism. However, a closer look at the values suggested that there was no apparent relationship between the half-life and lipophilicity (Table 3). This finding was consistent with the results from previous studies and published reviews that have demonstrated no apparent association of AO activity with lipophilicity (Pryde et al., 2010).
Further rationalization of the differences in metabolic stability of 1 and its analogs was attempted by comparing their electronic properties (partial atomic charge at the carbon atom and LUMO energies) with half-lives of compounds 1 to 16. The catalytic mechanism for AO or xanthine oxidase oxidation generally involves a nucleophilic attack of a water molecule on the electron-deficient carbon adjacent to the nitrogen atom in the heteroaromatic ring (Hille, 2005; Torres et al., 2007). Hence, reactivity of this carbon atom or overall electrophilicity of the heteroaromatic ring of the molecule could potentially influence AO-mediated oxidation. The ESP charge at the carbon atom adjacent to the nitrogen atom in the heteroaromatic ring and ELUMO are two descriptors that can predict electrophilicity at the site of attack and therefore possibly, the rate of AO metabolism (Chan et al., 2008; Sharma et al., 2011). An increasing positive value of ESP charges signifies an increase in electrophilicity of the atom and therefore its increased reactivity. Likewise, the molecule become more electrophilic as ELUMO becomes more negative (Cronin et al., 2001; Chan et al., 2008).
Because zoniporide and its analogs have two likely oxidation sites (Fig. 4A), the ESP charge of the two carbon atoms 1) adjacent to the quinoline or benzimidazole ring (C1) and 2) adjacent to the nitrogen atom in the pyrazole ring (C2), were estimated (Table 3). The computed values suggested that the carbon atom adjacent to the nitrogen atom in the quinoline ring (or benzimidazole ring of 16) was more electron-deficient than the one in the pyrazole ring (Table 3) and therefore was the likely site of oxidation. This was consistent with the site of AO oxidation that was identified after incubation with human S9 fractions and supported the previously proposed theory that a potential predictor of the site of AO metabolism for a compound is the most positively charged carbon atom in the molecule (Torres et al., 2007). The relationship between the partial atomic charge and turnover by AO was also assessed by comparing intensities of the atomic charge (ESP) with half-lives. The estimated ESP charge for most analogs ranged from 0.365 to 0.409. Compounds 8, 10, and 15, which showed rapid metabolism in human S9 fractions, displayed an increased partial positive charge of 0.387, 0.409, and 0.404, respectively, relative to that of 1 (0.374) (Table 3). Likewise, 2 and 16, which exhibited a low ESP charge relative to that of 1 (0.333 and 0.275, respectively), were poor substrates for AO. However, 14, which also exhibited better substrate properties had a low partial charge of 0.365 (relative to that of 1). Likewise, 9 and 11–13 showed higher ESP charge values despite the poor substrate properties exhibited by these compounds. Previous reports have also noted a lack of reliability of partial charges in the prediction of the site of metabolism (Torres et al., 2007). Although the site of metabolism could be predicted with great accuracy using partial charges in the present study, the success of the ESP charge to predict whether the compound was a good substrate for AO was only 60% (9 of 15 zoniporide analogs).
For comparing the half-life of zoniporide congeners with ELUMO values, lowest unoccupied molecular orbitals were first generated for all compounds, and the ELUMO values of the orbitals that were delocalized on the heteroaromatic ring (Fig. 4B) were used for subsequent comparison with t1/2 values. Although the benzimidazole (16) showed a higher ELUMO value of −0.037 au, all other quinoline-containing compounds had ELUMO values ranging from −0.054 to −0.065 au The relationship of half-life with ELUMO energies (Table 3) also showed a similar trend compared with the half-lives as was observed in the case of ESP charge and half-life comparison. By and large, there was only a 60% agreement between the computed ESP charges or LUMO energies and t1/2 values.
As a part of assessing the relationship between electronic and substrate properties of zoniporide and its structural variants, the formation energies of the putative tetrahedral intermediates (ΔG) resulting from AO-mediated oxygenation of the C1 carbon were also computed and compared with their respective half-lives. A recent report by Torres et al. (2007) has shown that this simple qualitative method can predict the site of hydroxylation on the heteroaromatic ring after biotransformation by AO. Given that these relative energies of the tetrahedral intermediate formation are proportional to the activation enthalpies of the AO oxidation, these can serve as an indicator for AO reactivity. Thus, the lower the ΔG values are, the higher the possibility of a favorable reaction. The energies of the likely tetrahedral intermediates (Fig. 4C) resulting from oxygenation reaction by AO are presented in Table 3. In nearly all instances, the compounds that were metabolized at rates that were equivalent to or faster than 1 formed tetrahedral intermediates with a ΔG value ≤20.11 kcal/mol (ΔG value for 1) and compounds that were resistant to AO oxidation displayed ΔG values significantly greater than 1 (Table 3). The exception was the methoxymethyl (9) and the benzimidazole analog (16). Even though the ΔG value of formation of the tetrahedral intermediate of 9 and 16 was lower than 1 (∼19.96 and 19.84 kcal/mol, respectively), these compounds were poor substrates for AO. Overall, in addition to predicting the site of metabolism reported previously, the tetrahedral intermediate formation energies were able to guess whether the analogs were better or weak AO substrates relative to 1 for 13 of 15 compounds (∼87%). It is important to note that even though the low ΔG values could predict whether the compound was a good substrate compared with 1, there was no relationship between the actual rate of metabolism and the estimated value. For instance, the estimated ΔG for 14, a weaker AO substrate (t1/2 = 14 min) yielded an energy value of 13.42 kcal/mol compared with that of 15 (19.31 kcal/mol), which had a 5 min half-life (Table 3).
Discussion
This study explored the effect of structural changes in zoniporide on its AO-mediated metabolism. The results presented herein indicated that subtle changes in the structure of 1 modified its metabolism, in some cases significantly. Because the relative energies of the likely tetrahedral intermediates showed reasonable confidence in predicting AO properties among the different in silico tools, these were used to discern the differences along with molecular docking.
The loss of AO activity of 2 and 3 was attributed to the weak interactions of the carboxy or the amide groups with Glu882 residue (observed in case of 1) (Fig. 6) and suggested that the guanidine moiety was required for AO-mediated metabolism. High ΔG values for 2 and 3 relative to that for 1, indicated a higher activation threshold for the reaction and correlated well with their poor substrate properties (Table 3). In contrast, comparable half-lives of diethylamide analog (4) and 1 (Table 1) implied that the two ethyl groups of 4 compensated for the electrostatic interactions between the acylguanidine of 1 and Glu882 probably via interactions with hydrophobic residues in the AO active site (Fig. 7A). However, a 5-fold lower affinity of 4 compared with that of 1 (Table 2) emphasized the importance of electrostatic interactions between Glu882 and the guanidine moiety. Good AO activity of 4 was also supported by its favorable tetrahedral intermediate formation energy (Table 3).

Docking of the (A) diethylamide analog (4), (B) methoxymethyl analog (9), (C) 3-cyclopropyl analog (10), (D) 6-hydroxyquinoline analog 12, (E) benzimidazole analog (16) of zoniporide, and positional isomers 14 (F) and 15 (G) in the AO homology model.
Substitution of a cyclopropyl group in 1 with small alkyl groups (5–7) did not affect the rates to a significant extent (Table 1), suggesting that replacement of a cyclopropyl group with other aliphatic side chains was tolerated and that these groups most likely occupied the same hydrophobic pocket that was occupied by the cyclopropyl group. The energy for their respective tetrahedral intermediates (Table 3) also corroborated well with these results. A trend of decreasing half-life with increasing alkyl chain length also suggested that the size/length of the alkyl group was probably important in modulation of AO activity. Replacement of the cyclopropyl with an isopropyl group (8) enhanced the rate of metabolism slightly. Although the reason for this is unclear, a lower ΔG (18.91 kcal/mol) compared with that for 1 was one possible explanation for this increased AO activity, especially because its binding affinity was similar to that of 1 (Km = 2.2 μM).
Weak AO activity of the methoxymethyl analog (9) despite the low ΔG value (19.96 kcal/mol) was probably attributed to its weak binding to AO (Km = 9.0 μM). Docking studies of 9 showed that its binding pose in the active site was different compared with that of 1 (Fig. 7B). The guanidine moiety oriented toward the substrate access entrance region, possibly causing energetically unfavorable interactions with the two phenylalanine residues, Phe655 and Phe885, and shifted the site of metabolism farther away from the molybdenum cofactor (4.0 Å). Thus, disruption of the hydrophobic interactions observed with 1 and suboptimal distance between the MoCo and the site of oxygenation in addition was possibly a cause for the low activity of 9. These results also emphasized the importance of orientation of the molecule in the active site, which should be considered along with computational analysis when metabolism is predicted.
The enhanced activity of 10, a 3-cyclopropyl isomer, was at least in part due to its ∼10-fold higher binding affinity in the AO active site compared with that of 1 (Table 2). Docking results indicated that the cyclopropyl group of 10 binds to residues Phe655 and Phe885 in the active site via strong hydrophobic interactions in addition to π-π stacking and electrostatic interactions (Fig. 7C). This juxtaposed the site of metabolism in proximity to MoCo at approximately 3.5 Å. Moreover, its lower energy for tetrahedral intermediate formation (Table 3) suggested a thermochemically favorable reaction (ΔG = 20.16 kcal/mol) with MoCo and supported its excellent substrate property.
For 2 and 3, any modifications of the quinoline ring also resulted in compounds that were resistant to AO-catalyzed metabolism. For 6-substituted analogs (11–13), addition of the electron-donating groups possibly perturbed the electronics of the quinoline ring and therefore their AO activity. Incorporation of these substituents increased the ΔG value of these analogs relative to that of 1, suggesting a higher activation threshold for AO metabolism. In addition, docking yielded binding modes in which these substituents were oriented toward the backbone region of the active site residues and resulted in sterically unfavorable interactions (Fig. 7D). Thus, thermochemical properties and steric effects together were a plausible cause for low AO activity of 11, 12, and 13.
The loss of AO activity after replacement of the quinoline ring with indole (17) and indazole (18) rings was attributed to the differences in the electronic properties of these rings. Although the lone electron pair of the quinoline nitrogen is not involved in the π-electron sextet of the aromatic ring (making this ring π-deficient), it is part of the aromatic sextet in the case of the indole and indazole rings, which makes these rings π-excessive. Consequently, it reduces the reactivity of the neighboring carbon atom and therefore its potential to undergo AO-mediated oxidation. Likewise, the weak activity of the benzimidazole analog (16) is attributed to the π-excessive nature of the benzimidazole rings. Reduced reactivity of the neighboring carbon atom in 16 was supported by ELUMO and the ESP charge at the site of attack, which were lower than those observed for analogs containing a quinoline ring (Table 3). However, because computation of ΔG yielded a value that was almost equivalent to that of 1 (19.84 kcal/mol), 16 was docked into the AO homology model (Fig. 7E). Although the orientation of 16 was similar to that of 1 in terms of π-π stacking with Phe923, the carbon undergoing hydroxylation was positioned relatively farther from the hydroxyl group of the molybdenum cofactor. In addition, the acylguanidine moiety oriented at a distance of ∼4.1 Å to Glu882, causing the electrostatic interactions to be weaker. The weak electrostatic interactions and the distal positioning of the site of attack were probably responsible for the weak activity of 16.
Of interest, positional isomers of 1, 14, and 15 displayed better substrate properties for AO with half-lives of 14 and 5 min, respectively. Not only did the energetics predict 14 and 15 to be better AO substrates, but both compounds also exhibited tighter binding with Km values that were 2- to 3-fold lower than those of 1 (Table 2). The orientation of 14 and 15 into the homology model suggested that the two compounds interacted with the active residues quite differently. In the case of 14 (Fig. 7F), the distance between the acylguanidine moiety and Glu882 residue was decreased relative to that of 1 (2.6 Å). Conversely, the guanidine group of 15 occupied a space outside the binding pocket (Fig. 7G). Despite this, the site of metabolism for both compounds was in proximity to MoCo (3.0–3.3 Å). Furthermore, in both cases, an additional π-π interaction between the pyrazole ring and Phe885 was observed, which was absent in all other analogs including 1. Thus, favorable thermochemical energies of 14 and 15 coupled with their orientation that juxtaposed the site of metabolism near to MoCo probably led to their better substrate properties. Overall, these results also reiterated the earlier observations that spatial arrangement of the molecule in the active site plays an important role in metabolism.
In conclusion, this study not only provided insights into the structural features responsible for AO metabolism of zoniporide but also assisted in evaluating different in silico tools for predicting AO liability. Structure-metabolism relationship studies suggested that the guanidine moiety and the quinoline ring were prerequisites for AO-catalyzed metabolism of 1 to M1 and their modification led to a drop in activity. This finding was supported by docking studies in the human AO homology model that revealed electrostatic and π-π stacking interactions with these two groups. Modeling studies with other analogs led to the inference that other poses were as productive as long as the site of attack was in close proximity to MoCo (2.8–3.5 Å). Although this was in agreement with the previously reported results, more structure-activity relationship studies are required to generalize these potential interactions and distances. In addition, given that all docking studies were performed using a homology model constructed from a XDH crystal structure, additional refinement of the homology model may help in assessing precise interactions with residues in the active site. Better yet, a crystal structure of human AO will be very valuable to understand these interactions.
The results also showed that ΔG values of likely tetrahedral intermediates formed during AO-mediated oxidation were not only better predictors of the site of metabolism as reported earlier but also had the potential to predict the AO substrate properties of analogs, in particular when the energy values were compared with those of the lead candidate. However, the effect of structural variation on AO activity could be rationalized using a combination of energy calculations and docking studies. This combination method can be incorporated into the strategy for mitigating AO liabilities observed in the lead candidate or studying the structure-metabolism relationships of other AO substrates.
Authorship Contributions
Participated in research design: Dalvie, Sun, Xiang, Hu, and Kang.
Conducted experiments: Dalvie, Sun, Xiang, Hu, and Jiang.
Contributed new reagents or analytic tools: Dalvie, Sun, Jiang, and Hu.
Performed data analysis: Dalvie, Sun, Xiang, and Hu.
Wrote or contributed to the writing of the manuscript: Dalvie, Sun, Xiang, and Kang.
Footnotes
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.ABBREVIATIONS:
- AO
- aldehyde oxidase
- LUMO
- lowest unoccupied molecular orbitals
- ELUMO
- energy of lowest unoccupied molecular orbitals
- DMSO
- dimethyl sulfoxide
- HPLC
- high-performance liquid chromatography
- MS
- mass spectrometry
- ESP
- electrostatic potential
- ΔG
- energy for the formation of tetrahedral intermediate in the oxygenation of substrate
- MoCo
- molybdenum pyranopterin cofactor moiety
- XDH
- xanthine dehydrogenase
- au
- atomic units.

- Received March 22, 2012.
- Accepted May 15, 2012.
- Copyright © 2012 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}