


Abstract
The effect of central nervous system inflammation on the levels and activity of hepatic and brain cytochrome P450 were examined in the rat. Brain ethoxyresorufin dealkylkase (EROD) was depressed during localized inflammatory responses evoked by lipopolysaccharide (LPS) injected into the lateral ventricle. This loss was accompanied by a concomitant loss of EROD activity and cytochrome P450 in liver. Similar losses in hepatic enzyme were observed for benzyloxy-resorufin and pentoxy-resorufin dealkylase (CYP2B) and chlorzoxazone hydroxylation (CYP2E). Protein levels of CYP2D and CYP2E1 but not CYP1A also were depressed. Similar i.p. doses of LPS had no effect on hepatic cytochrome P450, indicating that the hepatic effect was not caused by LPS leakage from the central nervous system. Also in support of this contention is that heat shock protein 27 was expressed throughout the brain by LPS given i.c.v. but was undetectable in the liver. Tumor necrosis factor-α given i.c.v. depressed EROD activity in the brain but this was not accompanied by a concomitant loss in the liver. Hepatic EROD did respond to the i.p. injection of tumor necrosis factor-α. The LPS-evoked loss in hepatic cytochrome P450 could not be prevented by blocking β-receptor-mediated sympathetic nerve activity. This study demonstrates that localized inflammatory responses in the brain cause a concomitant down-regulation of cytochrome P450 and drug-metabolizing activity in the liver and the brain. The effect on brain cytochrome P450 may be regulated via cytokine-mediated pathways but signaling to the liver does not involve a cytokine-mediated pathway nor a β-receptor-mediated sympathetic nerve pathway.
It is apparent that the system (host defense) that affords us protection from infectious organisms interacts with the principle system that protects us from chemicals or that generates reactive metabolites (drug-metabolizing enzymes). Numerous reports indicate that during host defense or inflammatory responses the levels of cytochrome P450 in the liver are modulated. The expression of constitutive enzymes, such as CYP3A2, CYP2C11, CYP2C12, and CYP2E1 (Craig et al., 1990; Morgan and Norman, 1990), in the rat and CYP1A2 and CYP2C6 (Stanley et al., 1991) in the mouse is known to be down-regulated by interferons. Endogenous interferon (IFN)-α/β also down-regulates all of the major inducible hepatic P450s, including CYP1A1, CYP1A2, CYP2E1, CYP2B isozymes, and CYP3A1 (Stanley et al., 1991; Delaporte et al., 1993). Cytokines such as interleukin (IL)-1, IL-2, IL-6, tumor necrosis factor (TNF)-α, and transforming growth factor have been shown to suppress the expression of CYP1A1 (Fukuda et al., 1992; Clark et al., 1995), CYP1A2 (Barker et al., 1992), CYP2B1/2 (Clark et al., 1995), and CYP3A3 (Fukuda et al., 1992). Tinel et al. (1995) have shown that the action of IL-2 on cytochrome P450 in rat hepatocytes requires the direct involvement of a cytokine receptor. Induction of a systemic inflammation by endotoxin or turpentine depresses the hepatic mRNAs for CYP2C11 and CYP2C12, presumably via cytokine production (Wright and Morgan, 1990). Several other reports indicate that the administration of lipopolysaccharide (LPS) depresses the expression of several cytochrome P450 forms in the liver in a number of species (Morgan, 1993; Monshouwer et al., 1996a,b;Barclay et al., 1999). The administration of LPS to human volunteers also can significantly decrease the elimination rates of drugs with cytochrome P450-dependent metabolism (Shedlofsky et al., 1997).
Even though the brain responds to inflammatory stimuli in a manner different from most tissues and is considered to be an immunologically privileged organ (due to blood-brain barrier, poor lymph drainage, and low antigen production), the tissue can and does respond to infection and injury with an inflammatory response that includes the production of cytokines, cell infiltration, and tissue damage (Perry et al., 1993;Rothwell et al., 1996). Cytokines, which are synthesized in glial cells, can produce a large number of responses involving the autonomic, endocrine, and behavioral systems (Rothwell et al., 1996). Recently, we have shown that the regulation of cytochrome P450 forms in the rat brain and in astrocyte cultures is disturbed during the activation of an inflammatory response by LPS (Nicholson and Renton, 1999; Renton et al., 1999). When localized inflammation occurs in response to brain injury a concomitant immunodepression or anti-inflammatory response can take place in peripheral organs (Woiciechowsky et al., 1998). When IL-1β or IFN-α are administered directly into the brain, the inflammatory and immune systems can be depressed in peripheral tissues, as measured by a suppression of IL-1 production, natural killer cell activity, and lymphocytic cell proliferation (Ichijo et al., 1994;Woiciechowsky et al., 1999). These reports indicate that insults to the central nervous system (CNS) can invoke signaling mechanisms of various types to cause changes to immune responses in peripheral organs. In this article, we provide evidence that LPS-evoked inflammatory responses in the brain evoke a depression of cytochrome P450 and its dependent drug metabolism in peripheral organs such as the liver. Because metabolism of drugs in the liver is largely responsible for the excretion and elimination of a large number of compounds, this suggests that drug clearance rates might be altered during inflammatory responses that are localized to the brain.
Experimental Procedures
Materials and Animal Treatment.
All reagents and chemicals were obtained from Sigma Chemical Co. (St. Louis, MO), except as noted below. Male Sprague-Dawley rats (175–200 g) obtained from Charles River Laboratories (Wilmington, MA) were housed on clay chips and allowed free access to Purina rat chow and water ad libitum. Rats were allowed to acclimatize in our facilities for 5 days before use. The animals were anesthetized with enflurane (3–4%) and LPS (Serotype 0127:B8) was injected into the lateral cerebral ventricle (i.c.v.) with a stereotaxic frame. The coordinates used for the injection were 1.5 mm lateral to the bregma and 4.7 mm below the skull surface. Volumes of injection were 10 μl of LPS (25 μg) dissolved in PBS given over 1 min and control animals received an equivalent volume of saline. After recovery from the anesthetic the animals were maintained with free access to water and food for various time periods. Other animals were given 25 μg of LPS by i.p. injection and compared with a group of animals given an equivalent volume of saline i.p. In some experiments TNF-α was administered by i.c.v. injection in a dose of 5 ng in 5 μl of saline or by i.p. injection in a dose of 0.4 μg. In the experiments involving β-receptor blockade, propranolol was administered at a dose of 1 mg/kg i.p. 5 min before LPS injection and at 8 and 24 h after LPS injection. Animals were then sacrificed 48 h after LPS administration.
Microsomal Drug Biotransformation.
At the end of the treatment time the whole brain was dissected, rinsed in ice-cold KCl (1.15%), and then homogenized in 8 ml of buffer containing Tris (0.1 M; pH 7.4), dithiothreitol (0.1 mM), EDTA (0.1 mM), KCl (1.15%), phenylmethylsufonyl fluoride (0.1 mM), butylated hydroxytoluene (22 μM), and glycerol (10%) with a glass-Teflon homogenizer (Bhagwat et al., 1995). The resulting homogenate was centrifuged for 10 min at 3000g, the supernatant was removed, and centrifuged at 100,000g for 40 min. The resulting pelleted membrane fraction was resuspended in 0.5 ml of buffer containing 20% glycerol with a mini glass/PTFE homogenizer. The membrane suspension was stored at −70°C in 50- or 100-μl aliquots. Hepatic microsomes were prepared by differential centrifugation as described (El-Masry et al., 1974) and were resuspendend in glycerol-phosphate buffer. Microsomes were stored until use at −70°C. Protein levels in the brain membrane fractions and in liver microsomes were measured by the method of Lowry et al. (1951). The oxidations of ethoxy, pentoxy, and benyloxy-resorufins (specific measures of CYP1A and CYP2B activity) were determined by the method of Burke et al. (1985). The hydroxylation of chlorzoxazone (a measure of CYP2E activity) was determined as described by Tindberg et al. (1996). Cytochrome P450 levels were determined by the method of Omura and Sato (1964).
Western Blot Immunoassay.
Microsomal proteins were diluted to 3 mg/ml protein, diluted 50% with Laemmili buffer, and 10 μl of each sample was separated by electrophoresis under nonreducing conditions with a 10% SDS-polyacrylamide gel (Smith, 1984). The protein bands were transferred to an Immobilon P membrane with a wet transfer apparatus (Bio-Rad, Hercules, CA). Protein bands were detected with goat monoclonal antibodies (Gentest, Woburn, MA) directed against rat CYP1A1/2 and CYP2E used at a 1:500 dilution. CYP2D was detected with a rabbit antibody directed against CYP2D forms (provided by Dr. A. Cribb, Merck Laboratories, West Point, PA) used at a 1:200 dilution. Bands were visualized with an anti-goat or anti-rabbit IgG conjugated to peroxidase (Sigma Chemical Co) used at in a dilution of 1/100,000 and Supersignal Ultra chemiluminescent substrate (Pierce, Rockford, IL). Band intensities were assessed with Molecular Analyst (Bio-Rad) and expressed as arbitrary units.
Immunohistochemistry.
At the end of the treatment period animals were deeply anesthetized with pentobarbital and perfused transcardially with 100 ml of 0.9% saline at room temperature followed by 60 ml of 4% paraformaldehyde as fixative. The brains and livers were dissected out and cryoprotected by placing each in 30% sucrose at 4°C until the organs became totally submerged. The tissues were then cut into 40-μm-thick coronal sections on a freezing microtome. Sections were then rinsed in 0.3% H2O2 in PBS for 10 min and blocked for 20 min with 10% rabbit serum in PBS at room temperature with gentle shaking. Sections were then placed in a solution of primary antibody [rabbit anti-mouse heat shock protein 27 (hsp27); Stressgene Biotechnologies, Victoria, Canada] diluted 1:2000 with PBS and 1% Triton X-100 and incubated for 48 h at 4°C. The tissues were then washed three times for 30 min each at room temperature with PBS and then incubated with biotinylated goat anti-rabbit-IgG (Vector Laboratories, Burlingame, CA) as secondary antibody (1:500) at 4°C overnight before incubation with Avidin biotin solution for 30 min. The tissues were then transferred to PBS for 12 h. The sections were then incubated with 0.05% 3,3′-diaminobenzidine tetrahydrochloride and 0.01% H2O2 solution for 5 min at room temperature. The tissues were then rinsed three times with PBS and mounted on glass slides.
Statistics.
Statistical analysis involving a comparison between two groups was carried out with a Student's t test. Comparison of four groups was carried out with ANOVA and a Student-Newman-Keuls test.
Results
Protein Content in Brain and Liver Membrane Fractions.
The total protein concentrations in the membrane fractions prepared from the brain and in the microsomes prepared from the liver were unaffected after 48 h LPS was administered as a single dose by either the i.c.v. or i.p. route as illustrated in Table1.
Total protein levels in brain and liver membrane fractions in rats given 25 μg LPS by i.c.v. or i.p. administration
Cytochrome P450 Levels and Activity in Liver and Brain Membrane Fractions.
Forty-eight hours after the administration of 25 μg of LPS directly into the lateral ventricle of the brain, the activity of ethoxyresorufin dealkylase (EROD) in whole-brain membrane fractions was significantly depressed by 38% compared with rats treated in a similar manner with saline (Fig. 1). In liver microsomes obtained from the same animals a similar loss (40%) in EROD activity was observed in the animals treated with 25 μg of LPS administered directly into the brain compared with the saline-treated control rats (Fig. 1). When the same dose of LPS (25 μg) was administered by the i.p. route the activity of EROD was unaffected in hepatic microsomes and in whole-membrane fractions obtained from the brain (Fig. 1). In animals treated with 25 μg of LPS administered directly into the brain, the levels of total cytochrome P450 in liver microsomes was depressed by 33% (Fig.2), whereas the levels remained unchanged when the LPS was administered by the i.p. route. The levels of total cytochrome P450 in the brain membrane fractions were below levels detectable by carbon monoxide-binding spectra and therefore could not be measured in these experiments. In animals treated with higher doses of LPS (0.5 mg/kg) given by the i.p. route, the activities of EROD in the brain and liver were depressed by 41 and 69%, respectively (Fig.3).

Effect of LPS given directly into the lateral ventricle of the brain or by i.p. injection on the activity of EROD in liver and brain. LPS (25 μg) was given for 48 h as a single i.c.v. or i.p. injection and control animals received an equivalent dose of saline. Saline-treated rats (▪) and LPS-treated rats (▨). Values are expressed as mean ± S.E., n = 4. ∗, significantly different from controls at P < .05.

Effect of LPS given directly into the lateral ventricle of the brain or by i.p. injection on the levels of cytochrome P450 in liver. LPS (25 μg) was given for 48 h as a single i.c.v. or i.p. injection and control animals received an equivalent dose of saline. Saline-treated rats (▪) and LPS-treated rats (▨). Values are expressed as mean ± S.E., n = 4. ∗, significantly different from controls at P < .05.
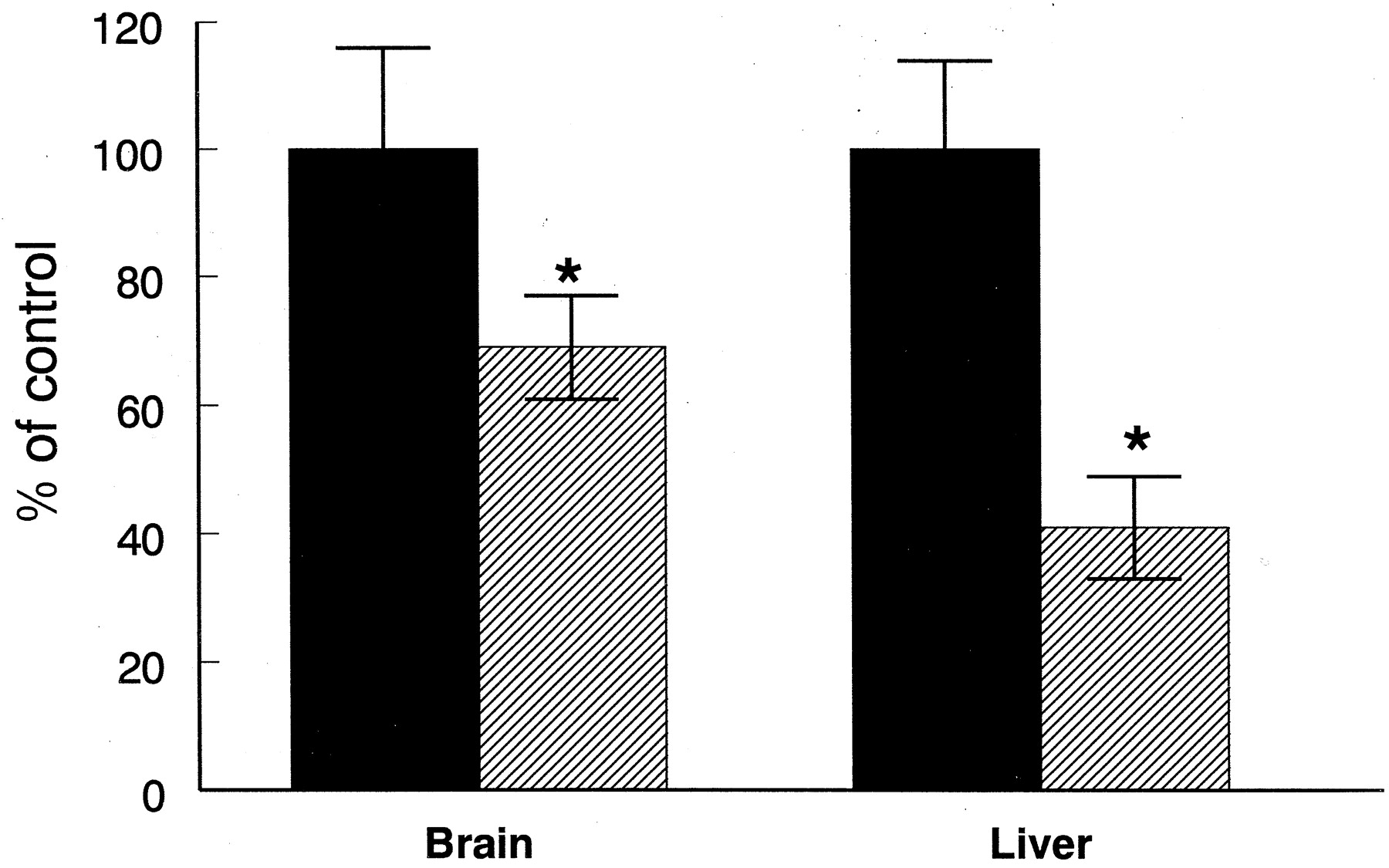
Effect a high dose of LPS given by i.p. injection the activity of EROD in liver and brain. LPS (0.5 mg/kg) was given for 48 h as a single i.p. injection and control animals received an equivalent dose of saline. Saline-treated rats (▪) and LPS-treated rats (▨). ∗, significantly different from controls atP < .05.
The dealkylation of two other alkyloxy resorufins was examined in the livers of rats treated i.c.v. with 25 μg of LPS as shown in Table2. In hepatic microsomes obtained from animals treated with LPS directly into the brain, pentoxyresorufin dealkylase (PROD) was significantly depressed by 49% and benzyloxyresorufin dealkylase (BROD) by 55%. The activities of BROD and PROD in hepatic microsomes were unaffected when the same dose of LPS was given by i.p. injection. The activity of CYP2E1 in hepatic microsomes, as measured by the hydroxylation of chlorzoxazone, was depressed by 33% after the administration of 25 μg of LPS by the i.c.v. route but this activity was unchanged when the LPS was administered by the i.p. route (Fig. 4). The levels of three individual cytochrome P450 forms were assessed by Western blotting in liver microsomes (Fig.5). The administration of LPS into the lateral ventricle of the brain resulted in a decrease in the levels of 31 and 36%, respectively, for CYP2D and CYP2E1 protein compared with saline-treated control animals. No significant loss in CYP1A protein was observed.
The activities of ethoxy-, pentoxy-, and benzyloxy-phenoxazone dealkylase in liver microsomes from rats given 25 μg LPS by i.c.v. or i.p. administration

Effect of LPS given directly into the lateral ventricle of the brain or by i.p. injection on the activity of chlorzoxazone hydroxylation in the liver. LPS (25 μg) was given for 48 h as a single i.c.v. injection and control animals received an equivalent dose of saline. Values are expressed as mean ± S.E.,n = 4. ∗, significantly different from controls at P < .05.
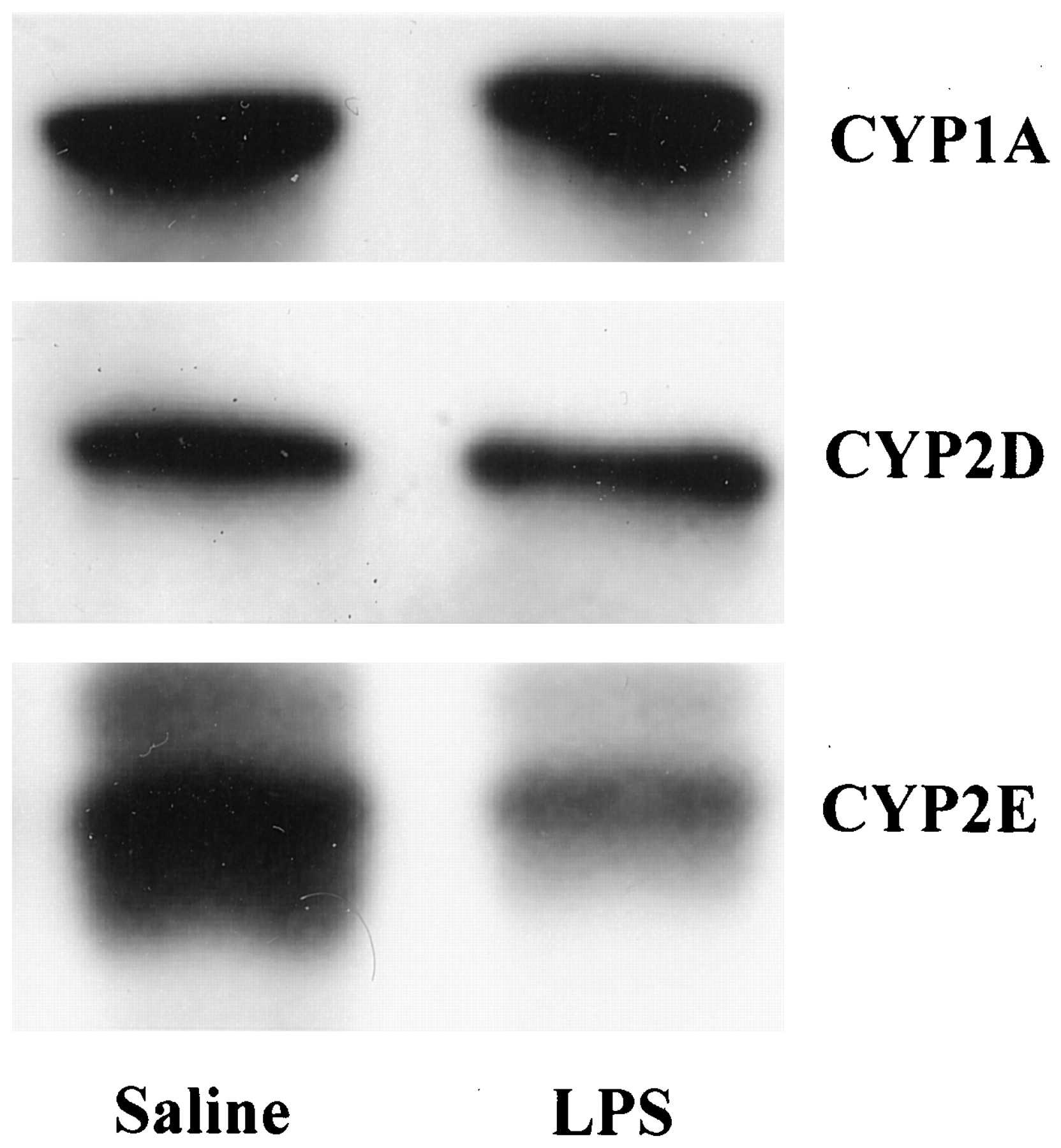
Effect of LPS given directly into the lateral ventricle of the brain on the levels of cytochrome P450 forms in the liver. Western blots of microsomes from rats given LPS (25 μg) as a single i.c.v. injection or saline for 48 h were probed with specific antibodies as described in Experimental Procedures. Representative blots from separate experiments for each cytochrome P450 form are illustrated and the following densitometer arbitrary values are expressed as mean ± S.E. for each blot, n = 4. CYP1A, saline 3.38 ± 0.78 versus LPS 2.96 ± 0.45; CYP2D, saline 2.53 ± 0.42 versus LPS 1.63 ± 0.43*; and CYP2E1, saline 6.45 ± 0.15 versus LPS 4.46 ± 0.18*. ∗, significantly different from controls atP < .05.
In animals given 5 ng of TNF-α directly into the lateral ventricle of the brain, the activity of EROD in whole-brain membrane fractions was significantly depressed by 27% but there was no concomitant decrease in the activity of EROD in liver microsomes prepared from the same rats (Fig. 6). When TNF-α was administered systemically in a dose of 0.4 μg by i.p. injection, the activity of EROD in hepatic microsomes was depressed by 43% (Fig. 6).

Effect of TNF-α given directly into the lateral ventricle of the brain or by i.p. injection on the activity of EROD in liver and brain. TNF-α was given for 24 h as a single i.c.v. (5 ng) or i.p.(0.4 μg) injection and control animals received an equivalent dose of saline. Saline-treated rats (▪) and TNF-α-treated rats (▨). Values are expressed as mean ± S.E.,n = 4. ∗, significantly different from controls at P < .05.
Sympathetic Nerve Blockade.
The ability of propranolol to block β-adrenergic receptors on the LPS evoked loss of cytochrome P450 is illustrated in Table 3. Propranolol itself had no effect on the activities of EROD or levels of cytochrome P450 in hepatic microsomes or on EROD activities in whole-brain membrane fractions (Table 3). Propranolol also was unable to prevent the loss in the activity of EROD and the loss in the levels of total cytochrome P450 in hepatic microsomes that occurred after the administration of LPS by i.c.v. (Table 3). In the brain similar results were obtained showing that propranolol was unable to block LPS-evoked losses in EROD activity (Table 3).
The effect of propranolol (1 mg/kg) on the loss of ethoxyresorufin dealkylase in the liver and brains of rats given 25 μg LPS by i.c.v. injection
Immunohistochemistry of hsp27.
The distribution of hsp27 in sections of brain and liver was used as a marker for LPS-evoked inflammatory responses. When coronal tissue sections obtained from the brain were examined with an antibody directed against hsp27, the protein had been extensively expressed throughout the brains of all four animals given LPS by i.c.v. injection. An example of the LPS-evoked expression of hsp27 in the cortex is shown in Fig.7B. In all animals treated with an equivalent volume of saline (n = 4) the expression of hsp27 was negligible (Fig. 7A) with only slight responses that could be observed in proximity to the injection needle track. In tissue sections taken from the livers of the same animals the expression of hsp27 was undetectable in all animals treated with LPS or saline. Examples of liver sections stained for hsp27 are shown in Fig. 7, C and D.

Effect of LPS (25 μg i.c.v.) on the expression of hsp27 in rat brain and liver. Coronal sections of brain and sections of liver were stained with an antibody directed against rat/mouse hsp27. Animals received saline (A and C) or LPS (B and D) 48 h previously. The brain sections (A and B) illustrate the response in the cortex and edge of the hippocampus. Similar responses were observed throughout the brain. No hsp27 expression could be observed in the liver (C and D) in either saline- or LPS-treated rats.
Discussion
This article demonstrates that during an inflammatory response caused by the injection of LPS into the lateral ventricle, there is a loss of cytochrome P450-dependent pathways of drug biotransformation in the brain with EROD as a marker enzyme for the CYP1A family. The activity, protein and mRNA for this cytochrome P450 family are located in discreet areas throughout the brain and are inducible by β-napthaflavone (McFadyen et al., 1997; Morse et al., 1998). The loss of brain EROD activity in these experiments is consistent with our previous reports that inflammation evoked by centrally administered LPS can down-regulate CYP1A in several different brain regions (Renton et al., 1999) and that LPS added to cultured astrocytes depresses the activity and protein levels of CYP1A were not altered (Nicholson and Renton, 1999).
This LPS-evoked loss of cytochrome P450 in the brain is accompanied by a concomitant decrease in several cytochrome P450-dependent pathways in the liver. Considering that this dose of LPS causes an inflammatory response localized to the brain the loss of EROD activity in the liver after the administration of LPS directly into the CNS is a surprising accompaniment to the loss of these enzymes in the CNS. The decrease in cytochrome P450 in the liver was not confined to the CYP1A family because the activity of BROD, PROD, and chlorzoxazone hydroxylation representing substrates metabolized by CYP2B and CYP2E and the protein levels of CYP2D and CYP2E also were depressed. The loss of several hepatic cytochrome P450 forms in response to the injection of an inflammatory agent directly into the CNS suggests that the enzyme system is modified via a common pathway. These findings are similar to those indicating a loss of several cytochrome P450 forms in the liver in response to the central injection of LPS (Shimamoto et al., 1998). When LPS is given systemically by i.p. injection it is known to cause an inflammatory response that results in a loss in hepatic cytochrome P450 (Wright and Morgan, 1990; Monshouwer et al., 1996a,b; Barclay et al., 1999); therefore, it is possible that these results are caused by a leakage of LPS from the CNS across the blood-brain barrier into the periphery. The doses of LPS given i.p. that were required to evoke a loss in hepatic cytochrome P450 are about 5 to 10 times greater per kilogram body weight than that administered by i.c.v. injection in this study. When the low doses of LPS were given i.p. there was no change to the enzymes in both the liver and the brain (Fig. 1). Leakage of LPS across the blood-brain barrier from the CNS to the peripheral system is, therefore, an unlikely explanation. We also show that high doses of LPS given i.p. modulate cytochrome P450 in the CNS and liver, which is consistent with reports indicating that high doses can induce immunoreactive responses in macrophages in the meninges and in microglial cells in various brain regions (van Dam et al., 1998). This probably results from a decrease in the integrity of the blood-brain barrier, which can break down in response to high doses of LPS (Banks et al., 1999). Also in support of the contention that the leakage of LPS from the brain does not cause the hepatic effects is that hsp27 was expressed throughout the brain in response to LPS given by i.c.v. injection but the levels of this protein in the liver were undetectable. This low molecular heat shock protein can be used to indicate inflammatory responses and is known to be induced in response to the administration of LPS (Beyaert et al., 1996). Collectively, these results suggest that the loss of cytochrome P450 in the liver that accompanies the direct administration of LPS into the CNS is caused by some type of signaling mechanism or the release of a mediator that is generated in the brain during the localized inflammatory response that is evoked in that organ by LPS.
Inflammatory responses in the brain are different from most other tissues because an intense inflammatory response might evoke irreversible tissue damage in an organ with a limited regenerative ability. Even though the brain is considered to be immunologically privileged (due to blood-brain barrier, poor lymph drainage, and low antigen production), the tissue can and does react to infection and injury with the production of cytokines, cell infiltration, and tissue damage (Perry et al., 1993; Rothwell et al., 1996). The release of TNF and other cytokines probably plays a role in the development of brain inflammation caused by infection, Alzheimer disease, multiple sclerosis, or physical injury (Arvin et al., 1995). Cytokines in the CNS have been implicated in responses to disease and injury involving neuroendocrine, immune, and behavioral responses (Rothwell et al., 1996) and LPS is well known to evoke an inflammatory response accompanied by the production of TNF-α and subsequently cytokines such as IL-1, IL-2, and IL-6 in the CNS (Gayle et al., 1998;Hauss-Wegrzyniak et al., 1998). We have suggested that CYP1A loss in the brain involves the synthesis of cytokines (Nicholson and Renton, 1999). In these experiments, the administration of TNF-α into the CNS modulated EROD activities in the brain without an accompanying loss of enzyme in the liver. This is consistent for an involvement of TNF and the subsequent cytokine cascade in the down-regulation of cytochrome P450 in the brain but does not support its involvement in the initiation of events in the brain that lead to the concomitant down-regulation of the enzyme in the liver. The liver is not insensitive to TNF because EROD activity was diminished when the cytokine was administered by i.p. in these experiments.
Numerous reports suggest that inflammatory responses localized to the CNS evoke responses in peripheral systems. Immune cytokines that are induced locally in the CNS produce autonomic, neuroendocrine, and behavioral responses that are external to the brain (Ichijo et al., 1994; Rothwell et al., 1996; Woiciechowsky et al., 1998, 1999). Central injections of cytokines also evoke effects in peripheral cells involved in immune responses (Ichijo et al., 1994). After brain trauma or elective brain surgery it has been reported that immunodepression occurs in the periphery and is associated with an increase risk of infection (Woiciechowsky et al., 1998). Because this suppression occurred with no evidence of systemic inflammation it was suggested that these peripheral responses involve splanchnic sympathetic nerve activity and/or the hypothalamic-pituitary-adrenal axis. Sympathetic nerve blockade by β-receptor antagonists can prevent these CNS-evoked peripheral cytokine-mediated effects, indicating that sympathetic nerve responses play a key role in communication between inflammatory responses in the brain and responses in peripheral systems (Take et al., 1993; Woiciechowsky et al., 1999). In these experiments, propranolol was unable to prevent the loss of cytochrome P450 in either the liver or the brain in response to LPS given by i.c.v. injection. We conclude that the loss of cytochrome P450 in the liver after CNS inflammation is unlikely to be due to a β-receptor-mediated sympathetic pathway. Although other peripheral neural pathways could be implicated this is unlikely because they have not been reported to participate in brain-peripheral immune responses (Ichijo et al., 1994;Rothwell et al., 1996; Woiciechowsky et al., 1998, 1999).
This study demonstrates that localized inflammatory responses in the CNS causes a concomitant down-regulation of cytochrome P450 and drug-metabolizing activity in the liver and the brain. Although the effect in the brain is probably regulated via cytokine-mediated pathways, the signaling to the liver does not appear to involve a cytokine-mediated pathway within the brain nor a β-receptor-mediated sympathetic nerve pathway. It is interesting to speculate that the loss in liver might be mediated by the induction of cytokines from a peripheral source (Rothwell et al., 1996; Reichlin, 1998) or to events involving the hypothalamic-pituitary-adrenal axis (Sternberg, 1998).
Although changes to cytochrome P450 in the brain might be important to the metabolism and activation of drugs, toxins, or endogenous compounds within the brain, the loss of drug metabolism in a peripheral organ such as the liver is a much more important aspect of this phenomenon. Such a loss during inflammation or infection of the brain would alter the metabolism and pharmacokinetics of any drugs dependent on hepatic cytochrome P450 for disposition. This could explain some of the untoward drug responses or contribute to the drug interactions that are commonplace in patients with infectious or inflammatory disease of the brain. By understanding the source of potential drug dosage problems and drug interactions a strategy can be developed to minimize or prevent these problems in this population that often receive an extraordinary number of different agents concomitantly.
Footnotes
-
Send reprint requests to: Dr. Kenneth W. Renton, Department of Pharmacology, Dalhousie University, Sir Charles Tupper Medical Bldg., Halifax Nova Scotia, Canada B3H 4H7. E-mail:Ken.Renton{at}dal.ca
-
↵1 This study was supported by the Medical Research Council of Canada.
- Abbreviations:
- IFN
- interferon
- IL
- interleukin
- TNF
- tumor necrosis factor
- LPS
- lipopolysaccharide
- CNS
- central nervous system
- hsp
- heat shock protein
- EROD
- ethoxyresorufin dealkylase
- PROD
- pentoxyresorufin dealkylase
- BROD
- benzyloxyresorufin dealkylase
- Received January 25, 2000.
- Accepted April 13, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}