


Abstract
The liver plays an important role in the disposition of acyl glucuronides by determining their extent of formation, biliary excretion, and efflux into blood. Thus, both intrahepatic and extrahepatic exposure to these reactive polar conjugates depends on the efficiency of hepatic transport mechanisms, which may be shared with other nonbile acid organic anions. Using the isolated perfused rat liver preparation, the hepatic disposition of the acyl glucuronide, 1-O-gemfibrozil-β-d-glucuronide, was examined in the presence of the organic anion dibromosulfophthalein (DBSP). Using a recirculating system, livers were perfused for 90 min with an erythrocyte-free perfusion medium containing 1% (w/v) albumin and 1-O-gemfibrozil-β-d-glucuronide (3 μM) alone (n = 6) or with DBSP (200 μM,n = 7). The glucuronide was avidly taken up by the liver, excreted into bile, and hydrolyzed within the liver to its aglycone, gemfibrozil. DBSP significantly (P < .05) lowered the conjugate’s mean hepatic clearance (8.98–5.17 ml/min), intrinsic clearance (44.0–17.7 ml/min), and fraction eliminated in bile (72.8–48.7% of the dose), while increasing perfusate gemfibrozil concentrations (0.52–0.92 μM at 90 min). Furthermore, DBSP significantly (P < .05) lowered the ratio of intrahepatic to unbound perfusate concentrations of 1-O-gemfibrozil-β-d-glucuronide (139.0–35.0) and showed a trend to lower the ratio of bile to intrahepatic concentrations (111.3–76.2, P = .05). Thus, the study demonstrated that DBSP inhibited both the sinusoidal uptake and canalicular transport of 1-O-gemfibrozil-β-d-glucuronide, suggesting that the hepatic membrane transport of acyl glucuronides is carrier mediated and shared with other organic anions.
Many drugs, including nonsteroidal anti-inflammatory agents and fibrate hypolipidemic agents, possess a carboxylic acid functional group and are metabolized to acyl glucuronide conjugates. These conjugates are chemically reactive and susceptible to nucleophilic attack. Depending on the reacting nucleophilic species, substitution at the carbonyl group may result in hydrolysis to regenerate the parent carboxylic acid, formation of rearrangement isomers via migration of the parent group from the 1-O-β-position of the glucuronic acid ring, and formation of adducts via covalent binding to nucleophilic residues on proteins (Spahn-Langguth and Benet, 1992) and DNA (Sallustio et al., 1997b).
Acyl glucuronide conjugates are essentially fully ionized at physiological pH and are highly polar (Vore, 1994). Therefore, their movement between their primary site of formation, hepatocytes, and blood, bile, or extrahepatic tissues may be restricted by their limited ability to passively diffuse across the membrane barriers that separate these compartments. Moreover, their movement across these membranes may involve carrier systems, the characteristics of which are poorly understood.
In vitro studies using isolated hepatocytes have shown that bromosulfophthalein (BSP), bilirubin, and bilirubin monoglucuronide, but not the bile acid taurocholate, inhibit the cellular uptake of preformed bilirubin diglucuronide (Adachi et al., 1991). Studies using canalicular membrane vesicles from TR− rats (Nishida et al., 1992), which retain normal bile acid transport (Kuipers et al., 1989) but have genetically defective canalicular ATP-dependent transport of nonbile acid organic anions such as dibromosulfophthalein (DBSP) (Jansen et al., 1987), have shown impaired canalicular transport of bilirubin diglucuronide. Both the ATP- and membrane potential-dependent canalicular transport of bilirubin diglucuronide were inhibited by BSP (Nishida et al., 1992). BSP and DBSP are cholephilic dyes that are commonly used as markers for hepatic sinusoidal and canalicular membrane transport systems of nonbile acid organic anions. A study with nafenopin acyl glucuronide demonstrated that its transport across the canalicular membrane in TR− rats also was impaired (Jedlitschky et al., 1994). In addition, canalicular transport of nafenopin acyl glucuronide in normal rats was ATP dependent and inhibited by the organic anions cystenyl leukotriene and S-(2,4-dinitrophenyl)-glutathione (DNP-SG) (Jedlitschky et al., 1994). These studies suggest that the carrier-mediated sinusoidal uptake and canalicular secretion of the acyl glucuronides of bilirubin and nafenopin are shared with other nonbile acid organic anions.
The acyl glucuronide 1-O-gemfibrozil-β-d-glucuronide (GG) is a major metabolite of the fibrate hypolipidemic agent gemfibrozil (Curtis et al., 1985; Knauf et al., 1990). Previous studies have shown that GG is efficiently excreted into the bile of rats (Curtis et al., 1985), and indirect evidence indicates its biliary excretion in humans (Knauf et al., 1990). Using the rat isolated perfused liver model, we previously investigated the disposition of GG and demonstrated that it undergoes avid hepatic uptake and biliary secretion and that the liver is involved in the conversion of GG to its aglycone (Sallustio et al., 1996). The aim of the present study was to investigate the hepatic disposition of preformed GG in the presence of a potentially competing organic anion, DBSP, using the rat isolated perfused liver. By using the intact liver, it was possible to investigate the effect of DBSP not only on the movement of GG across hepatic membranes but also on the extent of conversion of the conjugate to the aglycone.
Experimental Procedures
Materials.
Gemfibrozil, β-glucuronidase, sodium taurocholate, phenylmethylsulfonyl fluoride, andd-saccharic acid-1,4-lactone were purchased from Sigma Chemical Co. (St. Louis, MO). GG was biosynthesized and purified as previously described (Sallustio and Fairchild, 1995) and stored at −20°C. Bovine serum albumin (Pentex, Fraction V) was purchased from Miles Inc. (Kankakee, IL). DBSP was purchased from Societe d’Etutes et de Reserches Biologiques (Paris, France). All other reagents were of analytical grade.
Liver Perfusion.
The studies were approved by the animal ethics committees of the Queen Elizabeth Hospital, the University of Adelaide, and the University of South Australia. Male Sprague-Dawley rats (250–350 g) were used, and livers were perfused in situ as described previously (Sallustio et al., 1996). Perfusions were performed using erythrocyte-free Krebs-bicarbonate buffer (0.25 liters, pH 7.4) supplemented with glucose (3 g/liter), sodium taurocholate (4.5 mg/liter), and albumin (1% w/v); gassed with humidified carbogen (5% CO2/95% O2); and pumped through the portal vein at a constant flow rate of 30 ml/min. In this recirculating system, the hepatic outflow was returned to the perfusate reservoir via a cannula inserted into the superior vena cava. The common bile duct was cannulated to allow collection of bile, the flow rate of which was determined gravimetrically assuming a specific gravity of 1. Sodium taurocholate was continuously infused (7.74 mg/h) into the perfusion medium to maintain adequate concentrations of the bile acid to promote bile flow. The whole preparation was maintained at 37°C within a thermostatically controlled perfusion cabinet. The viability of the isolated perfused liver was assessed by monitoring oxygen consumption (>10 μmol/min), bile flow (>5 μl/min), percent recovery of perfusate (>95%), and the gross appearance of the organ. The livers were perfused for an initial equilibration period of 20 min before the addition of GG to the perfusion medium in the reservoir.
In control perfusions (n = 6), GG was added as a single bolus to the perfusion medium reservoir to achieve an initial concentration of 3 μM. Assuming that concentrations of the conjugate are approximately 2–4% of the aglycone, a concentration of 3 μM was chosen to approximate the range of GG concentrations that may be achieved clinically (Knauf et al., 1990). In studies with DBSP (n = 7), the organic anion was added to achieve an initial concentration of 200 μM, 10 min before addition of GG. In preliminary investigations, using the recirculating rat isolated perfused liver, DBSP, at an initial perfusate concentration of 200 μM, exhibited biexponential disposition kinetics with a terminal half-life (T1/2) of 126 min (data not shown). The biliary excretion of DBSP appeared to achieve a maximum value of 160 nmol/min (data not shown), which is of the same order as the transport maximum reported previously (195 ± 20 nmol/min) for female rats (Auansakul and Vore, 1982). Thus, under the conditions used in the present study, the hepatic membrane transport of DBSP was saturated.
Perfusion medium (1 ml) was collected from the reservoir before and at 1, 2, 5, 7.5, 10, 15, 20, 30, 40, 50, 60, 70, 80, and 90 min after the addition of GG, and collected samples were stabilized by the addition of 15 μl of 1.5 M phosphoric acid. Bile samples were collected at 10-min intervals before and throughout the 90 min of perfusion. All samples were immediately frozen after collection and stored at −20°C. At the end of each perfusion, the liver was blotted dry, frozen, and stored at −80°C. On the next day, bile samples were thawed and diluted (1:200) in 1.0 M glycine buffer (pH 3.0). The diluted bile samples were stored at −20°C until analysis.
Protein Binding of Gemfibrozil and GG in Perfusate.
GG or gemfibrozil was added to perfusion medium at 37°C to achieve concentrations of 2 to 133 μM and 20 to 200 μM, respectively. Stock solutions of GG and gemfibrozil were prepared in acetonitrile, and the amount of acetonitrile added to the perfusate represented <2% of the total final volume of perfusate. For displacement studies with DBSP, the organic anion was added to the perfusate at a concentration of 200 μM, before the addition of either GG or gemfibrozil. The binding of GG and gemfibrozil was determined at 37°C by rapid ultrafiltration of 1-ml aliquots, in duplicate, using a micropartition filter (Centrifree; Amicon Corporation, Beverly, MA) centrifuged at 2000g for 15 min using an angled rotor. A 500-μl aliquot of ultrafiltrate was immediately stabilized by the addition of 50 μl of 0.3 M phosphoric acid and stored at −20°C until analysis. The fraction unbound (fu) was calculated as the concentration ratio of the ultrafiltrate to the unfiltered perfusion medium. Previous studies in this laboratory (Sallustio et al., 1996) have demonstrated that there was no nonspecific binding of GG and gemfibrozil to the ultrafiltration equipment.
Analytical Methods.
Concentrations of GG, its rearrangement isomers, and gemfibrozil in perfusion medium, bile, and ultrafiltrate were determined by direct high performance liquid chromatography analysis as described previously (Sallustio and Fairchild, 1995). The limits of quantification for GG and gemfibrozil were 0.06 and 0.08 μM, respectively. To determine intrahepatic concentrations of GG and gemfibrozil, livers were thawed, weighed, and homogenized in 0.15 M phosphate buffer (2 ml/g tissue) containing 2 mM phenylmethylsulfonyl fluoride and 40 mM d-saccharic acid-1,4-lactone to prevent degradation of GG during analysis (Ojingwa et al., 1994a), and 0.5 ml of the homogenate was assayed in four replicates. Calibration curves spanning a concentration range of 0.4 to 220 nmol/g liver (GG) and 8 to 320 nmol/g liver (gemfibrozil) were prepared by spiking liver homogenates from drug-free perfused rat livers. Flurbiprofen (100 μl of 2.5 mg/liter) was added as internal standard. Acetonitrile (1.3 ml) was added to the liver homogenates (0.5 ml), and the samples were vortexed and centrifuged (1000g for 15 min). The supernatant was transferred to a clean tube, and 1.0 M glycine buffer (1 ml, pH 3.0) and ethyl acetate (4 ml) were added. Samples were gently mixed on a horizontal shaker and centrifuged (1000g for 15 min). The organic layer was dried in an evacuated centrifuge and the residue reconstituted in high performance liquid chromatography mobile phase (0.25 ml) before analysis (Sallustio and Fairchild, 1995). Ratios of GG concentrations in liver to perfusate (total and unbound) and bile to liver were calculated at the 90-min time point. An assay for measuring the extent of covalently bound adduct formation in the liver was used as described previously (Sallustio and Foster, 1995). The limit of quantification for covalently bound gemfibrozil was 0.2 nmol/g liver. DBSP did not interfere with the analysis of GG and gemfibrozil in perfusate, bile, or liver.
Pharmacokinetic Analysis.
TheT1/2 of GG was determined by regression analysis of the log concentration-versus-time profile, and the area under the perfusate concentration versus time curve from 0 to 90 min [AUC(0–90)] was calculated by the trapezoidal method, and this was added to the extrapolated area to determine the area under the curve to infinite time [AUC(0-∞)].
For each liver perfusion experiment, the total clearance of GG (CL) was calculated as:
The fraction of the eliminated dose of GG cleared unchanged via biliary excretion (Bgg) was calculated as:
The hepatic extraction ratio (E) of GG was calculated as the ratio of CL to perfusate flow rate (Q), and intrinsic clearance (CLint) was calculated, assuming a well-stirred model (Wilkinson and Shand, 1975), as:
Statistical Analysis.
All values are presented as mean ± S.D. The nonparametric, unpaired Mann-Whitney U test (Prism 2.0; GraphPAD Software, San Diego, CA) was used to determine whether there were differences between the control and DBSP groups with respect to the pharmacokinetic parameters describing the disposition of GG and gemfibrozil and their respective concentrations in perfusate and bile at individual sampling times. Two-way analysis of variance (Prism 2.0) was used for the statistical analysis of bile flow rates, oxygen consumption rates, and protein-binding data. For all statistical tests, a P value of less than .05 was taken to represent significance.
Results
Protein Binding of GG and Gemfibrozil in Perfusate.
Analysis of the ultrafiltrate samples from protein-binding experiments revealed minimal degradation of GG during ultrafiltration. Approximately 10% of the total ultrafiltrate glucuronide content was rearrangement isomers of GG, and this may have been partly due to minor contamination (∼5%) of the biosynthetic sample of GG. No gemfibrozil was detected in any GG ultrafiltrate samples, suggesting negligible hydrolysis.
GG exhibited a moderate degree of binding to albumin, whereas gemfibrozil was more highly bound (Fig.1). The binding of GG over the range of 2 to 133 μM was linear with a mean fu of 0.30 ± 0.01 (Fig. 1). In contrast, the binding of gemfibrozil was nonlinear over the range of 20 to 200 μM, with an fu of less than 0.05 (Fig. 1; P < .05). In the presence of 200 μM DBSP, the fu of GG was increased to 0.36 ± 0.03 (P < .05); however, DBSP did not alter the binding of gemfibrozil to albumin (Fig. 1).

The binding of GG and gemfibrozil in perfusate containing 1% albumin at 37°C, alone (○) or in the presence of DBSP (•). Top, fu for GG. Bottom, fu for gemfibrozil.
Liver Perfusion Experiments.
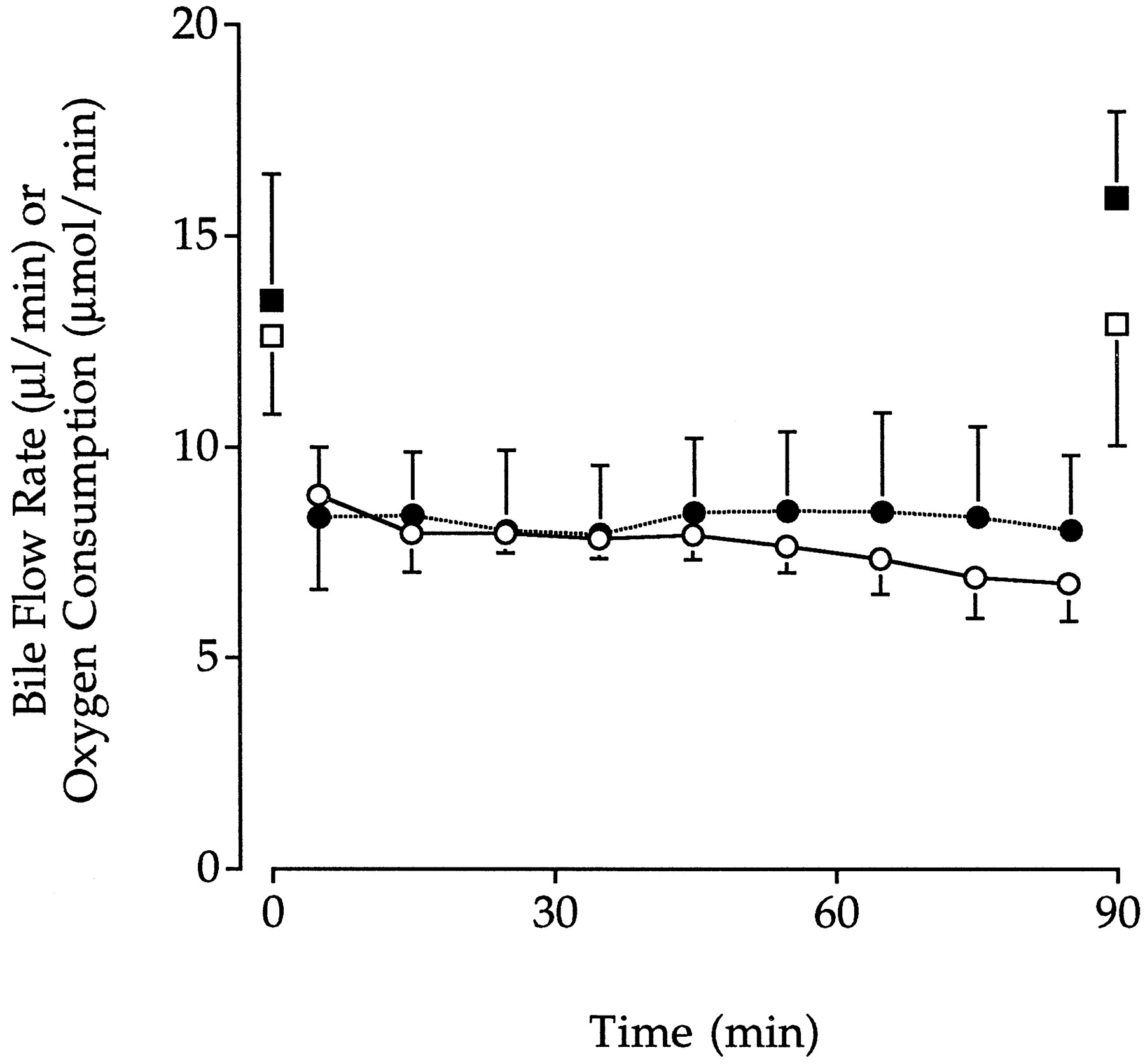
The viability of perfused livers was comparable between control and DBSP experiments. Throughout all perfusions, bile flow rate remained greater than 5 μl/min and did not change significantly over time (Fig. 2). Oxygen consumption, which was measured at the start and end of each perfusion, was greater than 10 μmol/min for both control and DBSP perfusions and did not change with time or between groups (Fig. 2).

Mean ± S.D. bile flow rates in control (○,n = 6) and DBSP (•, n = 7) groups and hepatic oxygen consumption rates in control (■,n = 6) and DBSP (▪, n = 7) groups.
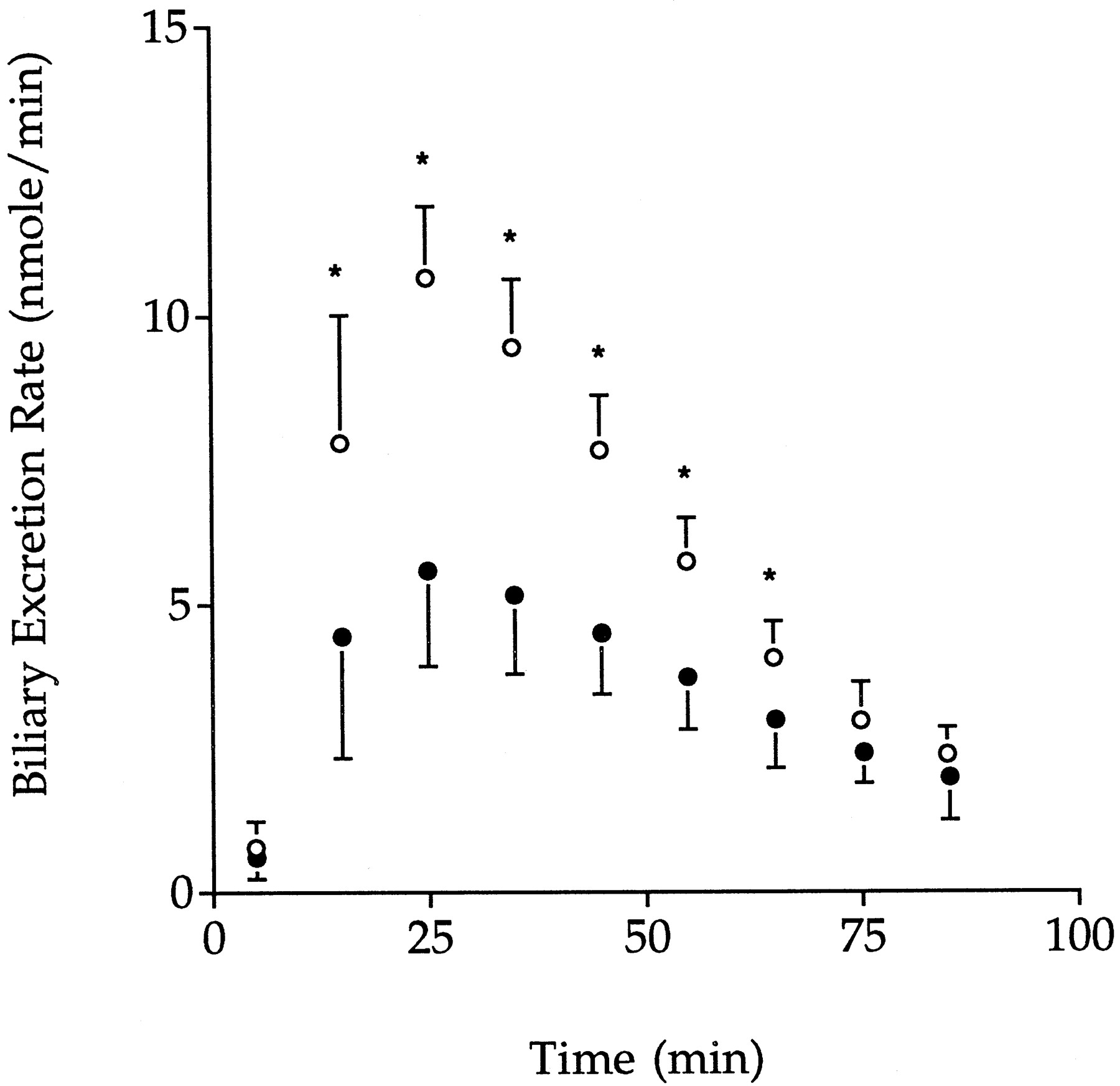
The perfusate concentration versus time profiles for GG and gemfibrozil are shown in Figs. 3 and4, respectively, and the biliary excretion rate versus time profiles for GG are shown in Fig.5. Under control conditions, perfusate concentrations of GG declined monoexponentially (Fig. 3) with a meanT1/2 of 16.0 min and a mean CL of 8.98 ml/min, corresponding to a hepatic extraction ratio of 0.30 and a mean CLint of 44.0 ml/min (Table1). Of the dose of GG that was cleared from perfusate, 72.8% was excreted into bile (Table 1). The appearance of gemfibrozil in perfusate was also observed (Fig. 4) reaching a maximum concentration of 0.62 ± 0.19 μM at 50.2 ± 0.04 min. The liver-to-perfusate and bile-to-liver concentration ratios were greater than unity (Table 1).

Mean ± S.D. perfusate concentration-time profiles of GG after administration of GG alone (○,n = 6) or in the presence of DBSP (•,n = 7). *Significant difference from control value (P < .05).
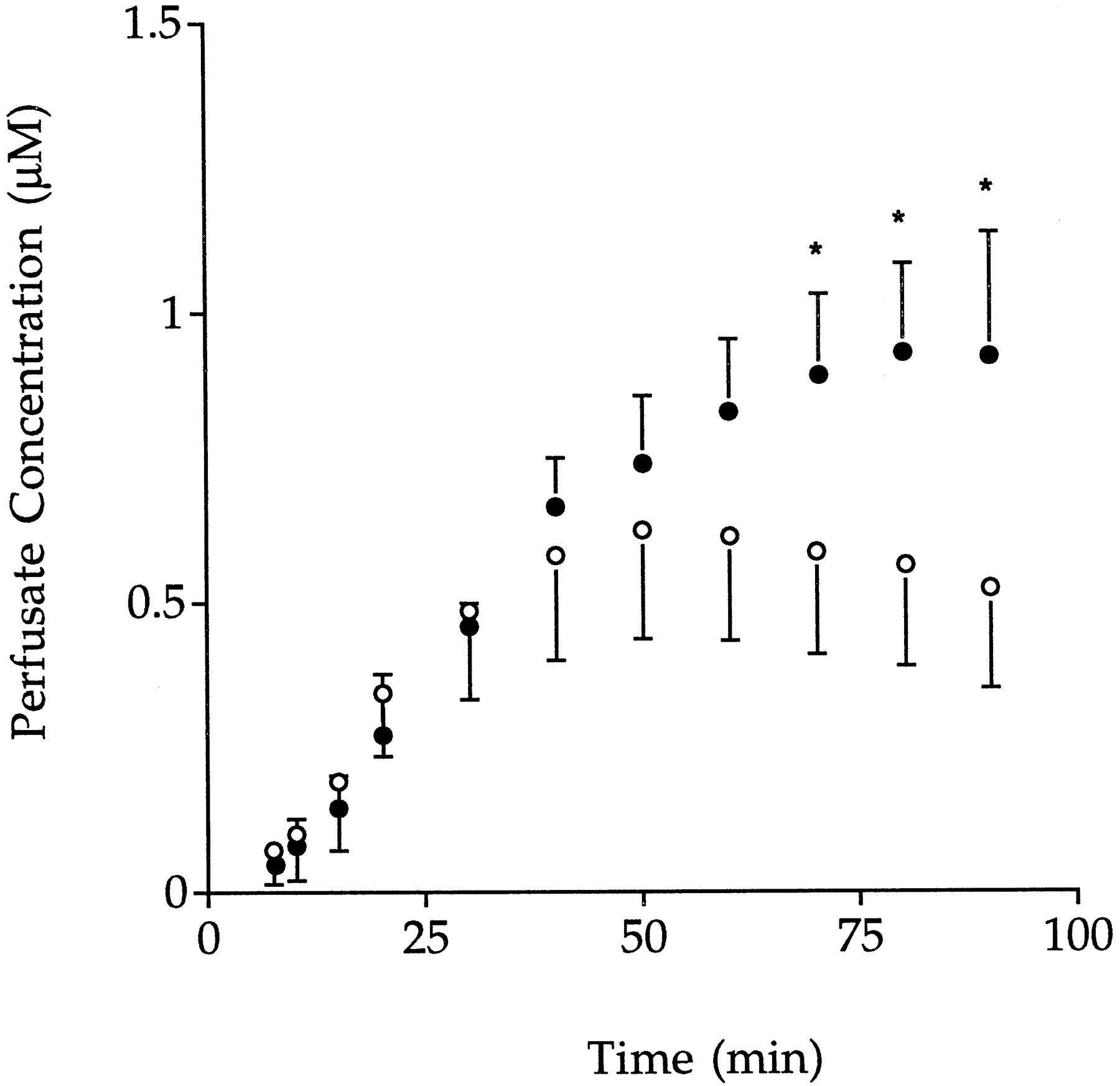
Mean ± S.D. perfusate concentration-time profiles of gemfibrozil after administration of GG alone (○,n = 6) or in the presence of DBSP (•,n = 7). *Significant difference from control value (P < .05).

Mean ± S.D. biliary excretion rate-time profiles for GG after administration of GG alone (○,n = 6) or in the presence of DBSP (•,n = 7). *Significant difference between groups (P < .05).
Pharmacokinetic parameters for the disposition of 1-O-gemfibrozil-β-d-glucuronide in the rat isolated perfused liver
The presence of DBSP significantly altered the pharmacokinetics of GG as shown in Figs. 3, 4, and 5 and Table1. Compared with control perfusions, perfusate concentrations of GG were higher (Fig. 3), reflecting a longerT1/2 and a lower CL, hepatic extraction ratio, and CLint (P < .05, Table1). Mean biliary excretion rates of GG in the presence of DBSP (Fig. 5) were significantly lower than the corresponding control values between 10 to 70 min of perfusion. The fraction of the dose of GG cleared from perfusate that was excreted into bile was 33% lower than for the control group (Table 1). In the presence of DBSP, the concentrations of gemfibrozil achieved in perfusate from 70 min were significantly higher than control values and, unlike control perfusions, continued to increase until 90 min (Fig. 4). The hepatic concentration of GG was not significantly different from the control (Table 1). The liver-to-perfusate (either total or unbound) concentration ratio of GG was significantly lower (P < .05) than the control value, and there also was a trend for the bile to liver concentration ratio to be lower (P = .05; Table 1).
In all experiments, no glucuronide rearrangement isomers were detected in either perfusate or bile, and no gemfibrozil was detected in bile. In addition, the concentration of covalently bound adducts in all livers was less than the limit of quantification of the assay.
Discussion
Consistent with a previous report from our laboratory (Sallustio et al., 1996), the present study demonstrates that GG is avidly taken up across the sinusoidal membrane into hepatocytes, wherein it is either transported across the canalicular membrane into bile or hydrolyzed to form its aglycone gemfibrozil. This study also confirms that the hepatically generated gemfibrozil is not excreted into bile but is subject to sinusoidal efflux into perfusate (Sallustio et al., 1996). An additional finding of the present study is the high concentration gradients for GG between the liver and perfusate and between bile and the liver. This is consistent with the concept that the movement of GG from perfusate into bile is a two-step concentrative process involving carrier-mediated systems at both the sinusoidal and canalicular membranes of hepatocytes.
The organic anion DBSP was found to cause a significant reduction in the hepatic intrinsic clearance of GG, leading in turn to a reduction in hepatic clearance and extraction ratio and a significant prolongation of T1/2. The reduction in hepatic intrinsic clearance of GG could conceivably be due to inhibition of sinusoidal uptake into hepatocytes, metabolism, and/or the movement of GG into bile. A schematic diagram of the disposition of GG in the rat isolated perfused liver and the potential sites at which DBSP could influence the disposition of GG is shown in Fig.6.

Schematic diagram of the disposition of GG in the isolated perfused liver preparation. GG is taken up into the liver across the sinusoidal membrane (1). Within the liver, GG is subject to hydrolysis to form gemfibrozil (G) (2), biliary excretion across the canalicular membrane (3), and sinusoidal efflux into perfusate (4). Hepatically generated G is subject to sinusoidal efflux into perfusate (5) and reuptake (6), reconjugation with glucuronic acid to form GG (7), and metabolism to other compounds (M) (8). *Potential site for interaction with DBSP.
The observed reductions in CLint and biliary excretion rate of GG in the presence of DBSP are unlikely to be due to an inherent toxic effect of DBSP on the liver as bile flow rate and hepatic oxygen consumption were unaffected by the addition of DBSP. Other studies that have used DBSP in vivo (Javitt, 1964; Klaassen and Plaa, 1968; Dhumeaux et al., 1974) or in the rat isolated perfused liver (Durham et al., 1985) have not reported changes in hepatic viability after DBSP administration. In addition, the observed effects of DBSP on the disposition of GG were unlikely to be due to altered cellular metabolism or depletion of cosubstrates because DBSP is excreted unchanged into bile (Javitt, 1964; Klaassen and Plaa, 1968). Furthermore, as the study was carried out with preformed glucuronide conjugate, a decrease in the glucuronidating capacity of the liver would not have been expected to markedly alter the hepatic disposition of the preformed conjugate. DBSP, however, is extensively bound to albumin (98% bound) (Meijer et al., 1977) and is a substrate for both the sinusoidal and canalicular organic anion transporter systems (Oude Elferink et al., 1995). Therefore, to identify the mechanism by which DBSP alters the disposition of GG, the potential competition for perfusate albumin binding sites and hepatic transport systems should be considered.
Both GG and gemfibrozil were bound to albumin, consistent with many findings for carboxylic acid drugs and their glucuronides such as zomepirac (Ojingwa et al., 1994b), tolmetin (Ojingwa et al., 1994b), ketoprofen (Hayball et al., 1992), carprofen (Iwakawa et al., 1990), and fenoprofen (Bischer et al., 1995). In the present study, it is clear that gemfibrozil had a higher affinity for albumin than did its glucuronide conjugate. Therefore, given the molar ratios of ligand to binding protein, it is not unexpected that gemfibrozil exhibited nonlinear binding to albumin. DBSP caused a significant increase in the fu of GG but had no effect on the binding of gemfibrozil. The different effects of DBSP on the binding of the two ligands to albumin are consistent with the lower affinity of GG for albumin compared with that of gemfibrozil. However, the result is also consistent with the presence of different binding sites on albumin for gemfibrozil and its glucuronide. Different binding sites on albumin have been reported for carprofen enantiomers and their acyl glucuronides (Iwakawa et al., 1990), whereas fenoprofen (Bischer et al., 1995), zomepirac (Ojingwa et al., 1994b), and tolmetin (Ojingwa et al., 1994b) share common binding sites with their respective acyl glucuronides.
With a hepatic extraction ratio of 0.3, GG may be classified as a low clearance compound, and thus its hepatic clearance should be dependent on fu and CLint (Wilkinson and Shand, 1975). The 42% lower CL of GG is the combined result of an increase in fu counteracted by a larger decrease in CLint. As discussed earlier, DBSP is unlikely to alter intrinsic metabolic activity; therefore, the observed reduction in CLint is likely to be due to an interaction with DBSP at the level of hepatic membrane transport.
A number of carrier-mediated transport systems have been identified for organic anions at both the sinusoidal and canalicular membranes of hepatocytes. At the sinusoidal membrane, a sodium-dependent transport system (Hagenbuch et al., 1990) and a sodium-independent transport system have been identified for the uptake of bile acids into the liver (Jacquemin et al., 1994), the latter system of which also mediates uptake of nonbile acid organic anions such as DBSP and BSP (Oude Elferink et al., 1995). In addition, at least three other putative carrier-proteins have been proposed to mediate the sinusoidal uptake of nonbile acid organic anions, including DBSP, BSP, and bilirubin (Groothuis and Meijer, 1996). Little information is available on the efflux of compounds from the liver across the sinusoidal membrane into the systemic circulation. However, the inhibition of sinusoidal efflux of morphine-3-glucuronide by probenecid (Evans et al., 1995) and of harmol sulfate by DBSP (De Vries et al., 1985) indicates that carrier-mediated efflux systems are present at the sinusoidal membrane. Transporters at the canalicular membrane include an ATP-dependent bile acid transporter and the ATP-dependent canalicular multispecific organic anion transporter (rat cmoat or human cMOAT), which exports nonbile acid organic anions such as cystenyl leukotriene, DBSP, bilirubin glucuronide conjugates, and DNP-SG (Keppler and Arias, 1997). As discussed earlier, the transport mechanisms for acyl glucuronides are poorly understood. Previous studies suggest that the transport mechanisms for acyl glucuronides at both the sinusoidal and canalicular membranes of hepatocytes may be shared with nonbile acid organic anions (Adachi et al., 1991; Jedlitschky et al., 1994).
In the present study, the lower CLint and lower biliary excretion rate of GG in the presence of DBSP may have been due to inhibition of sinusoidal uptake of GG into the liver and/or inhibition of canalicular transport into bile (Fig. 6). In the presence of DBSP, the liver-to-perfusate and bile-to-liver concentration ratios of GG were lower, indicating that DBSP inhibited both sinusoidal uptake and canalicular transport of GG. This is consistent with previous reports that DBSP inhibited the sinusoidal uptake of DNP-SG (Hinchman et al., 1993), pravastatin (Yamazaki et al., 1996), the glucuronide and sulfate conjugates of E3040 (Takenaka et al., 1997), and the canalicular transport of lithocholic acid 3-O-glucuronide (Kuipers et al., 1989) and liquiritigenin (ether) glucuronides (Shimamura et al., 1994) into bile. DBSP may also have inhibited sinusoidal efflux of GG (De Vries et al., 1985); however, this effect may have been masked by the significant inhibition of sinusoidal uptake.
It has previously been demonstrated that the liver is involved in the hydrolysis of GG to gemfibrozil (Sallustio et al., 1996). In the present study, higher perfusate concentrations of gemfibrozil were observed in the presence of DBSP, suggesting that a greater fraction of the dose of GG was hydrolyzed within the liver (Fig. 5). In the absence of an effect of DBSP on intrinsic cellular metabolism, this increase in gemfibrozil concentrations may have arisen from inhibition of canalicular transport of GG.
Reduced in vivo clearance of highly glucuronidated carboxylic acid drugs has previously been demonstrated as a consequence of decreased renal membrane transport of their acyl glucuronides (Meffin et al., 1983b). Reduced clofibric acid clearance in the presence of probenecid, in rabbits and humans, has been suggested to be due to competitive inhibition of the renal membrane transport of clofibric acid glucuronide, leading to reduced renal clearance and increased systemic hydrolysis to the aglycone (Meffin et al., 1983a). The present study shows that similar interactions between acyl glucuronides and organic anions may occur at the level of the hepatic transport systems, resulting in reduced hepatic clearance of the acyl glucuronide and increased systemic concentrations of the aglycone.
Acyl glucuronides are known to form covalently bound adducts with liver proteins (Hargus et al., 1994; Ojingwa et al., 1994a) As the major site of acyl glucuronide formation, the liver is a target site for adduct formation, and any factor that increases its exposure to acyl glucuronides may also increase the extent of adduct formation. In the present study, we were unable to detect any adduct formation, which is consistent with GG being one of the least reactive acyl glucuronides (Spahn-Langguth and Benet, 1992; Sallustio et al., 1997a).
In summary, the present study demonstrates that the hepatic membrane transport of the acyl glucuronide GG is carrier mediated and shared with other organic anions and that interactions at the level of hepatic sinusoidal and canalicular transport can lead to significant alterations in the hepatic disposition of acyl glucuronides. Furthermore, an indirect consequence of this interaction is the increased formation of the aglycone.
Footnotes
-
Send reprint requests to: Dr. B.C. Sallustio, Department of Clinical Pharmacology, The Queen Elizabeth Hospital, 28 Woodville Rd. Woodville South 5011, South Australia.
-
↵1 This study was supported in part by a University of South Australia Internal Research Development Grant and a National Health and Medical Research Foundation Grant. L.S. is funded by a Queen Elizabeth Hospital Postgraduate Research Scholarship.
- Abbreviations:
- Agg(0–90)
- amount excreted in bile over 90 min
- AUC(0-∞)
- area under the perfusate concentration versus time curve from 0 to infinity
- AUC(0–90)
- area under the perfusate concentration versus time curve from 0 to 90 min
- Bgg
- fraction cleared unchanged by biliary excretion
- BSP
- bromosulfophthalein
- CL
- total clearance
- CLint
- intrinsic clearance
- D
- dose
- DBSP
- dibromosulfophthalein
- DNP-SG
- S-(2,4-dinitrophenyl)-glutathione
- fu
- fraction unbound in perfusate
- GG
- 1-O-gemfibrozil-β-d-glucuronide
- Received March 6, 1998.
- Accepted August 7, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}