


Abstract
trans-Resveratrol (3,5,4′-trihydroxy-trans-stilbene) has been reported to confer chemoprotection against 7,12-dimethylbenz[a]anthracene (DMBA)-induced carcinogenicity in a murine model. A potential mechanism for this effect by trans-resveratrol is inhibition of DMBA-bioactivating cytochrome P450 (CYP) enzymes such as CYP1B1, CYP1A1, and CYP1A2. In the present study, we examined in detail the in vitro inhibitory effects of trans-resveratrol on these three human CYP enzymes. trans-Resveratrol decreased 7-ethoxyresorufin O-dealkylation activity catalyzed by human recombinant CYP1B1, CYP1A1, and CYP1A2 in a concentration-dependent manner and by a mixed type of inhibition. This direct inhibition was enzyme-selective, as judged by the differences in the apparent Ki values (0.8 ± 0.1 μM, 1.2 ± 0.1 μM, and 15.5 ± 1.1 μM for CYP1B1, CYP1A1, and CYP1A2, respectively). Preincubating recombinant CYP1A2 or human liver microsomes with trans-resveratrol and NADPH prior to the initiation of substrate oxidation resulted in a time- and concentration-dependent decrease in catalytic activity. The inactivation of liver microsomal CYP1A2 bytrans-resveratrol required NADPH, was not reversible by dialysis, and was not affected by the trapping agents glutathione,N-acetylcysteine, catalase, or superoxide dismutase, but was attenuated by a CYP1A2 substrate, imipramine. Analysis of a panel of individual human liver microsomes showed intersample differences in the response to the in vitro inactivation bytrans-resveratrol. In contrast to CYP1A2, CYP1B1 was not subject to inactivation by this compound and the reduction in CYP1A1 activity was time- but not concentration-dependent. In summary,trans-resveratrol differentially inhibited human CYP1 enzymes and this occurred by two distinct mechanisms: direct inhibition (mainly CYP1B1 and CYP1A1) and mechanism-based inactivation (CYP1A2).
Cancer chemoprevention is the use of drugs or nutraceuticals to prevent, inhibit, or reverse carcinogenesis (Sporn and Suh, 2000). There are several classes of cancer chemopreventive agents including blocking agents, which act at the initiation stage of carcinogenesis by inhibiting procarcinogen-activating enzymes, by inducing carcinogen-detoxifying enzymes, by enhancing anti-oxidant activity, or by inducing DNA repair enzymes (Stoner et al., 1997). Cytochrome P450 (CYP) enzymes are a superfamily of hemoproteins that catalyze the biotransformation of not only a wide array of drugs and endogenous substances, but also the bioactivation of many procarcinogens and toxins (Guengerich and Shimada, 1998). Consequently, specific CYP enzymes have been identified as potential targets for cancer chemoprevention (Yang et al., 1994).
Resveratrol (3,5,4′-trihydroxy-trans-stilbene) is a polyphenolic compound present in a variety of plant genera, includingPolygonum and Vitaceae (Fremont, 2000). The root extracts of the weed Polygonum cuspidatum, which contain resveratrol, have been used in traditional Asian medicine to treat a variety of diseases (Kimura et al., 1985). This naturally occurring compound is also ingested by humans through the consumption of grapes and wine, especially red wine (Fremont, 2000). Currently, resveratrol is available as a nutraceutical and this unregulated product is sold in health food stores (Creasy and Creasy, 1998).
Studies performed in vitro and in vivo have shown that resveratrol has a variety of biological activities (Fremont, 2000). For example, it prevents platelet aggregation, modulates eicosanoid synthesis, and down-regulates polymorphonuclear leukocyte function. Interestingly, cell culture and animal experiments have shown that resveratrol inhibits cellular processes associated with tumor initiation, promotion, and progression (Jang et al., 1997). The impetus for our current study came from a published report indicating that resveratrol reduced the number of preneoplastic lesions in mouse mammary gland cultures treated with the procarcinogen 7,12-dimethylbenz[a]anthracene (DMBA) and decreased the incidence of tumor formation in mice treated with DMBA as an initiator and 12-O-tetradecanoylphorbol-13-acetate as a promoter of carcinogenesis (Jang et al., 1997). DMBA is a procarcinogen that requires bioactivation to exert its effects, and several CYP enzymes including CYP1A1, CYP1A2, and CYP1B1 have been identified as active catalysts of DMBA bioactivation (Shou et al., 1996; Savas et al., 1997). A potential mechanism by which resveratrol confers protection against DMBA-induced carcinogenicity is by inhibiting the specific CYP enzymes involved in DMBA bioactivation. It is important to have a detailed understanding of the metabolic effects of nutraceuticals such as resveratrol on CYP1 enzymes because of their potential chemoprotective properties. Furthermore, the information may lead to the identification of novel nutraceutical-drug interactions because CYP1A2, CYP1A1, and CYP1B1 are also catalysts of drug biotransformation (Rendic and Di Carlo, 1997; Rochat et al., 2001).
Resveratrol exists as trans- and cis-isomeric forms (Fremont, 2000), and it is the combination of thetrans-conformation and the presence of the 4′-hydroxy group that confer biological activity (Stivala et al., 2001). In the present study, we conducted a detailed, systematic investigation to compare under the same experimental conditions the effect oftrans-resveratrol on the catalytic activity of human CYP1A1, CYP1A2, and CYP1B1. Our novel results indicate that this polyphenolic nutraceutical has differential inhibitory effects on human CYP1 enzymes not only at the level of direct enzyme inhibition but also with respect to mechanism-based inactivation.
Materials and Methods
Chemicals and Reagents.
trans-Resveratrol (lot 05491, 99.5% purity as determined by HPLC) was a gift from Pharmascience, Inc. (Montreal, Quebec, Canada). 7-Ethoxyresorufin, NADPH, glutathione, N-acetylcysteine, catalase, and superoxide dismutase were bought from Sigma Chemical Co. (St. Louis, MO). Authentic resorufin metabolite standard was obtained from Molecular Probes, Inc. (Eugene, OR). Microsomes from baculovirus-infected insect cells coexpressing NADPH-cytochrome P450 reductase and human CYP1A1 (catalog number P211), CYP1A2 (catalog number P203), or CYP1B1 (catalog number P220), control insect cell microsomes (catalog number P201), pooled human liver microsomes (catalog number H161), and individual human liver microsomes (catalog numbers HG3, HG6, HG23, HG30, HG42, HG43, HG56, HG66, HG70, HG89, HG93, and HG112) were purchased from GENTEST Corp. (Woburn, MA). The total CYP content and CYP1A2 protein levels in the microsome samples were provided by the supplier. Slide-A-Lyzer mini-dialysis units (10,000 molecular weight cut-off) were bought from Pierce Chemical Co. (Rockford, IL).
7-Ethoxyresorufin O-Dealkylation Assay.
Microsomal 7- ethoxyresorufin O-dealkylation activity was determined by a continuous spectrofluorometric method as described previously (Chang and Waxman, 1998), but with minor modifications. Briefly, each standard 2-ml incubation mixture contained 100 mM potassium phosphate (pH 7.4), 1.5 mM EDTA, 0.25 μM 7-ethoxyresorufin, 0.25 mM NADPH, enzymes (human recombinant CYP enzyme or human liver microsomes), and trans-resveratrol, at the concentrations indicated in each figure legend. Reactions were carried out at 37°C and fluorescence readings were recorded every 30 s for 3 min at an excitation wavelength of 530 nm (5-nm slit width) and an emission wavelength of 582 nm (5-nm slit width). Calibration curves were constructed with authentic resorufin and linear regression analysis was used to calculate the amount of resorufin formed in each incubation sample. Preliminary experiments indicated that the assay was linear with respect to incubation time and amount of CYP enzyme.
Determination of Apparent Km andVmax.
To determine the enzyme kinetics for CYP1A1-, CYP1A2-, and CYP1B1-catalyzed 7-ethoxyresorufinO-dealkylation, the enzyme assay was conducted at substrate concentrations ranging from 0.025 to 4 μM. The apparentKm andVmax values were estimated by nonlinear regression analysis (ENZFITTER software program; Elsevier-Biosoft, Cambridge, UK) of the enzyme activity (V)-substrate concentration [S] data using the Michaelis-Menten model:
Enzyme Inhibition Experiments.
To compare the inhibitory effect of trans-resveratrol on the catalytic activity of CYP1A1, CYP1A2, and CYP1B1, the 7-ethoxyresorufinO-dealkylation assay was conducted in the presence of varying concentrations of this nutraceutical, as indicated in each figure legend. trans-Resveratrol was dissolved in methanol and the final concentration of the solvent was 0.15% v/v. At this concentration, methanol did not affect CYP catalytic activity. To characterize the enzyme kinetics for the inhibition of CYP1A1, CYP1A2, and CYP1B1 by trans-resveratrol, experiments were conducted using four concentrations of trans-resveratrol and four concentrations of 7-ethoxyresorufin, as indicated in each figure legend. The apparent Ki value (the equilibrium dissociation constant for the enzyme-inhibitor complex) was determined from the x-intercept of a plot of apparentKm/Vmax(obtained from the slope of the Lineweaver-Burk plots) versus inhibitor concentration (Segal, 1975). The x-intercept, which is equal to −Ki, was calculated by linear regression using the ENZFITTER software program. Lineweaver-Burk plots and Dixon plots of the enzyme kinetic data were generated to determine the mode of inhibition.
Mechanism-Based Inactivation Experiments.
trans-Resveratrol (at the concentrations indicated in each figure legend) or methanol (vehicle control, 0.15% v/v, final concentration) was preincubated with recombinant CYP enzyme (or human liver microsomes) and 0.25 mM NADPH or distilled water (control) at 37°C for various amount of time in 100 mM potassium phosphate buffer (pH 7.4) containing 1.5 mM EDTA. Subsequently, an aliquot (0.2 ml) of the preincubation mixture was transferred to a 1.8-ml enzyme activity assay mixture prewarmed to 37°C containing the buffer, 0.25 μM 7-ethoxyresorufin, and 0.25 mM NADPH. 7-EthoxyresorufinO-dealkylation activity was determined as described above. The pseudo-first order rate constant for inactivation (kobs) was calculated by the next equation:
Effect of Nucleophilic Trapping Agents and Scavengers of Reactive Oxygen Species.
A nucleophilic trapping agent (5 or 10 mM glutathione or N-acetylcysteine) or a scavenger of reactive oxygen species (1000 or 2000 units of catalase or 500 or 1000 units of superoxide dismutase) was incubated at 37°C for 6 min withtrans-resveratrol (25 μM) or vehicle (0.15% methanol), pooled human liver microsomes (75 pmol of total CYP), and NADPH (0.25 mM) in 100 mM potassium phosphate (pH 7.4) buffer containing 1.5 mM EDTA. A 0.2-ml aliquot was transferred to a 1.8-ml enzyme activity assay containing 100 mM potassium phosphate (pH 7.4), 1.5 mM EDTA, 0.25 μM 7-ethoxyresorufin, and 0.25 mM NADPH. 7-EthoxyresorufinO-dealkylation activity was determined as described above.
Effect of Dialysis.
Pooled human liver microsomes (75 pmol of total CYP) were incubated for 6 min at 37°C with NADPH (0.25 mM) and trans-resveratrol (25 μM) or vehicle (0.15% methanol) in 50 mM potassium phosphate buffer (pH 7.4) containing 0.1 mM EDTA. Subsequently, the samples were transferred to a Slide-A-Lyzer mini-dialysis unit with a molecular weight cutoff of 10,000 (Pierce Chemical Co., Rockford, IL.). Dialysis was performed at 4°C for 4 or 24 h in 1 liter of 50 mM potassium phosphate buffer (pH 7.4) containing 20% glycerol and 0.1 mM EDTA. The dialysis buffer was changed after every 1 h during the first 3-h period. The dialyzed samples were assayed for 7-ethoxyresorufin O-dealkylation activity as described above.
Effect of a CYP1A2 Substrate.
Imipramine, a CYP1A2 substrate (Lemoine et al., 1993), was added to the inactivation mixture containing 100 mM potassium phosphate (pH 7.4), 1.5 mM EDTA, pooled human liver microsomes (75 pmol of total CYP),trans-resveratrol (25 μM), and NADPH (0.25 mM). The final concentrations of imipramine were 0, 125, 250, or 500 μM and they correspond to a molar ratio of imipramine totrans-resveratrol of 0, 5, 10, and 20, respectively. After the inactivation mixture was incubated at 37°C for 6 min, a 0.2-ml aliquot was transferred to the 7-ethoxyresorufinO-dealkylation enzyme activity assay, which was performed as described above.
Statistics
The significance of the difference between the means of the various groups was assessed by one- or two-way analysis of variance, and if applicable, was followed by the Student Newman-Keuls test, using the SigmaStat statistical software program (Jandel Scientific Co., San Rafael, CA). The level of significance was set a priori at p < 0.05.
Results
Direct Inhibition of Human Recombinant CYP1A1, CYP1A2, and CYP1B1 by trans-Resveratrol.
To compare directly the inhibitory effect of trans-resveratrol on CYP1A1, CYP1A2, and CYP1B1 activities, we employed 7-ethoxyresorufin as a substrate because a previous study has demonstrated that each of these enzymes is active in the oxidative metabolism of this compound (Shimada et al., 1997). As shown in Fig. 1,trans-resveratrol decreased CYP1A1-, CYP1A2-, and CYP1B1-catalyzed 7-ethoxyresorufin O-dealkylation activity in a concentration-dependent manner. The concentration-response curves for CYP1A1 and CYP1B1 were relatively close to each other, whereas the one for CYP1A2 was shifted to the right by an order of magnitude. To investigate the mode of inhibition of these CYP1 enzymes bytrans-resveratrol, enzyme kinetic experiments were performed with four inhibitor concentrations and four substrate concentrations. Lineweaver-Burk plots of the enzyme kinetic data indicated thattrans-resveratrol inhibited CYP1A1, CYP1A2, and CYP1B1 by a mixed type of inhibition (Fig. 2, A–C). Graphical analysis by Dixon plot also yielded the same conclusion (data not shown). To determine the apparentKi values, the slope of the Lineweaver-Burk plot (i.e., ratio of apparentKm/Vmax) was plotted against trans-resveratrol concentrations and the apparent Ki calculated from thex-intercept (Segal, 1975). As shown in Table1, the apparentKi for CYP1B1 (0.8 ± 0.1 μM, mean ± S.E.M.) and CYP1A1 (1.2 ± 0.1 μM) was significantly less than that for CYP1A2 (15.5 ± 1.1 μM). The ratios of apparent Ki/apparentKm, used to assess relative inhibition potency (Murray and Butler, 1996), were 10, 13, and 204 for thetrans-resveratrol inhibition of CYP1B1, CYP1A1, and CYP1A2, respectively (Table 1). Thus, trans-resveratrol was 20- and 16-fold more potent in inhibiting CYP1B1 and CYP1A1, respectively, when compared with CYP1A2.

Concentration-dependent inhibition of CYP1A1, CYP1A2, and CYP1B1 by trans-resveratrol. Human recombinant CYP1A1-, CYP1A2-, and CYP1B1-catalyzed 7-ethoxyresorufinO-dealkylation activity was determined in the presence of 0.15% methanol (vehicle control) or varying concentrations oftrans-resveratrol (0.1–100 μM) as described underMaterials and Methods. Data are expressed as mean ± S.E.M. percentage of control enzyme activity for three or four independent experiments. The mean ± S.E.M. CYP1A1, CYP1A2, and CYP1B1 enzyme activities for the control group was 43 ± 4, 3.5 ± 0.03, and 11 ± 1 nmol/min/nmol CYP, respectively.

Lineweaver-Burk plot for the inhibition of CYP1A1, CYP1A2, and CYP1B1 enzyme activity by trans-resveratrol. 7-Ethoxyresorufin O-dealkylation catalyzed by human recombinant CYP1A1 (A), CYP1A2 (B), and CYP1B1 (C) was determined at four concentrations of 7-ethoxyresorufin (CYP1A1 and CYP1B1, 0.025–0.25 μM; CYP1A2, 0.05–0.4 μM) and four concentrations oftrans-resveratrol (RESV) (CYP1A1, 0–6 μM; CYP1A2, 0–20 μM; CYP1B1, 0–3 μM). The symbols indicate mean of the transformed data for three independent experiments, and the lines were generated by linear regression analysis. In all cases, the coefficient of variation was ≤8.3%.
Enzyme kinetic analysis of the inhibition of human recombinant CYP1A1, CYP1A2, and CYP1B1 by trans-resveratrol
Differential Inactivation of Human Recombinant CYP1A1, CYP1A2, and CYP1B1 by trans-Resveratrol.
The results of the enzyme kinetic analyses described above indicate a preference for CYP1B1 and CYP1A1 in the direct inhibition bytrans-resveratrol. To determine whether there is selectivity with respect to enzyme inactivation by this compound, the 7-ethoxyresorufin O-dealkylation assay was conducted with the inclusion of a preincubation step. trans-Resveratrol (at varying concentrations) or methanol (0.15% final concentration, vehicle control) was preincubated at 37°C for up to 9 min (6 min in the case of CYP1A2) with NADPH and a CYP1 enzyme. An aliquot of the primary inactivation mixture was then diluted into the secondary reaction mixture containing buffer, substrate, and fresh NADPH. The preincubation of trans-resveratrol with enzyme and NADPH resulted in a time- and concentration-dependent decrease in the magnitude of CYP1A2 catalytic activity (Fig.3A). With respect to the kinetics of CYP1A2 inactivation by trans-resveratrol, the rate constant for maximal inactivation at saturation (kinactivation ) was 0.43 ± 0.02 min−1, the time required for half of the enzyme molecules to be inactivated (t1/2) was 1.6 ± 0.1 min, the concentration of trans-resveratrol required to produce one-half the maximal rate of CYP1A2 inactivation (KI ) was 2.4 ± 0.4 μM, and the ratio of kinactivation toKI , used to assess the efficiency of enzyme inactivation (Roberts et al., 1998), was 0.21 ± 0.05 min−1μM−1. In the case of CYP1A1 (Fig. 3B), the preincubation of trans-resveratrol with NADPH and this enzyme resulted in a time- but not a concentration-dependent decline in catalytic activity. By comparison,trans-resveratrol did not inactivate CYP1B1 (Fig. 3C).

Inactivation of CYP1A2, CYP1A1, and CYP1B1 bytrans-resveratrol. Human recombinant CYP1A2 (5 pmol, A), CYP1A1 (2.5 pmol, B), or CYP1B1 (2.5 pmol, C) was preincubated with 0.15% methanol (vehicle control) or varying concentrations oftrans-resveratrol (RESV) and NADPH (0.25 mM) at 37°C for 0, 3, 6, or 9 min (or 0, 2, 4, or 6 min in the case of CYP1A2) in 100 mM potassium phosphate buffer (pH 7.4) containing 1.5 mM EDTA. A 0.2-ml aliquot was transferred to a 1.8-ml enzyme activity assay mixture and 7-ethoxyresorufin O-dealkylation activity was determined as described under Materials and Methods. Each point represents the mean percentage of control activity of three independent experiments. The coefficient of variation was ≤7.8%.
Inactivation of Human Liver Microsomal 7- EthoxyresorufinO-Dealkylation Activity bytrans-Resveratrol.
As described above,trans-resveratrol was shown to be effective in inactivating human recombinant CYP1A2 (Fig. 3A). Because CYP1A2 is expressed in liver (Schweikl et al., 1993), we determined whethertrans-resveratrol was similarly effective in inactivating human liver microsomal CYP1A2, which can be assessed by the 7-ethoxyresorufin O-dealkylation activity, a commonly used catalytic monitor selective for CYP1A2 in human liver microsomes (Murray et al., 1993). As demonstrated in Fig.4A, trans-resveratrol inactivated this activity in a time- and concentration-dependent manner. The Kitz-Wilson plot (Fig. 4B) oft1/2 versus the inverse oftrans-resveratrol concentration shows that the regression line crosses the ordinate above zero, indicating that the inactivation of human liver microsomal CYP1A2-mediated 7-ethoxyresorufinO-dealkylation activity by trans-resveratrol was a saturable process with respect to inactivator concentration. Additional enzyme kinetic analysis indicated that thekinactivation of human liver microsomes-catalyzed 7-ethoxyresorufin O-dealkylation was 0.28 ± 0.01 min−1, thet1/2 was 2.5 ± 0.1 min, theKI was 8.5 ± 1.2 μM, and the ratio ofkinactivation /KI was 0.03 ± 0.01 min−1μM−1.

Inactivation of human liver microsomal 7-ethoxyresorufin O-dealkylation activity bytrans-resveratrol. Pooled human liver microsomes (75 pmol of total CYP) were preincubated with 0.15% methanol (vehicle control) or varying concentrations of trans-resveratrol (RESV) and NADPH (0.25 mM) at 37°C for 0, 2, 4, or 6 min in 100 mM potassium phosphate buffer (pH 7.4) containing 1.5 mM EDTA. A 0.2-ml aliquot was transferred to a 1.8-ml enzyme activity assay mixture and 7-ethoxyresorufin O-dealkylation activity was determined as described under Materials and Methods. A, a plot of the log of percentage of control activity versus preincubation time. Each point represents the mean of three independent experiments. The coefficient of variation was ≤7.3%. B, a Kitz-Wilson plot of the half-life of enzyme inactivation versus the inverse oftrans-resveratrol concentration.
Requirement for NADPH in the Inactivation of Human Microsomal CYP1A2 Activity by trans-Resveratrol.
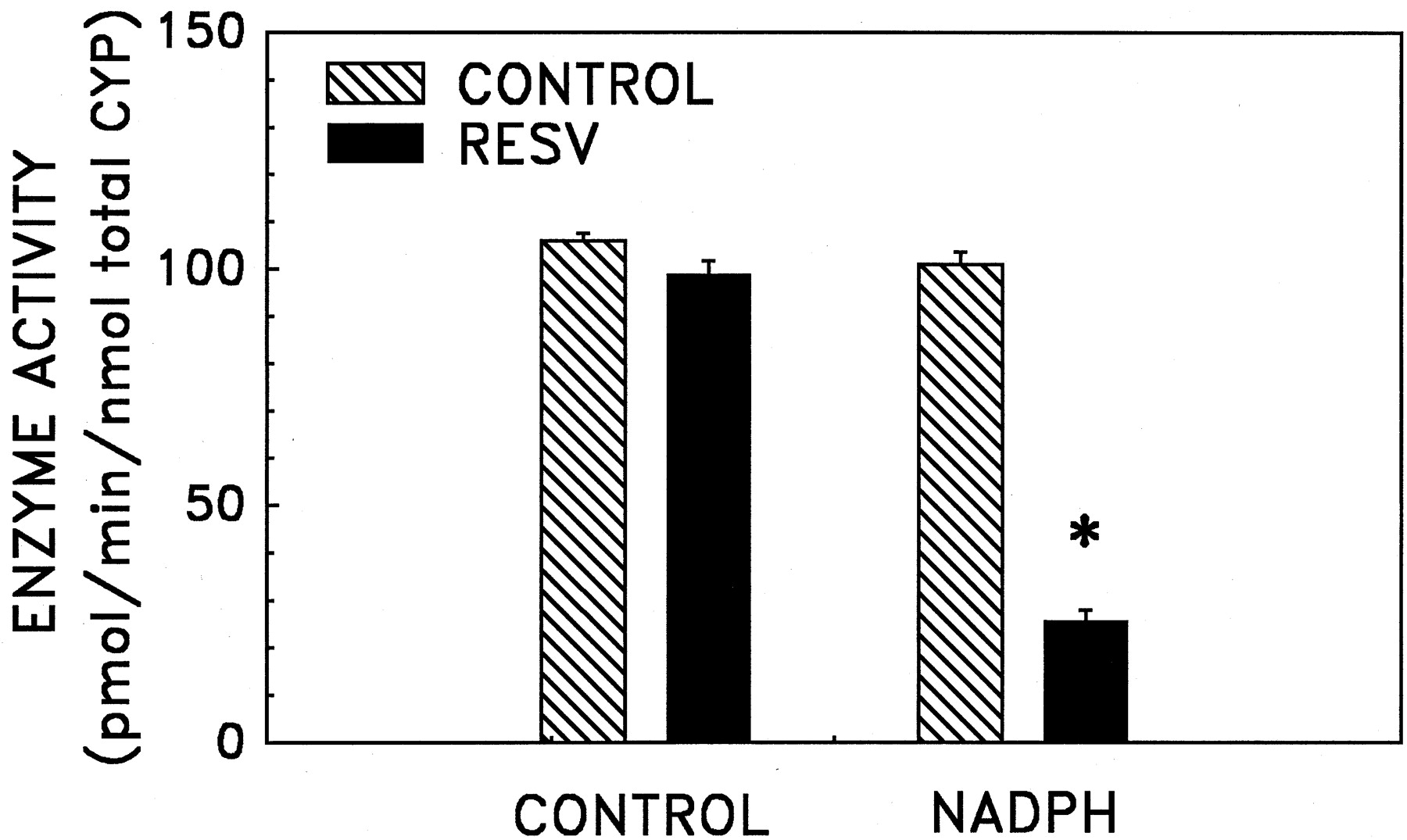
To determine whether the observed inactivation of human liver microsomal CYP1A2 bytrans-resveratrol required catalysis, the preincubation step was conducted in the presence and absence of NADPH. As shown in Fig.5, the preincubation of pooled human liver microsomes with trans-resveratrol or NADPH alone did not result in a decrease in 7-ethoxyresorufin O-dealkylation activity. However, the presence of both NADPH andtrans-resveratrol led to a reduction in enzyme activity. Therefore, inactivation of liver microsomal CYPA2 is mediated not by the parent compound, but by a metabolic product(s) oftrans-resveratrol.

Requirement for NADPH in the inactivation of human liver microsomal 7-ethoxyresorufin O-dealkylation activity by trans-resveratrol. Pooled human liver microsomes (75 pmol of total CYP) were preincubated with 0.15% methanol (vehicle control) or varying concentrations oftrans-resveratrol (RESV) in the presence or absence of NADPH (0.25 mM) at 37°C for 0, 2, 4, or 6 min in 100 mM potassium phosphate buffer (pH 7.4) containing 1.5 mM EDTA. A 0.2-ml aliquot was transferred to a 1.8-ml enzyme activity assay mixture and 7-ethoxyresorufin O-dealkylation activity was determined as described under Materials and Methods. Data are expressed as mean ± S.E.M. for four independent experiments. ∗, significantly different from the control group (p< 0.05).
Lack of an Effect by Trapping Agents ontrans-Resveratrol Inactivation of Human Liver Microsomal CYP1A2 Activity.
To examine whether inactivation of CYP enzymes bytrans-resveratrol was confined to the active site, experiments were performed in the presence of a nucleophilic trapping agent. As shown in Table 2, the inclusion of glutathione or N-acetylcysteine in the primary incubation mixture did not provide protection against the inactivation of human liver microsomal CYP1A2-mediated 7-ethoxyresorufinO-dealkylation activity by trans-resveratrol. Similarly, the presence of a scavenger of reactive oxygen species, such as catalase or superoxide dismutase, also did not attenuate the magnitude of the inactivation by trans-resveratrol.
Effect of trapping agents on the inactivation of human liver microsomal CYP1A2 activity by trans-resveratrol
Irreversibility of the trans-Resveratrol Inactivation of Human Liver Microsomal CYP1A2 Activity.
To determine whether the inactivation effects bytrans-resveratrol was reversible, this compound or the vehicle (control) was preincubated at 37°C for 6 min with pooled human liver microsomes and NADPH. The samples were then transferred to a mini-dialysis unit and dialyzed at 4°C for 4 or 24 h. For comparison, inactivation experiment was also conducted with samples that were not dialyzed. As indicated in Fig.6, dialysis did not affect the magnitude of the trans-resveratrol inactivation of human liver microsomal 7-ethoxyresorufin O-dealkylation activity.

Effect of dialysis on the inactivation of human liver microsomal 7-ethoxyresorufin O-dealkylation activity bytrans-resveratrol. Pooled human liver microsomes (75 pmol of total CYP) were preincubated with 0.15% methanol (vehicle control) or varying concentrations of trans-resveratrol (RESV) and NADPH (0.25 mM) at 37°C for 0, 2, 4, or 6 min in 100 mM potassium phosphate buffer (pH 7.4) containing 1.5 mM EDTA. A 0.2-ml aliquot was transferred to a Slide-A-Lyzer mini-dialysis unit and dialyzed at 4°C for 4 or 24 h prior to the determination of 7-ethoxyresorufin O-dealkylation activity as described under Materials and Methods. Parallel analysis was conducted in which trans-resveratrol or vehicle-treated samples were not dialyzed. Data are expressed as mean ± S.E.M. for three independent experiments. ∗, significantly different from the control group (p < 0.05).
Attenuation of trans-Resveratrol Inactivation of Human Liver Microsomal CYP1A2 Activity by a CYP1A2 Substrate.
Imipramine is a substrate for human CYP1A2 (Lemoine et al., 1993). To assess whether a CYP1A2 substrate influenced the extent of human liver microsomal CYP1A2 inactivation by trans-resveratrol, imipramine was added to the primary incubation mixture containing pooled human liver microsomes, trans-resveratrol, and NADPH. As shown in Fig. 7, the addition of imipramine at concentrations of 125, 250, or 500 μM (corresponding to a molar ratio of imipramine to trans-resveratrol of 5, 10, and 20, respectively) significantly attenuated the extent by whichtrans-resveratrol inactivated human liver microsomal 7-ethoxyresorufin O-dealkylation activity. At a molar ratio of 20, the reduction in enzyme activity changed from 25% of control activity to 46% of control activity.

Effect of imipramine, a CYP1A2 substrate, on the inactivation of human liver microsomal 7-ethoxyresorufinO-dealkylation activity bytrans-resveratrol. Pooled human liver microsomes (75 pmol of total CYP) were preincubated withtrans-resveratrol (25 μM), imipramine HCl (0, 125, 250, or 500 μM), and NADPH (0.25 mM) at 37°C for 6 min in 100 mM potassium phosphate buffer (pH 7.4) containing 1.5 mM EDTA. The ratio of imipramine to trans-resveratrol was 0:1, 5:1, 10:1, and 20:1. A 0.2-ml aliquot was transferred to a 1.8-ml enzyme activity assay mixture and 7-ethoxyresorufin O-dealkylation activity was determined as described under Materials and Methods. Data are expressed as mean ± S.E.M. for three independent experiments. ∗, significantly different from the control (0:1) group (p < 0.05).
Intersample Differences in the Inactivating Effect oftrans-Resveratrol on Human Liver Microsomal CYP1A2 Activity.
The above experiments with human liver microsomes were conducted with pooled samples. To investigate the response of individual samples to the inactivating effect oftrans-resveratrol, an experiment was performed with a panel of individual human liver microsomes. Of the 12 individual samples used in our analysis, inactivation of 7-ethoxyresorufinO-dealkylation activity by trans-resveratrol occurred in 11 samples (Fig. 8A). The mean (± S.E.M.) reduction in enzyme activity was 60% ± 6%, and the range was from 0% to 79%. In the one sample where inactivation was not evident, it had the lowest CYP1A2 protein content. Correlational analysis established a strong association between the extent of enzyme inactivation by trans-resveratrol and hepatic CYP1A2 protein content in the panel of individual human liver microsomes (Fig. 8B). The coefficient of determination (r2) was 0.81.

Inactivation of human liver microsomal 7-ethoxyresorufin O-dealkylation activity bytrans-resveratrol in a panel of individual human liver microsomes. trans-Resveratrol (RESV; 25 μM) or methanol (0.15%, vehicle control) was preincubated with individual human liver microsomes (10 pmol of CYP1A2 per microsome sample) and NADPH (0.25 mM) at 37°C for 6 min in 100 mM potassium phosphate buffer (pH 7.4) containing 1.5 mM EDTA. A 0.2-ml aliquot was transferred to a 1.8-ml enzyme activity assay mixture and 7-ethoxyresorufin O-dealkylation activity was determined as described under Materials and Methods. Results are shown as the mean of duplicate determinations (A). Correlation analysis was performed between the percentage decrease in enzyme activity bytrans-resveratrol and CYP1A2 protein content in each human liver microsome sample (B).
Discussion
The present study provides the first demonstration for the mechanism-based inactivation of human CYP1A2 bytrans-resveratrol. This conclusion is based on the findings that the inactivation of human liver microsomal CYP1A2-catalyzed 7-ethoxyresorufin O-dealkylation activity bytrans-resveratrol: 1) was time- and concentration-dependent; 2) was irreversible; 3) required the presence of NADPH; 4) was not affected by nucleophilic trapping agents such as glutathione andN-acetylcysteine or by scavengers of reactive oxygen species such as catalase and superoxide dismutase; 5) was attenuated by a CYP1A2 substrate, imipramine; and 6) was shown to exhibit saturation kinetics. The time- and concentration-dependent inactivation of human liver microsomal CYP1A2 activity by trans-resveratrol was also observed when the corresponding human recombinant enzyme was used in the inactivation experiments. As summarized in Table3, our experimentally obtained values ofkinactivation for thetrans-resveratrol inactivation of human microsomal CYP1A2 and human recombinant CYP1A2 were comparable with those reported for other known mechanism-based inactivators of CYP1A2, such as furafylline (Kunze and Trager, 1993; Clarke et al., 1994) and oltipraz (Langouet et al., 2000). In contrast, they are an order of magnitude greater than the published values of kinactivation for dihydralazine (Masubuchi and Horie, 1999) and desethylamiodarone (Ohyama et al., 2000). The potency of the trans-resveratrol inactivation of CYP1A2, as assessed by theKI, was similar to that of furafylline, oltipraz, and desethylamiodarone, whereastrans-resveratrol was at least 5-fold more potent than dihydralazine (Table 3). When compared with a previous human liver microsomal study in which 7-ethoxyresorufin was used as the substrate (Clarke et al., 1994), trans-resveratrol appear to be somewhat less efficient than furafylline in inactivating CYP1A2, as assessed by the ratio of kinactivation andKI (33 min−1mM−1 versus 90 min−1mM−1; see Table 3). Consistent with previous studies on the inactivation of recombinant CYP and the corresponding liver microsomal CYP enzyme by various chemicals (Kunze and Trager, 1993; Kanamitsu et al., 2000; Palamanda et al., 2001), we also found differences in the magnitude of the kinetic constants for the inactivation of liver microsomal CYP1A2 and recombinant CYP1A2 bytrans-resveratrol. The reason for this is not known, but may reflect the relative differences in the levels of NADPH-cytochrome P450 reductase. In the present study, the recombinant CYP1A2 was synthesized in baculovirus-infected insect cells cotransfected with NADPH-cytochrome P450 reductase cDNA (prepared by a commercial supplier; see Materials and Methods).
Comparison of enzyme inactivation kinetic constants for the various mechanism-based inactivators of human CYP1A2
The presence of either NADPH or trans-resveratrol alone in the primary reaction mixture was not sufficient to elicit CYP1A2 inactivation. Rather, both NADPH and trans-resveratrol were required. These findings lead to the conclusion thattrans-resveratrol is metabolized by CYP1A2. Consistent with this proposal, the presence of a CYP1A2 substrate imipramine (Lemoine et al., 1993) and trans-resveratrol in the primary reaction mixture attenuated the magnitude of CYP1A2 inactivation by this polyphenolic phytochemical. However, trans-resveratrol does not compete effectively with 7-ethoxyresorufin for CYP1A2, as suggested by the high apparent Ki/apparentKm ratio (Table 1). The requirement for NADPH in the trans-resveratrol inactivation of CYP1A2 also indicates that it is not trans-resveratrol, but a reactive intermediate that is responsible for the inactivation of this enzyme. The finding that trapping agents such as glutathione,N-acetylcysteine, catalase, and superoxide dismutase did not provide any protection against trans-resveratrol inactivation of CYP1A2 suggested that the intermediate was chemically highly reactive and was not released from the active site of the enzyme. However, the identity of the inactivating species is not known at the present time. A potential CYP1A2-catalyzed metabolite oftrans-resveratrol is an aromatic hydroxylation product, such as oxyresveratrol (Chun et al., 2001) or 3,4,5,4′-tetrahydroxystilbene (Lu et al., 2001), or a trans-resveratrol epoxide, which then generates a reactive p-benzoquinone methide derivative (Chan and Delucchi, 2000). Further studies will be required to investigate these possibilities.
In our experiments with a panel of individual human liver microsomes, intersample differences were found in the extent of the inactivation of CYP1A2 by trans-resveratrol. This reflected the interindividual differences in the expression of the CYP1A2 protein in this panel of microsome samples because a highly positive correlation was obtained between the percentage reduction in hepatic microsomal CYP1A2 activity by trans-resveratrol and hepatic CYP1A2 protein levels. Complete inactivation was not observed even in those human liver microsome samples with the greatest CYP1A2 protein levels, but this was due to the experimental conditions employed because based on our findings, maximal inactivation did not occur with 25 μMtrans-resveratrol and 6 min of preincubation (cf. Fig. 4A). Another possible contributing factor is that human liver microsomal 7-ethoxyresorufin O-dealkylation activity is not exclusively due to CYP1A2 because immunoinhibition experiments have shown that CYP1A2 accounts for approximately 80% of this activity in human liver microsomes (Murray et al., 1993), suggesting that the remainder of the activity is due to an enzyme(s) other than CYP1A2. Thus, the incomplete inactivation of human liver microsomal 7-ethoxyresorufinO-dealkylation activity could also suggest that these other CYP enzymes were not subject to inactivation bytrans-resveratrol.
The inclusion of trans-resveratrol in the primary reaction mixture did not result in mechanism-based inactivation of CYP1A1 or CYP1B1. Although the effect on CYP1A1 was time-dependent, it was not concentration-dependent, suggesting that trans-resveratrol does not affect CYP1A1 by mechanism-based inactivation (Silverman, 1988), in agreement with a previous conclusion (Chun et al., 1999). In the case of CYP1B1, no statistically significant reduction in catalytic activity was obtained when this compound was preincubated with CYP1B1 and NADPH in the primary reaction mixture prior to the initiation of substrate oxidation. In addition to CYP1A2, CYP3A4 is the only other CYP enzyme reported to date to be subject to mechanism-based inactivation by trans-resveratrol, as shown in a recent study with recombinant CYP3A4 (Chan and Delucchi, 2000). The values ofkinactivation andKI were 0.20 min−1 and 20 μM−1, respectively. In contrast to trans-resveratrol, rhapontigenin (3,5-dihydroxy-4′-methoxy-5′-hydroxystilbene), which is an analog of trans-resveratrol, was found to be a mechanism-based inactivator of human recombinant CYP1A1 (Chun et al., 2001), with a kinactivation of 0.06 min−1 and a KIof 0.09 μM−1. Thus, the replacement of the hydroxyl group with a methoxy group at the 4′ position and the addition of a hydroxyl group at the 5′ position rendered this resveratrol analog a mechanism-based inactivator of CYP1A1. However, the rate of CYP1A1 inactivation by this compound is still slower than the rate of CYP1A2 inactivation by trans-resveratrol, as demonstrated in the present study.
Another goal of the present study was to compare systematically and under the same general experimental conditions, the direct inhibitory effects of trans-resveratrol on the catalytic activity of CYP1A1, CYP1A2, and CYP1B1. This compound inhibited each of these three enzymes by a mixed type of mechanism. The apparentKi for thetrans-resveratrol inhibition of CYP1B1 (0.8 ± 0.1 μM) was similar to that for CYP1A1 (1.2 ± 0.1 μM), but it was 13- to 16-fold less when compared with the apparentKi for CYP1A2 (15.5 ± 1.1 μM). A recent study reported a 51-fold difference in the IC50 values for the inhibition of CYP1A1 and CYP1A2 by trans-resveratrol (Chun et al., 1999). In our study, the difference in the respective IC50values was only ∼10-fold. Collectively, these results indicate thattrans-resveratrol is selective not only for CYP1A1, as has been suggested (Chun et al., 1999), but also CYP1B1, as shown by the direct comparison in the present study. Other naturally occurring polyphenolic compounds such as galangin (3,5,7-trihydroxyflavone) and apigenin (5,7,4′-trihydroxyflavone) also inhibit CYP1A1 and CYP1A2 activities. In the case of CYP1A1, the apparentKi values for the inhibition of this enzyme by galangin and apigenin are 0.015 μM (Zhai et al., 1998) and 0.32 μM (Pastrakuljic et al., 1997), respectively, which are less than that for trans-reveratrol (1.2 ± 0.1 μM). In contrast to CYP1A1 and CYP1A2, much less is known about the inhibition of CYP1B1 catalytic activity by naturally occurring compounds. However,trans-resveratrol (apparentKi = 0.8 ± 0.1 μM) and homoeriodictyol (IC50 = 0.24 μM) (Doostdar et al., 2000), which is a bioflavonoid, appear to be the most potent inhibitors of CYP1B1 reported to date.
Currently, it is not known whether trans-resveratrol inhibits in vivo the catalytic activity of CYP1A1, CYP1A2, and CYP1A2. However, studies with rats have shown accumulation oftrans-resveratrol in various tissues (kidney > liver > plasma > heart) after in vivo administration of red wine (Bertelli et al., 1998). The potential in vivo effect oftrans-resveratrol on the bioactivation of CYP1 substrates, such as DMBA (Buters et al., 1999), may depend not only on the pharmacokinetics of trans-resveratrol but also the tissue of interest because of the known tissue-dependent expression of CYP1A1, CYP1A2, and CYP1B1 (Omiecinski et al., 1999). Studies are now in progress to investigate the in vivo effects oftrans-resveratrol on CYP catalytic activity and gene expression.
In summary, two distinct mechanisms exist for the in vitro inhibition of human CYP1 enzymes by trans-resveratrol: direct enzyme selective inhibition of CYP1B1 and CYP1A1 and mechanism-based inactivation of CYP1A2. The inactivation kinetics of CYP1A2 bytrans-resveratrol was comparable with those previously reported for furafylline, a known mechanism-based inactivator of CYP1A2 (Kunze and Trager, 1993; Clarke et al., 1994).
Acknowledgments
We thank Pharmascience, Inc., for the generous provision oftrans-resveratrol.
Footnotes
-
Supported by Grant MOP-42385 (to T.K.H.C.) from the Canadian Institutes of Health Research. T.K.H.C. is the recipient of a Research Career Award in the Health Sciences from the Canadian Institutes of Health Research and Rx & D Health Research Foundation.
- Abbreviations:
- CYP
- cytochrome P450
- DMBA
- 7,12-dimethylbenz[a]anthracene
- kinactivation
- rate constant for maximal inactivation
- kobs
- pseudo-first order rate constant for inactivation
- KI
- concentration of inactivator to produce one-half the maximal inactivation
- Ki
- the equilibrium dissociation constant for the enzyme-inhibitor complex
- t1/2
- time required for half of the enzyme molecules to be inactivated
- Received July 6, 2001.
- Accepted August 28, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}