


Abstract
Desloratadine (Clarinex), the major active metabolite of loratadine (Claritin), is a nonsedating antihistamine used for the treatment of seasonal allergies and hives. Previously we reported that the formation of 3-hydroxydesloratadine, the major human metabolite of desloratadine, involves three sequential reactions, namely N-glucuronidation by UGT2B10 followed by 3-hydroxylation by CYP2C8 followed by deconjugation (rapid, nonenzymatic hydrolysis of the N-glucuronide). In this study we assessed the perpetrator potential of desloratadine based on in vitro studies of its inhibitory effects on cytochrome P450 and UDP-glucuronosyltransferase (UGT) enzymes in human liver microsomes (HLM). Desloratadine (10 µM) caused no inhibition (<15%) of CYP1A2, CYP2C8, CYP2C9, or CYP2C19 and weak inhibition (32–48%) of CYP2B6, CYP2D6, and CYP3A4/5. In cryopreserved human hepatocytes (CHH), which can form the CYP2C8 substrate desloratadine N-glucuronide, desloratadine did not inhibit the CYP2C8-dependent metabolism of paclitaxel or amodiaquine. Assessment of UGT inhibition identified desloratadine as a potent and relatively selective competitive inhibitor of UGT2B10 (Ki value of 1.3 μM). Chemical inhibition of UGT enzymes in HLM demonstrated that nicotine (UGT2B10 inhibitor) but not hecogenin (UGT1A4 inhibitor) completely inhibited the conversion of desloratadine (1 µM) to 3-hydroxydesloratadine in HLM fortified with both NADPH and UDP-glucuronic acid. 3-Hydroxydesloratadine formation correlated well with levomedetomidine glucuronidation (UGT2B10 marker activity) with a panel of individual CHH (r2 = 0.72). Overall, the results of this study confirm the role of UGT2B10 in 3-hydroxydesloratadine formation and identify desloratadine as a relatively selective in vitro inhibitor of UGT2B10.
Introduction
Desloratadine (Clarinex) is a long-lasting, nonsedating, second-generation antihistamine (a selective H1-receptor histamine antagonist) commonly used for the treatment of seasonal allergies (allergic rhinitis) and chronic hives (chronic idiopathic urticaria) (Geha and Meltzer, 2001; Henz, 2001). Desloratadine is a major and pharmacologically active metabolite of the antihistamine loratadine (Claritin) and is formed primarily by CYP3A4 and to a lesser extent by CYP2D6 (Yumibe et al., 1995; Yumibe et al., 1996; Dridi and Marquet, 2013). In 2001, desloratadine was approved as a drug in its own right; however, at the time of its approval, the enzymology surrounding its metabolism was unknown (Clarinex label; www.accessdata.fda.gov/drugsatfda_docs/label/2001/21165lbl.pdf). In humans, desloratadine is converted to 3-hydroxydesloratadine (by hydroxylation of the pyridine ring) followed by O-glucuronidation to 3-hydroxydesloratadine O-glucuronide. Both 3-hydroxydesloratadine and its O-glucuronide conjugate are major in vivo metabolites excreted in approximately equal amounts in urine and feces (Ramanathan et al., 2007).Conventional in vitro systems such as recombinant P450 enzymes, human liver microsomes (HLM), and human liver S9 fractions do not convert desloratadine to 3-hydroxydesloratadine; hence, its enzymology and the basis for certain individuals being identified as poor metabolizers of desloratadine have remained a mystery for many years (Prenner et al., 2006; Ghosal et al., 2009). Previously, we demonstrated that cryopreserved human hepatocytes (CHH) can form 3-hydroxydesloratadine, as can HLM provided they are supplemented with both NADPH and UDP-glucuronic acid (UDP-GlcUA) (Kazmi et al., 2015). As shown in Fig. 1, formation of 3-hydroxydesloratadine involves three sequential reactions: 1) N-glucuronidation of desloratadine by UGT2B10; 2) 3-hydroxylation of desloratadine N-glucuronide by CYP2C8, and 3) deconjugation of 3-hydroxydesloratadine N-glucuronide (rapid nonenzymatic hydrolysis of the glucuronide). The N-glucuronide of desloratadine is highly unstable (both before and after 3-hydroxylation by CYP2C8). We were unable to detect desloratadine N-glucuronide or 3-hydroxydesloratadine N-glucuronide by LC-MS/MS in our previous study (Kazmi et al., 2015). Instability is a characteristic property of certain N-glucuronides (Ciotti et al., 1999).

Metabolic scheme for the conversion of desloratadine to 3-hydroxydesloratadine. Desloratadine is converted to an unstable N-glucuronide by UGT2B10 followed by CYP2C8-dependent hydroxylation to 3-hydroxydesloratadine as described previously (Kazmi et al., 2015).
The U. S. Food and Drug Agency's (FDA) 2012 draft Guidance for Industry on drug-drug interactions (DDIs) (www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm292362.pdf) and the European Medicine Agency’s 2012 Guideline on the Investigation of Drug Interactions (www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf) both recommend that the assessment of the perpetrator potential of investigational drugs include an evaluation of their ability to inhibit P450 and UGT enzymes in vitro. These regulatory agencies further require that, in the case of P450 enzymes, all investigational drugs be evaluated for their ability to cause time-dependent or metabolism-dependent inhibition (Grimm et al., 2009). The panel of enzymes recommended for testing and the types of inhibition to be evaluated have expanded since the FDA issued its first Guidance for Industry on drug interactions in 1997 (www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/UCM142439.pdf). Desloratadine was evaluated for its ability to inhibit P450 enzymes shortly after the FDA issued its first drug interaction guidance (Barecki et al., 2001). This study evaluated desloratadine and 3-hydroxydesloratadine as reversible inhibitors of CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, but it did not evaluate their potential to cause irreversible (time-dependent) inhibition, nor did it evaluate their potential to inhibit CYP2B6 or CYP2C8 (all of which were not recommended by the FDA at the time the study was performed). Furthermore, desloratadine was not evaluated as an inhibitor of UGT enzymes. In view of the role of CYP2C8 and UGT2B10 in the metabolism of desloratadine, it was of interest to evaluate desloratadine as a reversible and irreversible inhibitor of the seven P450 enzymes currently recommended by testing by the FDA (namely, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5) and as a reversible inhibitor of multiple UGT enzymes in HLM (namely, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B10, UGT2B15, and UGT2B17). In addition, because the N-glucuronide metabolites of desloratadine are highly unstable, we conducted additional studies to evaluate whether the N-glucuronidation of desloratadine is dependent largely or solely on UGT2B10.
Materials and Methods
Chemicals and Reagents.
Alamethicin, chenodeoxycholic acid, desloratadine, estradiol, hecogenin, morphine, 1-naphthol, nicotine, oxazepam, propofol, saccharic acid 1,4-lactone, testosterone, and trifluoperazine were purchased from Sigma-Aldrich (St. Louis, MO); levomedetomidine was purchased from Toronto Research Chemicals (Toronto, ON, Canada); amodiaquine was purchased from US Pharmacopeia (Rockville, MD); 3-hydroxydesloratadine was purchased from Santa Cruz Biotechnology (Dallas, TX); 3-hydroxydesloratadine-d4 was purchased from Medical Isotopes, Inc. (Pelham, NH). Testosterone 17β-O-glucuronide-d5, oxazepam N-glucuronide-d5, levomedetomidine glucuronide, and prochlorperazine glucuronide were prepared in house. Morphine 3-glucuronide-d3 was purchased from Cerilliant (Round Rock, TX). All other deuterated glucuronides were purchased from Toronto Research Chemicals (Toronto, ON, Canada). The sources of all other reagents have been described previously (Ogilvie et al., 2006; Parkinson et al., 2011; Kazmi et al., 2014).
Test System.
Pooled human liver microsomes (HLM, n = 200, mixed sex) and pooled cryopreserved human hepatocytes (CHH, n = 100, mixed sex) were prepared from nontransplantable livers and characterized at XenoTech, LLC (Lenexa, KS) as described previously (Pearce et al., 1996; LeCluyse et al., 2000; Parkinson et al., 2004). Demographic information on the individual preparations of CHH used for correlation analysis is summarized in Supplemental Table 1. Recombinant UGT enzymes were purchased from Corning Gentest (Woburn, MA).
In Vitro P450 Inhibition in Human Liver Microsomes.
Desloratadine was evaluated as an inhibitor of P450 enzymes as described previously (Parkinson et al., 2011). Briefly, 10 μM desloratadine was incubated at 37°C in 200-μl incubation mixtures (n = 2) containing pooled HLM (≤0.1 mg/ml), potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM, pH 7.4), an NADPH-generating system (consisting of 1 mM NADP, 5 mM glucose 6-phosphate, and 1 unit/ml glucose-6-phosphate dehydrogenase), and a P450 marker substrate at a concentration approximately equal to its Km. Substrates included phenacetin (CYP1A2; 40 μM), bupropion (CYP2B6; 50 μM), paclitaxel (CYP2C8; 5 μM), diclofenac (CYP2C9; 6 μM), S-mephenytoin (CYP2C19; 40 μM), dextromethorphan (CYP2D6; 7.5 μM), and midazolam (CYP3A4/5, 3 μM). Before the addition of P450 marker substrate, desloratadine was preincubated with HLM for 0 or 30 minutes with and without NADPH to assess direct, time-dependent, and metabolism-dependent inhibition (Parkinson et al., 2011). Reactions with the P450 marker substrate were terminated after 5 minutes by the addition of 200 μl of acetonitrile containing an internal standard (the metabolite of interest labeled with a stable isotope). Precipitated protein was removed by centrifugation (920 relative centrifugal force for 10 minutes at 10°C) followed by analysis by liquid chromatography tandem mass spectrometry (LC-MS/MS) as described below.
In Vitro CYP2C8 Inhibition in Cryopreserved Human Hepatocytes.
Desloratadine was evaluated as an inhibitor of CYP2C8 with and without a preincubation step to assess the potential for time-dependent inhibition (i.e., a time-dependent decrease in IC50) as described previously with minor modifications (Kazmi et al., 2014). In this so-called IC50 shift experiment, desloratadine was incubated at 0.1, 0.3, 1, 3, 10, 30, and 100 μM in 100-μl incubation mixtures (n = 2) containing pooled CHH (n = 100; 0.5 million cells/ml), Williams’ E medium, supplemented with 2 mM glutaMAX (Gibco, Grand Island, NY) and 0.1 mM HEPES, and 10 μM amodiaquine or paclitaxel. Reactions were initiated by the addition of hepatocytes. Desloratadine was preincubated with the test system for 0, 30, or 120 minutes at 37°C with 95% humidity and 5% CO2 on an orbital shaker (∼150 rpm). After preincubation, CYP2C8 marker substrates were added (individually) and samples were incubated for an additional 10 minutes (amodiaquine) or 30 minutes (paclitaxel). An equal volume of acetonitrile containing the appropriate internal standard (isotopically labeled metabolite) was added to terminate the reactions, followed by LC/MS-MS analysis of the deproteinated sample as described below.
In Vitro Inhibition of UGT Enzymes in HLM and rUGT.
Desloratadine was evaluated as an inhibitor of UGT enzymes as described previously (Kazmi et al., 2014). Briefly, 10 or 100 μM desloratadine was incubated at 37°C in 150-μl incubation mixtures (n = 3) containing pooled HLM (≤0.1 mg/ml) or rUGT (≤0.25 mg/ml), Tris-HCl (100 mM, pH 7.7 at 37°C), MgCl2 (10 mM), EDTA (1 mM) d-saccharic acid 1,4-lactone (0.1 mM), UDP-GlcUA (10 mM), and a UGT marker substrate at a concentration approximately equal to its experimentally determined Km value, as described in Table 1. HLM and rUGT enzymes were activated with 25 μg/ml alamethicin for 15 minutes on ice before incubations. For UGT2B10, a Ki determination was conducted in HLM (0.1 mg/ml) with desloratadine as an inhibitor at 0.1, 0.3, 0.6, 1, 2, 4, and 10 μM and levomedetomidine as the substrate at 1, 3, 6, and 15 μM (n = 2). Reactions were initiated by the addition of UDP-GlcUA and terminated after 5 to 30 minutes by the addition of 150 μl of acetonitrile containing an appropriate internal standard (generally an isotopically labeled metabolite). Precipitated protein was removed by centrifugation (920 relative centrifugal force for 10 minutes at 10°C) followed by liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis as described in Table 1 and below. In the case of UGT2B15, HLM and rUGT2B15 were incubated with racemic oxazepam and the glucuronides of R- and S-oxazepam were resolved by HPLC. UGT2B15 activity is based on the rate of formation of only S-oxazepam glucuronide.
Experimental conditions for measuring UGT activity for enzyme inhibition studies in human liver microsomes and recombinant enzymes
Chemical Inhibition of 3-Hydroxydesloratadine Formation in HLM.
The involvement of UGT1A4 and UGT2B10 in desloratadine metabolism was assessed by examining the effects of UGT inhibitors on 3-hydroxydesloratadine formation by HLM fortified with both NADPH and UDP-GlcUA. Briefly, pooled HLM (n = 200) at 0.1 mg/ml were incubated at 37°C in 200-μl incubation mixtures (n = 3) containing 1 or 10 μM desloratadine, 20 μM trifluoperazine (UGT1A4 substrate), or 5 μM levomedetomidine (UGT2B10 substrate) in the absence or presence of 100 μM hecogenin (UGT1A4 inhibitor), 500 μM nicotine (UGT2B10 inhibitor), or both hecogenin and nicotine. Reactions were performed in Tris-HCl (100 mM, pH 7.7 at 37°C), MgCl2 (10 mM), EDTA (1 mM) d-saccharic acid 1,4-lactone (0.1 mM), UDP-GlcUA (10 mM), and NADPH (1 mM). Reactions were initiated by the addition of a mixture of UDP-GlcUA with NADPH and terminated after 5 minutes (trifluoperazine), 10 minutes (levomedetomidine), or 2 hours (desloratadine) by the addition of 200 μl of acetonitrile containing internal standards followed by LC-MS/MS analysis of the deproteinated samples.
Correlation Analysis of Individual CHH with UGT2B10 Activity.
CHH from nine individual donors were assessed for levomedetomidine glucuronidation (a marker of UGT2B10) and desloratadine 3-hydroxylation. Briefly, nine individual lots of hepatocytes (1 million cells/ml) were incubated with 1 or 10 μM levomedetomidine or desloratadine in 160-μl incubation mixtures (n = 3) containing Williams’ E medium supplemented with 2 mM glutaMAX (Gibco) and 0.1 mM HEPES. Reactions were initiated by the addition of hepatocytes and conducted for 30 minutes (levomedetomidine) or 2 hours (desloratadine) at 37°C with 95% humidity and 5% CO2 on an orbital shaker (approximately 150 rpm). Reactions were quenched by the addition of an equal volume of acetonitrile containing internal standard followed by LC-MS/MS analysis of the deproteinated samples.
Analytical Methods.
Samples were analyzed by LC-MS/MS for CYP2C8 activity toward amodiaquine and paclitaxel and 3-hydroxydesloratadine formation as described previously (Kazmi et al., 2015). Briefly, samples were analyzed by LC-MS/MS with a Shimadzu LC system (Columbia, MD) interfaced by electrospray ionization (ESI) with an AB Sciex API4000 QTrap mass spectrometer (Foster City, CA) in positive mode with the multiple reaction monitoring (MRM) scan type. For analysis of desloratadine and its metabolites, a gradient elution method comprising 0.2% formic acid in water (A) and acetonitrile (B) ramping from 10 to 95% over 10.5 minutes was applied to a Waters XBridge C18 column (5 μm, 4.6 × 100 mm). For the CYP2C8 marker assays, short gradient elution methods were applied to a Waters Atlantis dC18 column (5 μm, 2.1 × 100 mm; Milford, MA) with mobile phase systems comprising 0.2% formic acid in water and methanol (amodiaquine metabolite) or 0.1 mM ammonium acetate in 95:5 (v/v) water: methanol and methanol (paclitaxel metabolite). AB Sciex API2000 or 3000 triple quadrupole mass spectrometers in positive mode with ESI and MRM scanning were used for analyte detection. Methods are described in more detail elsewhere (Paris et al., 2009). For all other P450 substrates, analysis was conducted as described previously (Parkinson et al., 2011).
For UGT analytes, samples were analyzed by LC-MS/MS with LC gradients applied to a Waters Atlantis dC18 column (5 μm, 2.1 × 100 mm). Mobile phases comprised either 0.2% formic acid in water (A) and methanol (B) or 0.1 mM ammonium acetate in 95:5 (v/v) water: methanol (A) and methanol (B). Shimadzu Prominence or Nexera LC systems were interfaced to AB Sciex API3000 triple quadrupole or API4000 or 5500 triple quadrupole linear ion trap mass spectrometers by ESI. The corresponding internal standard compounds (generally an isotopically labeled metabolite), mass spectrometry modes, and MRM transitions employed for each specific glucuronide metabolite monitored are shown in Table 1.
Data Analysis.
All data processing and statistical analysis were conducted with Microsoft Excel 2010 (Microsoft, Redmond, WA) or SigmaPlot (Systat Software, San Jose, CA). For IC50 assays and the Ki determination, the data were processed by nonlinear regression with GraFit 7.0.2 (Erithacus Software Ltd., Horley, Surrey, UK). For IC50 assessment, this software uses a damped least squares algorithm to fit a nonlinear regression (sigmoidal) curve to IC50 data based on the following equation: (1)where Max (range) is 100 (complete inhibition) and Min (background) is zero (no inhibition). For Ki assessment, the software carries out a full analysis of multiple inhibition models (competitive, non-competitive, mixed or uncompetitive) and uses a reduced χ2 value to determine the best mechanistic fit. An Eadie-Hofstee plot was generated with the same software.
(1)where Max (range) is 100 (complete inhibition) and Min (background) is zero (no inhibition). For Ki assessment, the software carries out a full analysis of multiple inhibition models (competitive, non-competitive, mixed or uncompetitive) and uses a reduced χ2 value to determine the best mechanistic fit. An Eadie-Hofstee plot was generated with the same software.
Three-dimensional correlation analysis was conducted with SigmaPlot (Systat Software, San Jose, CA) by using the regression wizard and processing the data with a three-dimensional scatter plot. The regression plane was determined by the embedded software algorithm using the 3-hydroxydesloratadine formation data as the independent variable and a coefficient of determination (r2) was generated.
Results
Assessment of Desloratadine as an Inhibitor of Seven P450 Enzymes.
To evaluate desloratadine as an inhibitor of P450 enzymes in vitro, desloratadine (10 μM) was incubated with HLM (≤0.1 mg/ml) with and without a 30-minute preincubation step in the presence or absence of NADPH. P450 activity was measured with P450-selective substrates (at a final concentration roughly equal to Km), as described in Materials and Methods. As shown in Fig. 2 and Table 2, desloratadine did not inhibit (<15%) CYP1A2, CYP2C8, CYP2C9, or CYP2C19 but caused partial inhibition of CYP2B6 (48%), CYP2D6 (32%), and CYP3A4/5 (44%). Preincubating desloratadine for 30 minutes with HLM in the absence or presence of NADPH caused little or no increase in P450 inhibition (i.e., there was no evidence of time-dependent or metabolism-dependent inhibition).

Assessment of the inhibition of P450 enzymes by desloratadine. The P450 inhibition potential of desloratadine (10 μM) was assessed in HLM (≤0.1 mg/ml) in duplicate with and without a 30-minute preincubation step in the presence or absence of NADPH, followed by a 5-minute incubation with a P450-selective marker substrate (at a concentration approximately equal to its Km) as indicated in the figure. Details of the experimental procedures are described in Materials and Methods.
Assessment of desloratadine as an inhibitor of P450 enzymes in pooled human liver microsomes (HLM)
Assessment of Desloratadine as an Inhibitor of CYP2C8 in Human Hepatocytes.
As shown in the preceding section (Fig. 2 and Table 2), desloratadine did not inhibit CYP2C8 in NADPH-fortified HLM. This enzyme does not metabolize desloratadine directly but it does metabolize desloratadine N-glucuronide. Unfortunately, this N-glucuronide is too unstable to perform direct tests of its inhibitory potential. Therefore, desloratadine was evaluated as an inhibitor of CYP2C8 in CHH, which can convert desloratadine to the N-glucuronide that is subsequently metabolized by CYP2C8. In this assay, desloratadine (0.1−100 µM) was incubated with CHH (0.5 million cells/ml) for up to 2 hours, after which the activity of CYP2C8 was measured with paclitaxel (10 µM) and amodiaquine (10 µM) as described in Materials and Methods. As shown in Fig. 3 and Table 2, desloratadine was not a direct or time-dependent inhibitor of CYP2C8 in CHH (IC50 values > 100 μM).

Evaluation of desloratadine as an inhibitor of CYP2C8 activity toward amodiaquine (left) and paclitaxel (right) in cryopreserved human hepatocytes. As described in Materials and Methods, the inhibition of CYP2C8, as measured by amodiaquine N-dealkylation (left panel) and paclitaxel 6α-hydroxylation (right panel), was performed with 0.1, 0.3, 1, 3, 10, 30, and 100 μM desloratadine in cryopreserved human hepatocytes (0.5 million cells/ml) in duplicate. Desloratadine was preincubated with CHH for 0, 30, and 120 minutes, after which CYP2C8 activity was measured with a 10-minute incubation with amodiaquine (10 µM) or 30-minute incubation time with paclitaxel (10 µM).
Assessment of Desloratadine as an Inhibitor of 13 UGT Enzymes.
Desloratadine was evaluated as an inhibitor of 9 UGT enzymes in HLM (≤0.1 mg/ml) and 13 recombinant UGT enzymes (≤ 0.1 mg/ml) as described in Table 1 and Materials and Methods. As shown in Table 3 and Fig. 4, desloratadine (10 μM) caused no inhibition (<15%) of UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, UGT2B15, UGT1A7, and UGT1A8 and caused weak-to-moderate inhibition of UGT2B17 (24% HLM, 38% rUGT), UGT1A10 (46%), and UGT2B4 (38%). In contrast, UGT2B10 activity, as measured by levomedetomidine N-glucuronidation, was strongly inhibited (84% HLM, 74% rUGT) by desloratadine. When 100 μM desloratadine was assessed, most UGTs were inhibited by >20% (data not shown). The UGT2B10 inhibition assay was repeated with a wide range of concentrations of desloratadine (0.1–10 µM) and multiple levomedetomidine concentrations spanning Km, which established that desloratadine inhibited UGT2B10 in a competitive manner with a Ki of 1.3 μM, as shown in Fig. 5.
Assessment of desloratadine as an inhibitor of UGT enzymes in pooled human liver microsomes (HLM) and recombinant enzymes (rUGT)

Evaluation of desloratadine (10 µM) as an inhibitor of UGT enzymes in human liver microsomes and recombinant enzymes. The UGT inhibition potential of desloratadine (10 μM) was assessed in HLM (≤0.1 mg/ml) or rUGT (≤0.25 mg/ml) in triplicate samples with UDP-GlcUA and the UGT-selective substrates identified in Table 3 (at a final concentration approximately equal to its Km). The incubation time was 5 to 30 minutes. Experimental details are described in Table 1 and Materials and Methods.

Ki determination of the UGT2B10 inhibition potential by desloratadine in HLM. Desloratadine (0, 0.1, 0.3, 0.6, 1, 2, 4, and 10 μM) was evaluated as an inhibitor of UGT2B10 (levomedetomidine N-glucuronidation) in HLM (0.1 mg/ml) in duplicate samples. Multiple substrate concentrations were evaluated (0.3 Km, 1 Km, 2 Km, and 5 Km), and the incubation time was 5 minutes, as described in Materials and Methods.
Further Characterization of the UGT Enzymes Involved in 3-Hydroxydesloratadine Formation.
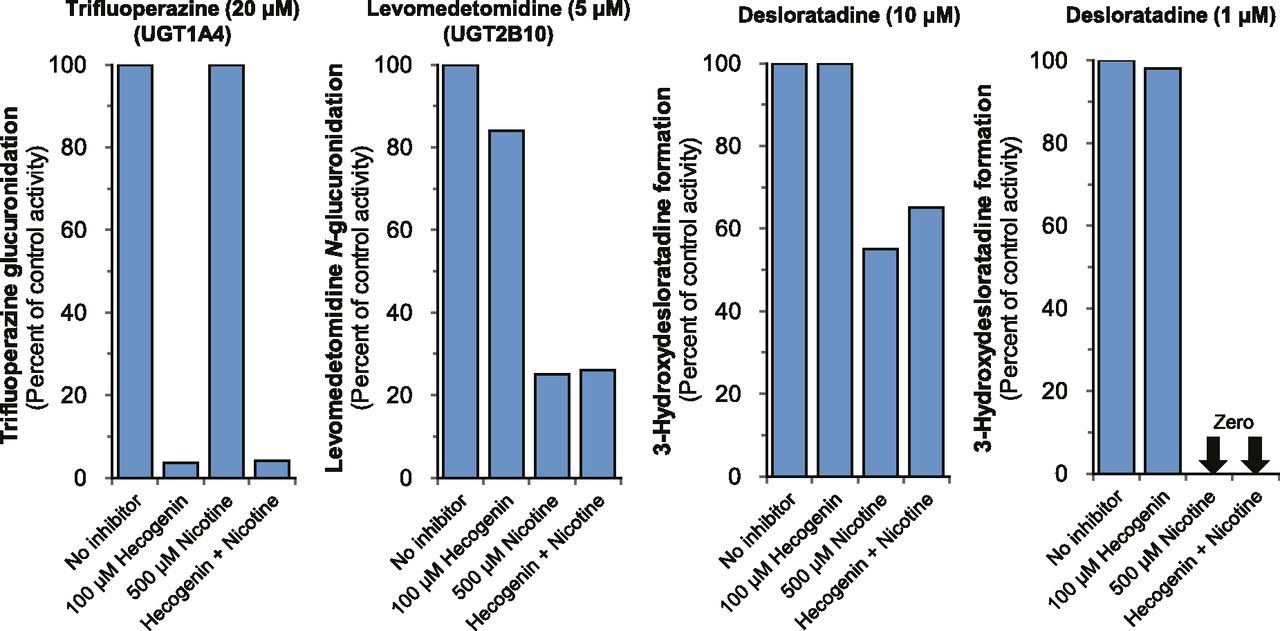
We previously identified recombinant UGT2B10 as the only UGT enzyme capable of supporting the CYP2C8-dependent formation of 3-hydroxydesloratadine (Kazmi et al., 2015). However, this assessment was based on studies with a panel of recombinant UGT enzymes (each of which was coincubated with recombinant CYP2C8). Although these studies implicated UGT2B10 in the formation of 3-hydroxydesloratadine, they do not exclude the possibility that HLM contain other UGT enzymes that contribute to this reaction. UGT2B10 is one of two enzymes renowned for its ability to form N-glucuronides; the other is UGT1A4 (Kaivosaari et al., 2011; Kato et al., 2013; Parkinson et al., 2013). To evaluate the role of UGT2B10 and UGT1A4 in the formation of 3-hydroxydesloratadine, inhibitors of either UGT1A4 (hecogenin; 100 μM) or UGT2B10 (nicotine; 500 μM) were added separately or in combination to HLM (0.1 mg/ml) in the presence of NADPH and UDP-GlcUA, followed by assessment of 3-hydroxdesloratadine formation and/or measurement of UGT1A4 activity (trifluoperazine N-glucuronidation) and UGT2B10 activity (levomedetomidine N-glucuronidation), as described in Materials and Methods. As shown in Fig. 6, trifluoperazine N-glucuronidation was inhibited only by the UGT1A4 inhibitor hecogenin, whereas levomedetomidine N-glucuronidation was inhibited only by the UGT2B10 inhibitor nicotine. When NADPH- and UDP-GlcUA-fortified HLM were incubated with 10 μM desloratadine, hecogenin did not inhibit 3-hydroxydesloratadine formation, whereas nicotine and a combination of hecogenin and nicotine caused moderate inhibition of 45 and 35%, respectively. When the concentration of desloratadine was lowered to 1 μM, complete inhibition of 3-hydroxydesloratadine formation was observed with nicotine and hecogenin + nicotine, whereas hecogenin caused no inhibition of 3-hydroxydesloratadine formation. These results suggest that UGT2B10 is the only UGT enzyme in HLM capable of catalyzing the N-glucuronidation of desloratadine to support its 3-hydroxylation by CYP2C8.

The effect of the UGT1A4 inhibitor hecogenin and the UGT2B10 inhibitor nicotine on the formation of 3-hydroxydesloratadine by NADPH- and UDP-GlcUA-fortified human liver microsomes (HLM). Inhibitors of UGT1A4 (hecogenin; 100 μM) and UGT2B10 (nicotine; 500 μM) or the combination of both were examined for their ability to inhibit the conversion of desloratadine (1 and 10 µM) to 3-hydroxydesloratadine by human liver microsomes (0.1 mg/ml) supplemented with NADPH and UDP-GlcUA (in triplicate). The activity of UGT1A4 and UGT2B10 were measured as trifluoperazine N-glucuronidation and levomedetomidine N-glucuronidation, respectively. Experimental details are described in Materials and Methods.
Correlation of 3-Hydroxydesloratadine Formation with Levomedetomidine Glucuronidation and CYP2C8 Activity.
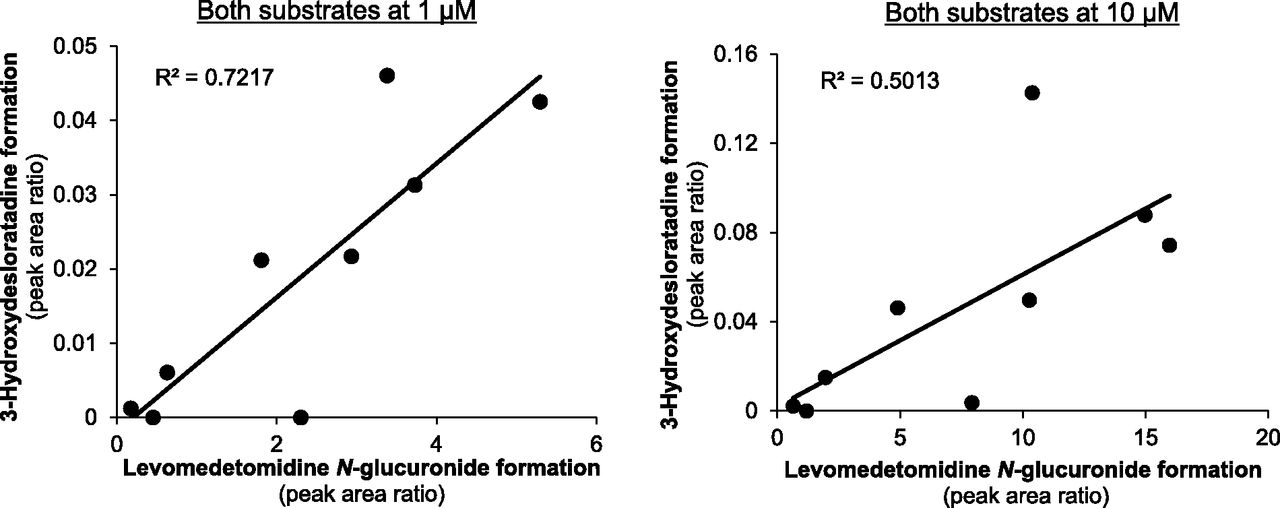
In our previous study we examined the sample-to-sample variation in 3-hydroxydesloratadine formation and known CYP2C8 activities in CHH (Kazmi et al., 2015). In the present study we compared the sample-to-sample variation of 3-hydroxydesloratadine formation in CHH with levomedetomidine N-glucuronidation, a marker of UGT2B10 activity. Hepatocytes from the same nine individual donors as our previous study were assessed for their ability to form levomedetomidine N-glucuronide, as described in Materials and Methods. As shown in Fig. 7, the sample-to-sample variation in levomedetomidine glucuronidation (at 1 μM levomedetomidine) correlated well with 3-hydroxydesloratadine formation (at 1 μM desloratadine), with an r2 value of 0.72. At 10 μM for both substrates, the correlation was poorer, with an r2 value of 0.50. Compared with CYP2C8 activity (determined in our previous study), all three activities (CYP2C8, UGT2B10, and 3-hydroxydesloratadine formation) correlated well at 1 μM, with r2 values of 0.75 (amodiaquine, levomedetomidine, and desloratadine 3-hydroxylation) and 0.73 (paclitaxel, levomedetomidine, and desloratadine 3-hydroxylation), as shown in Fig. 8.

Correlation of 3-hydroxydesloratadine formation with levomedetomidine N-glucuronide formation in a panel of individual donor cryopreserved human hepatocytes (CHHs). As described in Materials and Methods, nine individual donor CHH lots were assessed (at 1 million cells/ml) for levomedetomidine glucuronidation (UGT2B10) activity and 3-hydroxydesloratadine formation with 1 or 10 μM levomedetomidine and desloratadine for 30 minutes and 2 hours, respectively (in triplicate).

Correlation between CYP2C8 activity, UGT2B10 activity and 3-hydroxydesloratadine formation in individual CHH. CYP2C8 activity toward amodiaquine or paclitaxel (data from Kazmi et al., 2015), UGT2B10 activity toward levomedetomidine and 3-hydroxydesloratadine formation (at 1 μM desloratadine) were correlated (in triplicate), and the regression plane was determined as described in Materials and Methods. The two panels show the correlation with two CYP2C8 substrates, namely amodiaquine (left) paclitaxel (right).
Discussion
Desloratadine (marketed as Clarinex in the United States and Aerius in Europe) received FDA approval in 2001 for the treatment of allergic rhinitis and chronic idiopathic urticaria (Geha and Meltzer, 2001; Henz, 2001). The major human circulating metabolite of desloratadine is 3-hydroxydesloratadine, a metabolite whose enzymology remained a mystery for over 20 years due, in large part, to the inability of conventional in vitro test systems, such as subcellular fractions, to form 3-hydroxydesloratadine (Ghosal et al., 2009). As described previously, we demonstrated that CHH are capable of forming 3-hydroxydesloratadine, with a Km of 1.6 μM (Kazmi et al., 2015). Studies with P450 inhibitors in CHH and comparisons of the sample-to-sample variation in 3-hydroxydesloratadine formation with the variation in P450 activities in CHH (correlation analysis) implicated CYP2C8 in the conversion of desloratadine to 3-hydroxydesloratadine (Kazmi et al., 2015). These findings in CHH made the inability of HLM to form 3-hydroxydesloratadine all the more puzzling because HLM contain functional CYP2C8. Furthermore, recombinant CYP2C8 was also incapable of converting desloratadine to 3-hydroxydesloratadine despite catalyzing high rates of metabolism of substrates like paclitaxel and amodiaquine. The seemingly paradoxical findings were resolved with the discovery that the substrate for CYP2C8 is not desloratadine itself but its N-glucuronide, as shown in Fig. 1. When individual recombinant UGT enzymes were coincubated with recombinant CYP2C8, it was found that UGT2B10 was the only UGT enzyme capable of supporting the CYP2C8-dependent conversion of desloratadine to 3-hydroxydesloratadine (Kazmi et al., 2015).
In the present study, we examined the perpetrator potential of desloratadine based on its ability to inhibit P450 and UGT enzymes in HLM and a panel of recombinant UGT enzymes, some of which are not expressed in HLM [namely, UGT1A7, UGT1A8, and UGT1A10 (Parkinson et al., 2013)]. The direct but not time- or metabolism-dependent P450 inhibition potential of desloratadine was previously evaluated by Barecki et al. (2001). Since this study was published, subsequent regulatory guidelines expanded the panel of P450 enzymes recommended for testing (CYP2B6 and CYP2C8 were added), and they further recommended an evaluation of both reversible and irreversible inhibition of P450 enzymes. Inhibition of CYP2B6 or CYP2C8 is the basis for certain clinically relevant DDIs, such as the irreversible inhibition of CYP2B6 by clopidogrel and ticlopidine, leading to clinical interactions with bupropion, efavirenz, and ketamine (Richter et al., 2004; Turpeinen et al., 2005; Peltoniemi et al., 2011; Jiang et al., 2013). Likewise, the irreversible inhibition of CYP2C8 by gemfibrozil glucuronide or clopidogrel glucuronide leads to a clinical interaction with cerivastatin that resulted in the withdrawal of cerivastatin from the market (Backman et al., 2002; Ogilvie et al., 2006; Tornio et al., 2014).
In the present study, the inhibition of seven P450 enzymes, namely CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5, was evaluated with a single concentration of desloratadine (10 μM; approximately 10 times Cmax) at low HLM concentrations (≤0.1 mg/ml) with a short probe-substrate incubation time (5 minutes) and included an assessment of reversible inhibition (no preincubation), time-dependent inhibition (30-minute preincubation of desloratadine and HLM without NADPH) and metabolism-dependent inhibition (30-minute preincubation of desloratadine and HLM with NADPH). There was no evidence that desloratadine caused significant time- or metabolism-dependent inhibition of any of the P450 enzymes examined, but it did cause reversible inhibition of certain enzymes. Consistent with the data reported by Barecki et al. (2001), the results of this study (Fig. 2 and Table 2) showed that desloratadine is not an inhibitor of CYP1A2, CYP2C9, and CYP2C19 but is a moderate inhibitor of CYP2D6 and CYP3A4/5 The slightly greater degree of CYP2D6 and CYP3A4 inhibition observed in the current study (32 and 44%, respectively) compared with the Barecki study (14 and 16–20%, respectively) can reasonably be attributed to differences in experimental design. Barecki et al. (2001) used relatively high protein concentrations (up to 10 times those used in the current study), which would have lowered the unbound concentration of desloratadine to a greater extent. In addition to inhibiting CYP2D6 and CYP3A4/5, desloratadine also inhibited CYP2B6 (∼48%).
The ability of desloratadine to inhibit CYP2C8 was evaluated in HLM and CHH. Hepatocytes were used to evaluate the possibility that CYP2C8 could be inhibited by desloratadine N-glucuronide. No inhibition of CYP2C8 was observed in either HLM or CHH (Table 2, Figs. 2 and 3). These results are consistent with a clinical report demonstrating no effect of desloratadine on serum levels of the CYP2C8 substrate montelukast (Cingi et al., 2013). Few clinical studies have been performed to evaluate desloratadine as an inhibitor of CYP2D6 or CYP3A4. Desloratadine does not affect the pharmacokinetics of fluoxetine or azithromycin to a clinically significant extent (Gupta et al., 2001; Gupta et al., 2004). The previously reported requirement of UDP-GlcUA in 3-hydroxydesloratadine formation led us to investigate the perpetrator potential of desloratadine toward 10 UGT enzymes expressed in liver (namely, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B4, UGT2B7, UGT2B10, UGT2B15, and UGT2B17) and three UGT enzymes expressed in the gastrointestinal tract (namely, UGT1A7, UGT1A8, and UGT1A10) (Parkinson et al., 2013). Desloratadine was identified as a potent and relatively selective inhibitor of UGT2B10 (Fig. 4 and Table 3), consistent with its involvement in 3-hydroxydesloratadine formation. Desloratadine inhibited UGT2B10 with a Ki of 1.3 μM (Fig. 5), which is comparable to the reported clinical plasma Cmax value of 1.3 μM (Clarinex label; www.accessdata.fda.gov/drugsatfda_docs/label/2001/21165lbl.pdf).
In accordance with regulatory guidelines, the potential clinical relevance of P450 inhibition in vitro can be evaluated based on the [I]/Ki ratio, (where [I] is the total unbound plasma Cmax concentration and Ki is the unbound inhibition constant) and is used for in vitro to in vivo extrapolation to predicted the fold change in area under the curve (AUC) of a victim drug (where AUC with inhibitor/AUC without inhibitor = 1 + [I]/Ki) (Ito et al., 1998). In the absence of an experimentally determined Ki value, the Ki value can be estimated from the IC50 value using the Cheng-Prusoff equation, where Ki = IC50/2 for competitive inhibition, when the substrate concentration is equal to Km (Cheng and Prusoff, 1973; Haupt et al., 2011). This same approach has also been applied for the prediction of UGT-mediated DDIs (Williams et al., 2004; Miners et al., 2010). The fuinc for desloratadine would be 0.84 based on a logP value of 4 (from www.drugbank.ca) using the equation from Hallifax and Houston (2006). With this approach, the unbound Ki value is 1.1 μM (based on our experimentally determined Ki value); therefore, desloratadine is predicted to cause a 2.2-fold increase in the plasma AUC of a drug that is cleared exclusively by UGT2B10. To our knowledge, there is no known drug exclusively cleared through UGT2B10. Many drugs that are substrates of UGT enzymes are commonly cleared through other pathways, and many substrates of UGT2B10 are also N-glucuronidated by UGT1A4 (Kaivosaari et al., 2011; Kato et al., 2013; Parkinson et al., 2013). Nevertheless, the discovery that desloratadine at 10 μM is a relatively selective UGT2B10 inhibitor is important for in vitro UGT reaction phenotyping studies; it can be used as a chemical inhibitor to determine the fraction of a drug metabolized through UGT2B10.
Both UGT1A4 and UGT2B10 are well known for their ability to N-glucuronidate drugs such as amitriptyline, imipramine, ketotifen, pizotifen, olanzapine, diphenhydramine, tamoxifen, ketoconazole, and midazolam (Kaivosaari et al., 2011; Kato et al., 2013). Previously, we determined that recombinant UGT2B10 was the only UGT enzyme capable of supporting the CYP2C8-dependent conversion of desloratadine to 3-hydroxydesloratadine (Kazmi et al., 2015). This finding does not establish that UGT2B10 is the only enzyme in HLM capable of supporting this reaction. It is possible, for example, that in a more complete test system such as HLM, UGT1A4 may play a role in addition to UGT2B10. To evaluate this possibility, we performed a chemical inhibition study using the UGT1A4 inhibitor hecogenin and the UGT2B10 inhibitor nicotine (alone or in combination) with HLM (supplemented with both UDP-GlcUA and NADPH) and examined the formation of 3-hydroxydesloratadine as well as the activity UGT1A4 (with trifluoperazine) and UGT2B10 (with levomedetomidine) (Fig. 6). The formation of 3-hydroxydesloratadine was inhibited by the UGT2B10 inhibitor nicotine but not by the UGT1A4 inhibitor hecogenin, confirming that UGT2B10 is solely responsible for the formation of 3-hydroxydesloratadine, consistent with the results of our previous study with recombinant enzymes (Kazmi et al., 2015). Furthermore, previously we had correlated known CYP2C8 activities with 3-hydroxydesloratadine formation in hepatocytes from nine individual donors. We sought to correlate the UGT2B10 marker activity of these same donor hepatocytes with 3-hydroxydesloratadine formation and found that good correlation was achieved at 1 μM concentrations (r2 = 0.72; Fig. 7). A comparison of CYP2C8, UGT2B10, and 3-hydroxydesloratadine formation (Fig. 8) yielded good correlations (r2 = 0.73–0.75), confirming that both CYP2C8 and UGT2B10 are involved in 3-hydroxydesloratadine formation.
In conclusion, desloratadine was identified as a potent and relatively selective inhibitor of UGT2B10, the enzyme implicated in the N-glucuronidation of desloratadine. Desloratadine was a weak reversible inhibitor of a few P450 enzymes and showed no significant time- or metabolism-dependent inhibition of any of the P450 enzymes examined. Because of the instability of desloratadine N-glucuronide, desloratadine is unsuitable as a probe substrate for UGT2B10. However, as an inhibitor of UGT2B10, desloratadine can be used as part of standard in vitro chemical inhibition studies for the reaction phenotyping of UGT substrates, allowing for the determination of fractional metabolism by UGT2B10. These findings contribute to our understanding of the role of UGT enzymes in drug clearance and provide greater insight into the potential for desloratadine-mediated DDIs.
Acknowledgments
The authors thank the Analytical Sciences Department at XenoTech, LLC for technical assistance, Matt Beck for figure formatting, and Dr. David Buckley for helpful discussions and support.
Authorship Contributions
Participated in research design: Kazmi, Parkinson.
Conducted experiments: Kazmi, Yerino.
Performed data analysis: Kazmi, Yerino.
Wrote or contributed to the writing of the manuscript: Kazmi, Barbara, Parkinson.
Footnotes
- Received April 17, 2015.
- Accepted July 1, 2015.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the plasma concentration-time curve
- CHH
- cryopreserved human hepatocytes
- Cmax
- maximum plasma concentration
- DDI
- drug-drug interaction
- ESI
- electrospray ionization
- FDA
- U.S. Food and Drug Administration
- HLM
- human liver microsomes
- IC50
- inhibitor concentration causing 50% enzyme inhibition
- Km
- Michaelis-Menten constant (substrate concentration supporting half the maximum rate of an enzymatic reaction)
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- MRM
- multiple reaction monitoring
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}