


Abstract
Metabolism-dependent inhibition (MDI) of cytochrome P450 (P450) enzymes has the potential to cause clinically relevant drug-drug interactions. In the case of several alkylamine drugs, MDI of P450 involves formation of a metabolite that binds quasi-irreversibly to the ferrous heme iron to form a metabolic intermediate (MI) complex. The specific metabolites coordinately bound to ferrous iron and the pathways leading to MI complex formation are the subject of debate. We describe an approach combining heme iron oxidation with potassium ferricyanide and metabolite profiling to probe the mechanism of MI complex-based CYP3A4 inactivation by the secondary alkylamine drug lapatinib. Ten metabolites formed from lapatinib by CYP3A4-mediated heteroatom dealkylation, C-hydroxylation, N-oxygenation with or without further oxidation, or a combination thereof, were detected by accurate mass spectrometry. The abundance of one metabolite, the N-dealkylated nitroso/oxime lapatinib metabolite (M9), correlated directly with the prevalence or the disruption of the MI complex with CYP3A4. Nitroso/oxime metabolite formation from secondary alkylamines has been proposed to occur through two possible pathways: (1) sequential N-dealkylation, N-hydroxylation, and dehydrogenation (primary hydroxylamine pathway) or (2) N-hydroxylation with dehydrogenation to yield a nitrone followed by N-dealkylation (secondary hydroxylamine pathway). All intermediates for the secondary hydroxylamine pathway were detected but the primary N-hydroxylamine intermediate of the primary hydroxylamine pathway was not. Our findings support the mechanism of lapatinib CYP3A4 inactivation as MI complex formation with the nitroso metabolite formed through the secondary hydroxylamine and nitrone pathway, rather than by N-dealkylation to the primary amine followed by N-hydroxylation and dehydrogenation as is usually assumed.
Introduction
Inhibition of the cytochrome P450 (P450) drug-metabolizing enzymes is a major cause of drug-drug interactions (DDIs) (Grimm et al., 2009; Nettleton and Einolf, 2011). It has the potential to result in black box warnings for approved drugs or denial of regulatory approval of drugs in development. Clinically relevant P450 inhibition can be caused by the drug itself (direct inhibition) or by a metabolite (metabolism-dependent inhibition, MDI) (Nettleton and Einolf, 2011). MDI can be divided into three categories: reversible inhibition (where a metabolite is a potent direct inhibitor), irreversible inhibition (where a metabolite binds covalently to the heme or apoprotein and inactivates the enzyme), and quasi-irreversible inhibition [where the inhibitory metabolite forms a stable metabolic intermediate (MI) complex through a coordinate bond with the ferrous heme iron] (Orr et al., 2012). In some cases, MI complex-based inactivation can be reversed by oxidation of the ferrous heme iron to the ferric state with ferricyanide or a similar oxidizing agent (Orr et al., 2012). Reversibility of MDI by ferricyanide is often used to differentiate irreversible from quasi-irreversible enzyme inactivation. In addition, MI complexes are commonly associated with a characteristic Soret peak in the visible spectrum at ∼455 nm (Orr et al., 2012).
Quasi-irreversible inhibition has been described for numerous alkylamine-containing compounds and for compounds containing a benzodioxyphenyl (phenylmethylenedioxy) structural motif (Orr et al., 2012). MI complex formation has been reported with alkylamines from a variety of drug classes, including macrolide antibiotics, antidepressants (both selective serotonin reuptake inhibitors and tricyclics), antihistamines, and calcium-channel blockers, and with several cytochrome P450 enzymes (McNeil and Murray, 1996; Trieu and Murray, 2000; Murray and Murray, 2003; Fontana et al., 2005; Polasek and Miners, 2008). In the case of alkylamines, the ∼455-nm absorbing complex is generally assumed to involve coordinate binding of a nitroso metabolite to the ferrous heme iron (similar to the carbon monoxide complex that absorbs maximally at ∼450 nm), although direct evidence for the formation of a nitroso metabolite is often lacking. It is also generally assumed that formation of the nitroso metabolite from a tertiary or secondary alkylamine involves N-dealkylation to the primary amine followed by N-hydroxylation and then dehydrogenation to the nitroso metabolite.
The secondary alkylamine drug lapatinib, administered orally for the treatment of advanced or metastatic breast cancer, is a tyrosine kinase inhibitor of both epidermal growth factor receptor and human epidermal growth factor receptor-2 (Takakusa et al., 2011). There are several reports linking lapatinib use with drug-induced liver injury (Gomez et al., 2008; Peroukides et al., 2011; Spraggs et al., 2012), and lapatinib carries a black box warning. The metabolism of lapatinib in vivo involves both N- and O-dealkylation by CYP3A4, CYP3A5, and to a lesser extent, CYP2C19 and CYP2C8 (Castellino et al., 2012). Lapatinib was recently identified as an irreversible inhibitor of CYP3A5 (Chan et al., 2012) and a quasi-irreversible inhibitor of CYP3A4; the latter is associated with the formation of a ∼455-nm absorbing MI complex (Takakusa et al., 2011). In addition, several stable N-oxygenation metabolites of lapatinib have been reported both in vitro (Takakusa et al., 2011) and in vivo (Castellino et al., 2012). These findings suggest that lapatinib is converted to a nitroso metabolite that complexes with ferrous CYP3A4 (but not CYP3A5) and that the intervening metabolites might be sufficiently stable to provide information on the metabolic pathway leading to nitroso formation.
In the present study, an ultracentrifugation-based reversibility assay (Parkinson et al., 2011) coupled with heme iron oxidation by potassium ferricyanide was combined with metabolite profiling of lapatinib and stable isotope-labeled lapatinib (Fig. 1) in human liver microsomes (HLM) and recombinant CYP3A4. Liquid chromatography (LC) with accurate tandem mass spectrometry (MS/MS) was used to identify the metabolite complexed with ferrous CYP3A4 and elucidate the pathway leading to MI complex formation. Analogous experiments with CYP3A5, associated with MDI but not MI complex formation from lapatinib (Takakusa et al., 2011; Chan et al., 2012), were included to facilitate elucidation of the mechanism of quasi-irreversible inhibition of CYP3A4.

Chemical structures of unlabeled and stable isotope-labeled lapatinib.
Materials and Methods
Materials.
Lapatinib and 13C2,15N-labeled lapatinib were purchased from Toronto Research Chemicals (Toronto, ON, Canada). 2H4-Lapatinib was purchased from TLC Pharmachem (Vaughan, ON, Canada). Pooled HLMs (mixed sex; n = 200) were prepared and characterized in-house. Potassium ferricyanide was purchased from Sigma-Aldrich (St. Louis, MO). Recombinant (high-reductase) CYP3A4 + b5 and CYP3A5 + b5 were purchased from Cypex (Dundee, Scotland) or BD Gentest (Woburn, MA). The sources of all other reagents have been described elsewhere (Paris et al., 2009; Parkinson et al., 2011).
Incubation Procedures.
Lapatinib or stable isotope-labeled lapatinib (50 μM) was incubated for 30 minutes at 37°C in 100 mM potassium phosphate buffer (pH 7.4) with HLM (1 mg/ml; 4-ml incubation volume), recombinant CYP3A4 + b5, or CYP3A5 + b5 (100 pmol/ml; 4-ml incubation volume). An aliquot of 8 μl of lapatinib (25 mM) was added to the incubation in dimethylsulfoxide. Controls were incubated with the same volume of solvent but no substrate. Reactions were initiated by the addition of reduced NADPH (1 mM final concentration).
MDI Reversibility Assessments.
After the incubation described above, one set of samples was reserved for enzyme activity measurements (as described below). The remaining samples were treated with potassium ferricyanide (final concentration, 2 mM) or water for the control samples for 10 minutes at 37°C to dissociate the MI complex. The microsomes or recombinant enzymes were re-isolated by ultracentrifugation (100,000g, 4°C, 60 minutes), after which the supernatant fraction was decanted to remove excess substrate and noncomplexed metabolite(s). The protein pellets were rinsed with wash buffer (150 mM potassium chloride and 10 mM EDTA, pH 7.4) and resuspended in 400 μl of 250 mM sucrose. Protein concentration was determined using the Pierce BCA Protein Assay (Pierce Chemical, Rockford, IL). Residual CYP3A4 and CYP3A5 activity toward midazolam (30 μM; 37°C; 5 minutes) was measured at 0.05 mg/ml by LC/MS/MS as previously described for midazolam 1′-hydroxylation (Parkinson et al., 2011), with minor method modifications to enable detection of both 1′- and 4-hydroxymidazolam.
Chemical Oxidation with Metabolite Profiling Experiments.
The overall experimental approach is summarized in Fig. 2. The microsomes or recombinant enzymes were re-isolated by ultracentrifugation (100,000g, 4°C, 60 minutes), after which the supernatant fraction was decanted to remove excess substrate and noncomplexed metabolite. The protein pellets were rinsed with wash buffer (150 mM potassium chloride and 10 mM EDTA, pH 7.4) and resuspended in 500 μl of 100 mM potassium phosphate buffer (pH 7.4). Samples were split and treated with potassium ferricyanide (final concentration, 2 mM) or water for the control samples for 10 minutes at 37°C to dissociate the MI complex. At 10 minutes, samples were quenched with an equal volume of acetonitrile, and samples were analyzed for metabolite profiling by high-resolution liquid chromatography with accurate mass spectrometry.

Sample preparation procedure combining the reversibility assay (ultracentrifugation with and without potassium ferricyanide) with metabolite profiling and characterization.
Metabolite Profiling by Accurate Mass Spectrometry.
A Waters Ultra-performance Acquity LC coupled to a Waters Synapt G2 High Definition Mass Spectrometry system quadrupole time-of-flight (qTOF) mass spectrometer (Waters, Milford, MA) was operated in a positive, resolution elevated energy mass spectrometry (MSE) or MS/MS mode with electrospray ionization. Lapatinib and metabolites were separated on a Waters BEH Phenyl column (1.7 μm, 2.1 × 100 mm) with 0.1% formic acid in water or acetonitrile (0.4 ml/min) using the following gradient: 10% organic held for 1 minute, increased to 90% over 13 minutes, and held at 90% for 0.5 minute. Fexofenadine was used for real-time mass calibration. Data processing used MetaboLynx XS and QuanLynx subroutines of Waters MassLynx version 4.1 and metabolite structural elucidation was performed manually.
MI Complex Spectrophotometric Assessments.
Additional samples were prepared under similar experimental conditions (50 μM lapatinib; 100 pmol/ml recombinant protein; 30 minutes; 37°C; pH 7.4; 1 mM NADPH) to confirm MI complex formation and disruption by observation of a Soret absorbance peak. A Varian Cary 100 BIO UV/visible spectrophotometer (Agilent Technology, Santa Clara, CA) equipped with a circulating water bath was set to scan 400–500 nm every 30 seconds for 30 minutes. Reactions were initiated by the addition of substrate. At 15 minutes, a 10-μl aliquot of potassium ferricyanide (2 mM final concentration) was added to the sample cuvette while 10 μl of water was added to the reference cuvette.
Results
As shown in Table 1 and Fig. 3, HLM, rCYP3A4, and rCYP3A5 all converted lapatinib to 10 components (M1 through M10). Consistent with the results of previous in vitro (Teng et al., 2010) and in vivo studies (Castellino et al., 2012), the primary metabolites of lapatinib (M1, M3, M4, M8, and M10) were formed by N-dealkylation (M4 and M10), O-dealkylation (M1), N-hydroxylation (M8), or C-hydroxylation (M3). These and other oxidation reactions converted the five primary metabolites to four secondary metabolites (M2, M5, M6, and M7) and one tertiary metabolite (M9), as shown in Fig. 3. C-Hydroxylation of the fluorophenyl moiety (to form M3, which was further oxidized to M5) is a previously unidentified pathway of lapatinib metabolism, but its observation may be attributable to the high substrate concentration used. The relative abundance of each metabolite formed by rCYP3A4 compared with rCYP3A5, and vice versa, was used to assess which reaction was most efficiently catalyzed by each of the enzymes (Table 2). Inconsistent with published research (Chan et al., 2012), in our experiments rCYP3A4 and rCYP3A5 both formed all of the detected metabolites, but consistent with those studies, higher levels of N-dealkylation and N-oxygenation metabolites were observed in rCYP3A4 than in rCYP3A5 while higher levels of O-dealkylation metabolites were observed in the rCYP3A5 samples.
Metabolite profiling data for lapatinib (50 μM) incubated with NADPH-fortified human liver microsomes (1 mg/ml), recombinant CYP3A4, or recombinant CYP3A5 (100 pmol/ml) for 30 minutes and re-isolated by ultracentrifugation (100,000g; 60 minutes)

Proposed biotransformation scheme for lapatinib (50 mM) incubated for 30 minutes with NADPH-fortified HLM (1 mg/ml), rCYP3A4 + b5, or rCYP3A5 + b5 (100 pmol/ml). The predominant enzyme involved in each biotransformation is shown in boldface.
Relative abundance of metabolites formed from lapatinib (50 μM) incubated with NADPH-fortified rCYP3A4 and rCYP3A5 (100 pmol/ml) for 30 minutes, followed by ultracentrifugation
Under the experimental conditions used, MI complex formation in incubations of lapatinib with rCYP3A4 and HLM was initially established by visible spectrophotometry (λmax ∼455 nm) (data not shown). The quasi-reversibility of MDI of CYP3A4 was assessed in HLM and recombinant CYP3A enzymes by a combination of ultracentrifugation and ferricyanide treatment. Following incubation with lapatinib, residual CYP3A4 and CYP3A5 activity, both before and after ultracentrifugation with or without potassium ferricyanide, was measured by midazolam 1′-hydroxylation (Fig. 4A) and 4-hydroxylation (Fig. 4B). These activities were normalized to those observed in solvent control samples. Midazolam 1′-hydroxylase activity (Fig. 4A) was inhibited (40–60%) with HLM and rCYP3A4 but weakly inhibited (<15%) with rCYP3A5. Ultracentrifugation alone restored little to no midazolam 1′-hydroxylase activity in HLM or rCYP3A4, but ferricyanide oxidation with ultracentrifugation almost completely restored enzyme activity in both test systems, consistent with quasi-irreversible inhibition of CYP3A4. Midazolam 4-hydroxylase activity was inhibited (45–73%) by lapatinib in HLM, rCYP3A4, and rCYP3A5. Ferricyanide treatment partially restored midazolam 4-hydroxylase activity in HLM and rCYP3A4 but not rCYP3A5, consistent with some contribution from quasi-irreversible inhibition to enzyme inactivation with HLM and rCYP3A4 but only irreversible inactivation with rCYP3A5.

Relative CYP3A activity determined by (A) midazolam 1′-hydroxylation and (B) midazolam 4-hydroxylation for human liver microsomes (0.05 mg/ml), rCYP3A4 + b5, or rCYP3A5 + b5 (100 pmol/ml) preincubated with lapatinib (50 μM) for 30 minutes followed by incubation with midazolam (30 μM) for 5 minutes showing the effects of ultracentrifugation (100,000g; 60 minutes) alone and accompanied by oxidation with potassium ferricyanide (2 mM, 10 minutes).
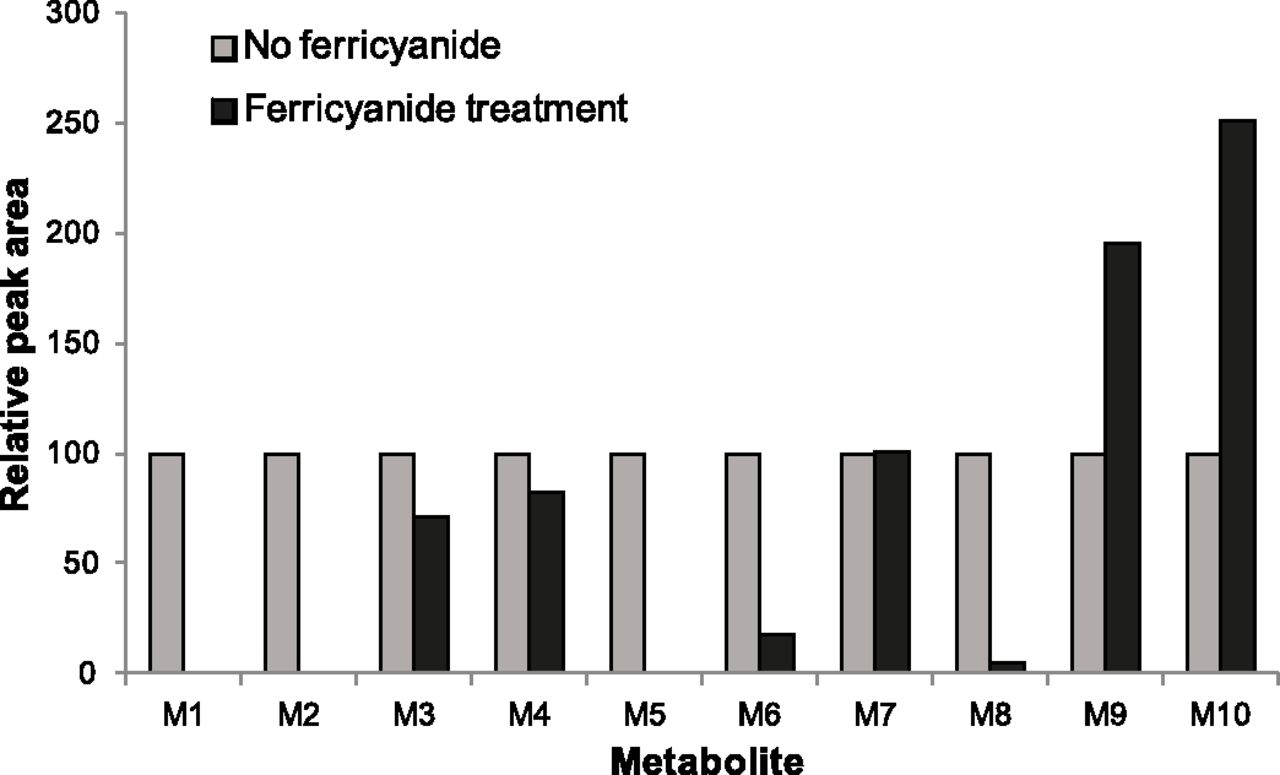
The effect of potassium ferricyanide treatment on the abundance of each detected metabolite (M1–M10) in the re-isolated microsomal membranes was assessed by calculating the relative abundance of each metabolite in the ferricyanide-treated samples compared with the nontreated samples in the same test system (HLM, rCYP3A4, or rCYP3A5). Although CYP3A5 forms all of the same lapatinib metabolites as CYP3A4, CYP3A5 preferentially catalyzes the O-dealkylation reaction to the para-aminophenol M1, which is associated with irreversible inactivation of CYP3A5 due to formation of an electrophilic quinoneimine by dehydrogenation of M1 (Chan et al., 2012). The inactivation of CYP3A5 by lapatinib does not involve quasi-irreversible inactivation through MI complex formation (Takakusa et al., 2011; Chan et al., 2012). For this reason, rCYP3A5 was used as a control to distinguish changes in metabolite abundance due to chemical oxidation by ferricyanide from abundance changes attributed to the release of the metabolite bound to ferrous CYP3A4 (i.e., MI complex disruption). Initially, the effect of ferricyanide treatment on metabolite abundance in HLM was assessed (Fig. 5). After ferricyanide treatment, the majority of lapatinib metabolites decreased in abundance and several of them disappeared, presumably due to chemical oxidation of these metabolites by ferricyanide. However, two metabolites (M9 and M10) increased in abundance following ferricyanide treatment. The abundance of M9 in the HLM sample treated with ferricyanide increased to approximately 195%, whereas the abundance of M10 increased to approximately 250% relative to the negative controls. Next, abundance changes for each metabolite caused by ferricyanide treatment were evaluated with rCYP3A4 and rCYP3A5 (Table 3). Prior to ferricyanide treatment, M9 was more abundant in rCYP3A4 than in rCYP3A5 samples, whereas M10 was equally abundant in the two recombinant enzymes. M9 increased in abundance upon addition of ferricyanide to the rCYP3A4 samples, consistent with a metabolite released from the MI complex with CYP3A4, but decreased slightly upon ferricyanide treatment of the rCYP3A5 samples (Fig. 6A). In contrast, the abundance of M10 increased considerably upon ferricyanide treatment, irrespective of the recombinant enzyme (Fig. 6B), and therefore irrespective of MI complex disruption.
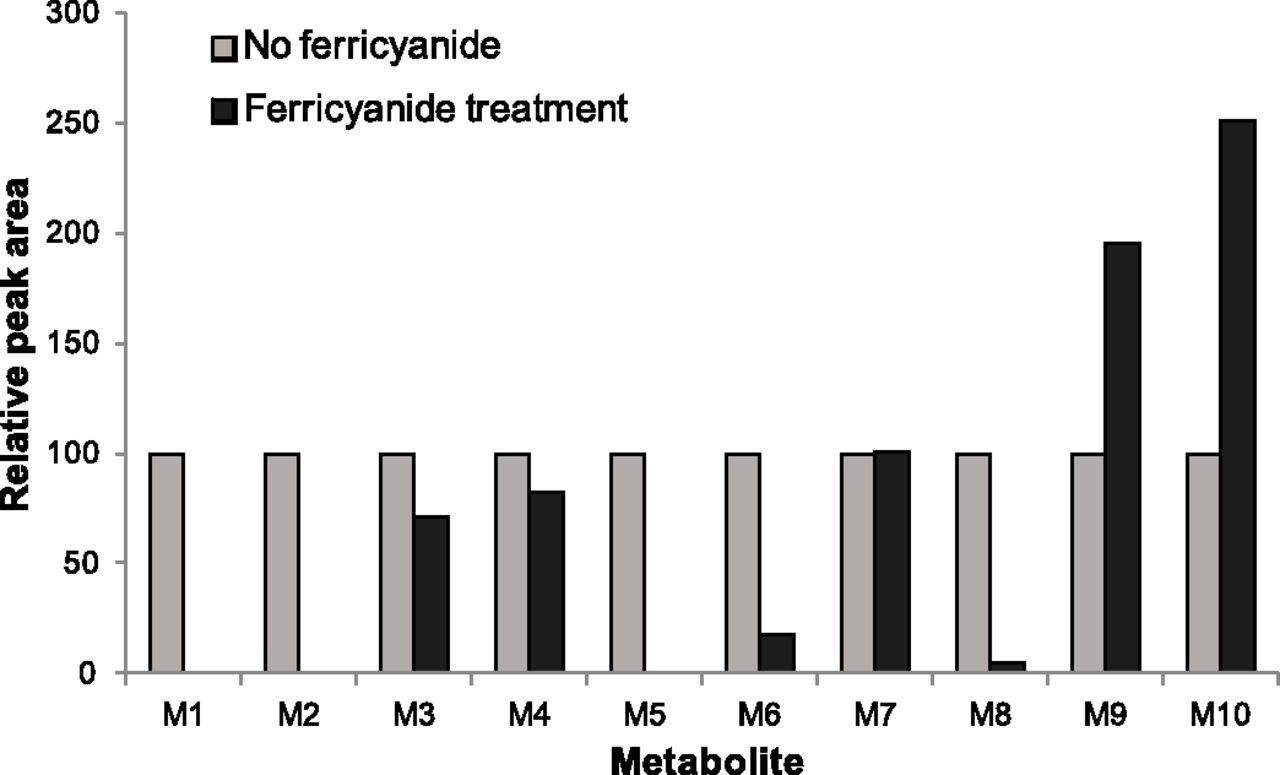
Abundance of metabolites formed from lapatinib (50 μM) incubated for 30 minutes with NADPH-fortified human liver microsomes (1 mg/ml) isolated by ultracentrifugation (100,000g; 60 minutes) and treated with potassium ferricyanide oxidation (2 mM, 10 minutes) relative to abundance with no oxidation.
Abundance of metabolites formed from lapatinib (50 μM) incubated with NADPH-fortified human liver microsomes (1 mg/ml), rCYP3A4, or rCYP3A5 (100 pmol/ml) for 30 minutes after ultracentrifugation and treatment with potassium ferricyanide (2 mM; 10 minutes) relative to abundance after ultracentrifugation in the absence of potassium ferricyanide

Relative abundance of (A) M9 and (B) M10 formed from lapatinib (50 μM) incubated for 30 minutes with NADPH-fortified HLM (1 mg/ml), rCYP3A4 + b5, and rCYP3A5 + b5 (100 pmol/ml) isolated by ultracentrifugation (100,000g; 60 minutes) and treated with or without potassium ferricyanide oxidation (2 mM, 10 minutes).
In experiments where metabolite profiling was conducted without the ultracentrifugation step but incorporating ferricyanide oxidation, an additional component (designated MB) structurally related to lapatinib was detected with 1.12 relative retention time (component retention time/lapatinib retention time). MB was detected at accurate m/z 503.1287, consistent with a component formed by N-dealkylation of lapatinib combined with the addition of one carbon and one oxygen atom. Upon ferricyanide oxidation, MB increased to >200% in HLM, >4000% in rCYP3A4, and >150% in rCYP3A5. MB showed several characteristics of the metabonate (a product of chemical breakdown of an enzymatically formed metabolite) formation that we have previously established as a complication of in vitro metabolite profiling of alkylamine compounds (Barbara et al., 2012). The formation of MB was extensive and rapid and involved the primary amine product of lapatinib N-dealkylation (M4) as an intermediate. The formation of MB may involve a reaction between N-desalkyl-lapatinib (M4, a major metabolite formed by CYP3A4) and formic acid to form a formamide. Formation of MB was inhibited by semicarbazide (data not shown), which supports a role for formaldehyde in MB formation. MB was detected when metabolite profiling was performed in the absence of the ultracentrifugation step, which underscores the importance of the ultracentrifugation step to the experiment.
Structural assignments for all 10 lapatinib metabolites were made on the basis of accurate mass product ion data and the detailed in vivo metabolite profiling studies by Castellino et al. (2012). A proposed biotransformation scheme for lapatinib is presented in Fig. 3. The elemental compositions derived from the accurate mass data for M9 and M10 (Table 1), the two metabolites that increased following ferricyanide treatment (and, hence, the two potential candidates for the complexed metabolite), showed that formation of M9 and M10 both involved N-dealkylation, but N-dealkylation occurred on opposite sides of the alkylamine nitrogen. Consequently, M9 is a large primary amine (with retention of the nitrogen), whereas M10 is a large aldehyde (with loss of the nitrogen). These data support M9 as the most likely candidate for a metabolite involved in MI complex formation and released through oxidation of the ferrous heme iron. In the low energy MSE data set, M9 was associated with a protonated molecule of accurate m/z 489.1125, which corresponds to a mass shift of −92.0300 amu from the theoretical accurate mass of the protonated molecule of unlabeled lapatinib. This is consistent with a metabolite formed by a combination of three sequential reactions, namely N-dealkylation, oxygenation, and dehydrogenation, although not necessarily in that order. M10 was associated with a protonated molecule of accurate m/z 474.1016, corresponding to a mass shift of −107.0409 amu from lapatinib, consistent with the aldehyde product of lapatinib N-dealkylation. The increase in abundance of M10 after ferricyanide treatment may involve oxidation of the secondary metabolite M7 back to the primary metabolite M10 or direct N-dealkylation of lapatinib by ferricyanide, for which there is precedent in the literature (Thyagarajan, 1958).
Quadrupole selection product ion spectra for lapatinib (Fig. 7A) and M9 (Fig. 7B) were acquired to probe the sites of the oxygenation and dehydrogenation involved in the formation of M9. The fragment ions of m/z 350.0699 and 380.0681 in the M9 product ion spectrum were consistent with biotransformation of the secondary amine. Two specific neutral losses were also observed, one of 17.0031 amu, corresponding to a hydroxyl radical (theoretical mass 17.0002 amu), and the other of 29.9982 amu, most likely associated with a nitric oxide radical (theoretical mass 29.9975 amu). These neutral losses implicated N-oxygenation rather than C-oxidation, but there are four potential sites of N-oxygenation in lapatinib. To determine which site of N-oxygenation leads to M9 formation, studies were performed with 2H4- and 13C2,15N-lapatinib.
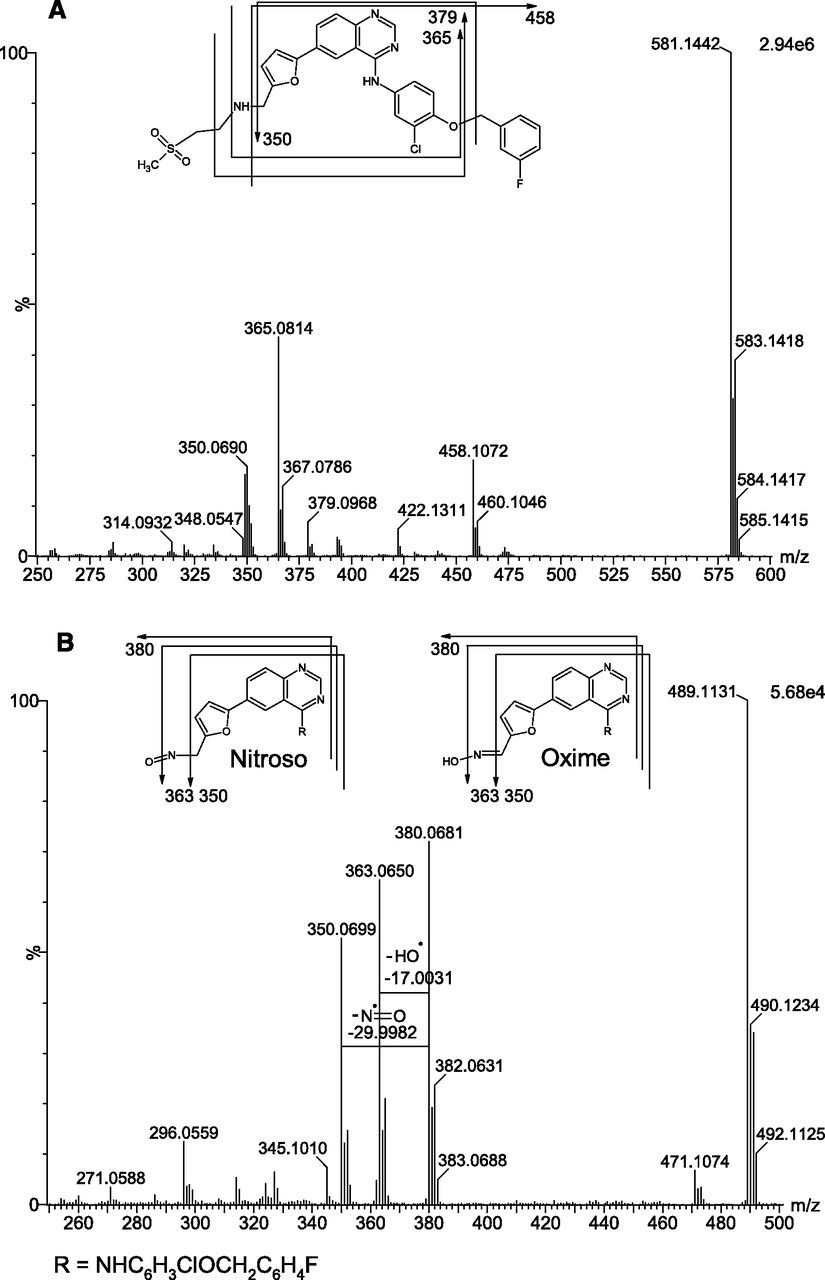
Product ion spectra for (A) lapatinib and (B) M9 formed from lapatinib (50 μM) incubated for 30 minutes with NADPH-fortified human liver microsomes (1 mg/ml) with proposed structures and diagnostic fragment assignments inset.
Interpretation of the mass spectral data for M9 formed from 2H4-lapatinib established that formation of M9 involved the loss of the methylsulfonyl(ethyl) group (data not shown). The data did not unequivocally establish the sites of oxygenation or dehydrogenation leading to M9 formation, but this information was obtained from studies with 13C2,15N-lapatinib. Product ion spectra for 13C2,15N-lapatinib (Fig. 8A) and M9 formed from 13C2,15N-lapatinib (Fig. 8B) established that formation of M9 involved N-dealkylation, N-oxygenation, and dehydration (but not necessarily in that order). The protonated molecule associated with M9 increased in m/z by 0.9979, consistent with incorporation of 15N, proving that the 15N remained intact during M9 formation. The neutral loss associated with loss of the hydroxyl radical remained unchanged, but the neutral loss proposed to correspond to the loss of a nitric oxide radical increased by 0.9982 amu. This confirmed the methylene amine nitrogen as the site of oxygenation. The only available sites for enzymatic dehydrogenation on the N-dealkylated, N-oxygenated lapatinib metabolite are either side of the alkylamine. Therefore, the only possibilities for M9 consistent with established biotransformation reactions are the N-dealkylated nitroso metabolite or its oxime tautomer, as shown in Fig. 3. Monomeric nitroso compounds are well established to tautomerize to their oxime forms if a hydrogen atom is present on the α-carbon (Lindeke 1982; Jonsson and Lindeke, 1992). In light of the presence of hydrogen on the α-carbon of N-dealkylated lapatinib and the observed stability of M9 in the incubation samples, and consistent with published literature (Takakusa et al., 2011; Castellino et al., 2012), metabolite M9 was proposed as the N-des-(ethylmethylsulfone) lapatinib oxime.

Product ion spectra for (A) 13C2,15N-lapatinib and (B) M9 formed from 13C2,15N-lapatinib (50 μM) incubated for 30 minutes with NADPH-fortified rCYP3A4 + b5 (100 pmol/ml) with proposed structures and diagnostic fragment assignments inset.
Discussion
Inhibition of P450 enzymes upon metabolism of alkylamine drugs through noncovalent complex formation between the ferrous heme iron of the enzyme and an unknown metabolite capable of donating an electron pair to form a coordinate bond (i.e., MI complex formation) is well established. Since enzyme inactivation has the potential to cause clinically relevant DDI with coadministered therapeutics, elucidation of the mechanism of MI complex-based P450 enzyme inhibition by alkylamine drugs could aid structural refinement of lead candidates to mitigate the associated DDI risk for drugs in development (Taxak et al., 2012). This information could also help refine methods for rational DDI prediction (Hanson et al., 2010). In the 1970s, N-dealkylation of the prototypical tertiary alkylamine P450 inhibitor SKF 525-A (Proadifen) to yield the secondary alkylamine was established as a first step toward generating the metabolite capable of binding with heme to yield the MI complex detected by a Soret absorbance peak at 455 nm (Schenkman et al., 1972). In addition, N-dealkylation of the tertiary alkylamine macrolide antibiotics troleandomycin and erythromycin was demonstrated as the initial step in their MI complex-based inactivation of CYP3A4 (Danan et al., 1981; Pessayre et al., 1981, 1982). These findings illustrate the importance of secondary alkylamines in mechanistic considerations for MI complex-based P450 inactivation for tertiary alkylamines as well as for the secondary alkylamines themselves.
We hypothesized that correlating changes in the metabolite profile of the secondary alkylamine lapatinib following MI complex disruption could identify the metabolite responsible for the quasi-irreversible inhibition of CYP3A4. A positive relationship between reversal of MI complex-based inactivation of CYP3A4 and the prevalence of only one metabolite (M9) was established with rCYP3A4 and rCYP3A5 experiments, despite the fact that CYP3A4 formed ten oxidative metabolites from lapatinib, seven involving oxidation of the alkylamine nitrogen or α-carbon. M9 was identified as N-des-(ethylmethylsulfone) lapatinib oxime (a tautomer of the nitroso metabolite). Its increase in abundance upon MI complex disruption is proposed as direct evidence of the involvement of the nitroso metabolite in the CYP3A4-MI complex formed from lapatinib, consistent with the hypothesis of Takakusa et al. (2011). We propose that, upon oxidation of the ferrous heme iron, CYP3A4 releases the coordinately bound nitroso metabolite, which tautomerizes to the oxime (M9), which is sufficiently stable for detection by mass spectrometry.
Tautomerization of a nitroso metabolite is not the only mechanism by which a lapatinib oxime metabolite could form; other possible routes of M9 formation merit consideration. Oxime formation from a hydroxylamine metabolite of methylamphetamine has been proposed as an example of metabonate formation, although this study could not exclude the possibility of oxime formation by sequential N-dealkylation and N-hydroxylation to the primary hydroxylamine followed by enzymatic dehydrogenation (Beckett, 1971). Lapatinib, a secondary alkylamine, was N-hydroxylated to M8 (Fig. 3). However, the primary amine of lapatinib (M4) did not undergo N-hydroxylation. We observed no evidence of the primary hydroxylamine metabolite (i.e., N-hydroxy-M4) throughout our experiments, so neither of the options proposed for methylamphetamine is likely to account for M9 formation. It is theoretically possible that M9 could have formed so efficiently from N-hydroxy-M4 that this hydroxylamine intermediate was undetectable but, for the reasons outlined below, this scenario is unlikely.
We surmise that M9 is formed by tautomerization of the nitroso metabolite and that the nitroso intermediate is the most likely candidate for the lapatinib metabolite complexed with the ferrous heme of CYP3A4. This interpretation is supported by several lines of evidence. Nitroso compounds are known to rapidly tautomerize to their oximes in protic solvents and at low concentrations (Lindeke, 1982), consistent with our incubation conditions. In addition, nitroso compounds are isoelectronic with molecular oxygen, the physiologic ligand of P450 enzymes (Lindeke, 1982). While Jonsson and Lindeke proposed that the MI complex formed from the primary amine amphetamine involved an amphetamine nitroxide metabolite (Jonsson and Lindeke, 1976), Mansuy and colleagues proposed concurrently that the electron pair donor stabilizing the MI complex formed from amphetamine was a nitroso metabolite (Mansuy et al., 1976). This hypothesis was subsequently supported with evidence from heme model- and model protein-based studies (Mansuy et al., 1977a,b; Mansuy, 1981) and with compelling evidence that the metabolite involved in MI complex formation could be formed either by oxidation of a hydroxylamine or by reduction of a nitro group (Mansuy et al., 1978). All of these results established a relationship between MI complex formation and metabolites formed by N-oxygenation. Furthermore, a recent comprehensive computational study on the mechanism of nitroso metabolite and MI complex formation from a model tertiary alkylamine (dimethylamine) established that a complex formed from the nitroso metabolite bound to the ferrous heme iron would theoretically be very stable. The overall reaction through sequential oxidation of the parent to the nitroso group, including MI complex formation, was exothermic with an energy release of approximately 60 kcal mol−1 (Taxak et al., 2012).
The Taxak study (2012) was based on a traditional view of nitroso formation—one that involves N-dealkylation of the tertiary amine to the secondary amine and then to the primary amine (R-NH2), which is N-hydroxylated to the primary hydroxylamine (R-NHOH) and further oxidized (by dehydrogenation) to the nitroso (R−N=O). This pathway is consistent with the proposed route of nitroso formation from primary amine compounds (Mansuy et al., 1976; Lindeke et al., 1982), which has been proposed as the route of nitroso formation from secondary and tertiary alkylamines (Orr et al., 2012). However, strong evidence that the major pathway to MI complex formation with nitroso metabolites formed from secondary alkylamine drugs involves N-hydroxylation of the secondary amine and not N-dealkylation of the secondary amine to the primary amine has been reported (Hanson et al., 2010). In fact, N-dealkylation to the primary amine was proposed by Hanson et al. (2010) as a competitive pathway inhibiting MI complex formation from the secondary alkylamines desipramine, (S)-fluoxetine, and nordiltiazem. Therefore, it is notable that the computational study identified N-oxidation of the primary amine as the rate-determining step in MI complex formation (Taxak et al., 2012), in agreement with previously reported literature (Buening and Franklin, 1976). MI complex formation from alkylamines involving the secondary hydroxylamine pathway has been established for both methamphetamine (Lindeke et al., 1979) and benzphetamine (Jeffery and Mannering, 1983).
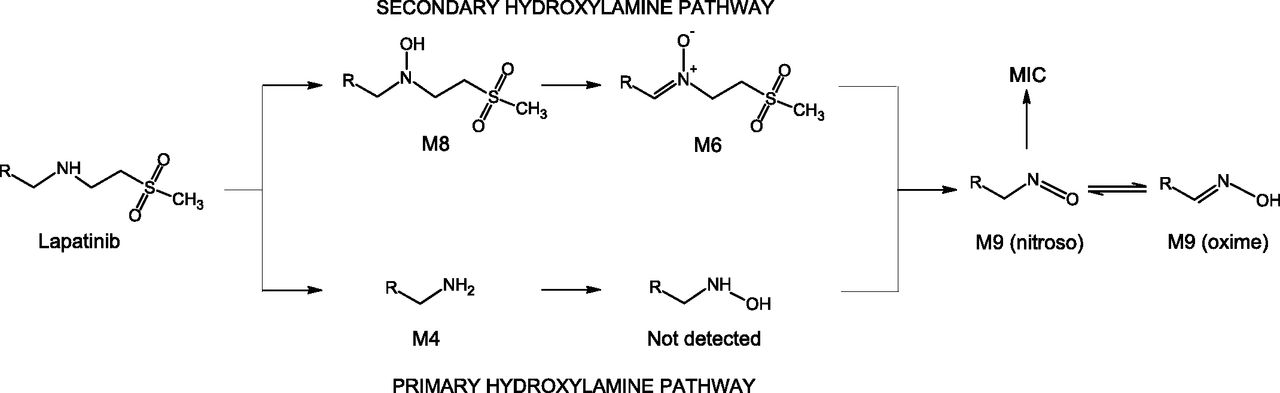
We compared the possible routes of formation of the nitroso/oxime tautomeric pair from lapatinib (Fig. 9) and conclude that our results for lapatinib support the secondary hydroxylamine mechanism generally proposed by Hanson et al. (2010) and not the primary amine hydroxylation mechanism. Both the primary hydroxylamine and the secondary hydroxylamine pathways (Fig. 9) to nitroso metabolite formation from lapatinib involve multiple P450-mediated sequential oxidation steps. The primary hydroxylamine pathway is initiated by N-dealkylation to the primary amine, which we detected as M4. Hydroxylation of the amine to the primary hydroxylamine metabolite should be the next step, but we saw no evidence of an N-dealkylated hydroxylamine lapatinib metabolite (i.e., N-hydroxy-M4). Theoretically, this primary hydroxylamine could be oxidized (dehydrogenated) to yield the nitroso metabolite, which tautomerizes to the oxime metabolite M9. The initial step in the secondary hydroxylamine pathway would be N-hydroxylation of lapatinib to yield the secondary hydroxylamine that we detected as M8. This secondary hydroxylamine is subsequently oxidized to the nitrone that we detected as M6. Dealkylation of the lapatinib nitrone would yield the nitroso/oxime tautomer M9. We observed all of the intermediates required for the secondary hydroxylamine pathway to nitroso formation, but we did not detect the N-dealkylated hydroxylamine metabolite (N-hydroxy-M4) necessary for nitroso formation by the primary hydroxylamine pathway. Our findings support the secondary hydroxylamine pathway to CYP3A4-mediated nitroso metabolite formation from lapatinib, consistent with the proposal of Takakusa et al. (2011). Given that the undetected primary hydroxylamine metabolite could be converted chemically or enzymatically to the observed lapatinib oxime metabolite, it could be argued that the primary hydroxylamine itself is bound in the MI complex, accounting for its lack of detection as a free metabolite. However, MI complex formation from primary hydroxylamines has been shown with amphetamine and analogs to be both relatively slow (Lindeke et al., 1982) and NADPH dependent (Lindeke et al., 1979), negating the possibility that the complexed metabolite is the primary hydroxylamine. Furthermore, even when the CYP3A4-MI complex was disrupted with ferricyanide, we did not detect N-hydroxy-M4.
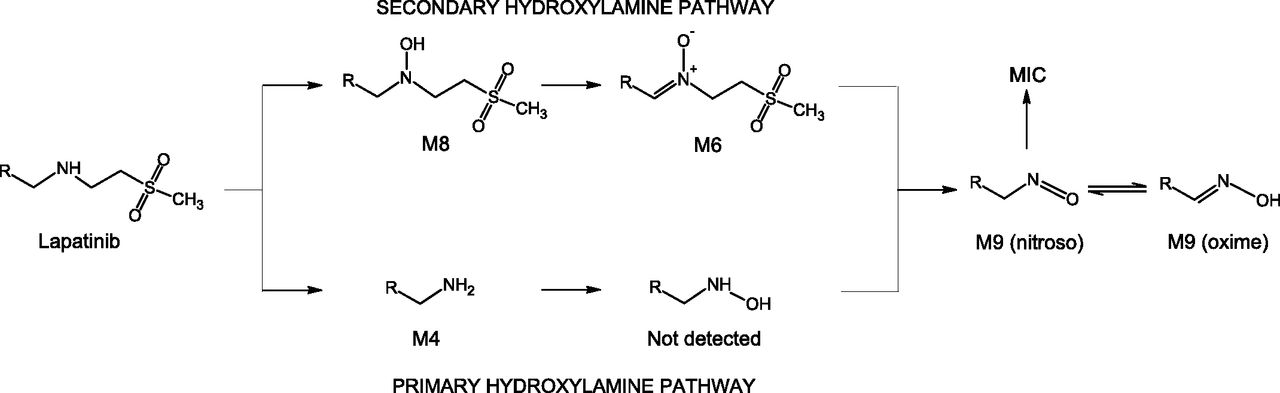
Mechanistic pathways for the formation of nitroso/oxime tautomeric metabolites from lapatinib.
Our findings show that lapatinib is converted to ten metabolites by CYP3A4 (mainly N-dealkylation to M4 and M10 and N-hydroxylation to M8) and CYP3A5 (mainly O-dealkylation to M1 and C-hydroxylation to M3). We established a relationship between MI complex formation with CYP3A4 and the N-dealkylated lapatinib oxime metabolite M9, providing evidence that a nitroso metabolite is the complexed species responsible for MI complex-based inactivation of CYP3A4 by lapatinib. Furthermore, we showed evidence supporting the secondary hydroxylamine pathway through a nitrone intermediate as the route to formation of the nitroso metabolite and surmise that lapatinib nitrone is N-dealkylated to yield the nitroso metabolite subsequently bound in the complex. Our studies with lapatinib implicate the pathway to nitroso and MI complex formation as N-hydroxylation, dehydrogenation, and N-dealkylation, rather than the traditional primary hydroxylamine pathway. This secondary hydroxylamine pathway is likely to be applicable to MI complex formation by other secondary and tertiary alkylamine drugs.
Acknowledgments
The authors thank Phyllis Yerino for assistance with incubations, Brian Ogilvie for scientific discussion, Mark Horrigan for resource support, and Kammie Settle for assistance with figure preparation.
Author Contributions
Participated in research design: Barbara, Kazmi, Parkinson.
Conducted experiments: Barbara.
Contributed new reagents or analytic tools: Barbara, Buckley.
Performed data analysis: Barbara, Kazmi, Parkinson, Buckley.
Wrote or contributed to the writing of the manuscript: Barbara, Kazmi, Parkinson, Buckley.
Footnotes
- Received January 15, 2013.
- Accepted February 12, 2013.
This work was previously presented: Barbara JE, Parkinson A, Yerino P, Buckley DB, and Kazmi F (2012) Metabolism-dependent inhibition of cytochrome CYP3A4 by lapatinib is due to metabolic intermediate complex (MIC) formation with a nitroso metabolite. Eighteenth North American Regional ISSX Meeting; 2012 Oct 14–18; Dallas, TX. International Society for the Study of Xenobiotics, Washington, DC.
Abbreviations
- DDI
- drug-drug interaction
- HLM
- human liver microsome
- LC
- liquid chromatography
- MDI
- metabolism-dependent inhibition
- MI
- metabolic intermediate
- MSE
- elevated energy mass spectrometry
- MS/MS
- tandem mass spectrometry
- P450
- cytochrome P450
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}