


Abstract
Estradiol-17β-d-glucuronide (E2-17G) induces an acute but reversible inhibition of bile flow after its intravenous administration to rats, due in part to the endocytic retrieval of the canalicular multidrug resistance-associated transporter protein 2 and the bile salt export pump, transporters that contribute to bile flow. Decreased bile salt-independent bile flow (BSIF) is also involved and persists during the phase of recovery from cholestasis. Because glutathione and  are major contributors to BSIF, we evaluated changes in their biliary excretion and the hepatic content of total glutathione during E2-17G-induced cholestasis. E2-17G acutely decreased bile flow and biliary excretion of total glutathione by about 80%; glutathione excretion was still inhibited at 80 min and 120 min, even though bile flow was partially and totally restored, respectively. Neither liver glutathione content nor the proportions of oxidized glutathione in bile and liver were affected by E2-17G at any time.
are major contributors to BSIF, we evaluated changes in their biliary excretion and the hepatic content of total glutathione during E2-17G-induced cholestasis. E2-17G acutely decreased bile flow and biliary excretion of total glutathione by about 80%; glutathione excretion was still inhibited at 80 min and 120 min, even though bile flow was partially and totally restored, respectively. Neither liver glutathione content nor the proportions of oxidized glutathione in bile and liver were affected by E2-17G at any time.  concentrations in bile were unchanged, so that secretion paralleled variations in bile flow. In the isolated perfused liver, addition of E2-17G decreased both bile flow and the biliary concentration of glutathione, whereas addition of its noncholestatic isomer estradiol-3-d-glucuronide (E2-3G) did not inhibit bile flow, but significantly reduced the concentration of glutathione in bile. The bile:liver concentration ratios of glutathione were significantly decreased in vivo by E2-17G and in the perfused liver by E2-17G and E2-3G. These data indicate that E2-17G cis-inhibits the canalicular transport of glutathione and thus contributes to the cholestatic effect by inhibiting BSIF.
concentrations in bile were unchanged, so that secretion paralleled variations in bile flow. In the isolated perfused liver, addition of E2-17G decreased both bile flow and the biliary concentration of glutathione, whereas addition of its noncholestatic isomer estradiol-3-d-glucuronide (E2-3G) did not inhibit bile flow, but significantly reduced the concentration of glutathione in bile. The bile:liver concentration ratios of glutathione were significantly decreased in vivo by E2-17G and in the perfused liver by E2-17G and E2-3G. These data indicate that E2-17G cis-inhibits the canalicular transport of glutathione and thus contributes to the cholestatic effect by inhibiting BSIF.
Bile secretion serves two main functions: 1) elimination of endogenous and exogenous compounds from blood, and 2) secretion of bile acids for emulsification and digestion of dietary fats. Bile formation in vertebrates is primarily dependent on transport of osmotically active solutes such as bile salts, glutathione, and  across the canalicular membrane of the hepatocyte, followed by the passive movement of water (Nathanson and Boyer, 1991; Trauner et al., 1998). The bile salt export pump (Bsep) mediates the concentrative transport of bile salts across the canalicular membrane (Gerloff et al., 1998), and thus generates the bile salt-dependent (BSDF) component of bile flow. Transport of different glutathione species into bile is considered critical to the generation of bile salt-independent bile flow (BSIF) (Ballatori and Truong, 1992). Whereas multidrug resistance-associated protein 2 (Mrp2, ABCC2) mediates the active transport of oxidized glutathione (GSSG) and glutathione conjugates (Konig et al., 1999), the data in support of Mrp2-mediated GSH transport are largely indirect (Ballatori and Rebbeor, 1998). Either Mrp2 or more specific, as yet unidentified GSH transporters, are proposed to mediate biliary secretion of GSH (Oude Elferink et al., 1995; Kaplowitz et al., 1996; Ballatori and Rebbeor, 1998; Konig et al., 1999; Yang and Hill, 2001). However, recent data provide direct evidence that Mrp2 in rat canalicular membranes can transport GSH in an ATP-dependent manner (Rebbeor et al., 2002). An electroneutral
across the canalicular membrane of the hepatocyte, followed by the passive movement of water (Nathanson and Boyer, 1991; Trauner et al., 1998). The bile salt export pump (Bsep) mediates the concentrative transport of bile salts across the canalicular membrane (Gerloff et al., 1998), and thus generates the bile salt-dependent (BSDF) component of bile flow. Transport of different glutathione species into bile is considered critical to the generation of bile salt-independent bile flow (BSIF) (Ballatori and Truong, 1992). Whereas multidrug resistance-associated protein 2 (Mrp2, ABCC2) mediates the active transport of oxidized glutathione (GSSG) and glutathione conjugates (Konig et al., 1999), the data in support of Mrp2-mediated GSH transport are largely indirect (Ballatori and Rebbeor, 1998). Either Mrp2 or more specific, as yet unidentified GSH transporters, are proposed to mediate biliary secretion of GSH (Oude Elferink et al., 1995; Kaplowitz et al., 1996; Ballatori and Rebbeor, 1998; Konig et al., 1999; Yang and Hill, 2001). However, recent data provide direct evidence that Mrp2 in rat canalicular membranes can transport GSH in an ATP-dependent manner (Rebbeor et al., 2002). An electroneutral  exchanger, the anion exchanger 2 (AE2), which functions as a counterpoint to the acid-extruding system (Na+/H+ exchanger and
exchanger, the anion exchanger 2 (AE2), which functions as a counterpoint to the acid-extruding system (Na+/H+ exchanger and  symport), is responsible for
symport), is responsible for  secretion into bile (Meier et al., 1985) and also contributes to BSIF formation (Hardison and Wood, 1978; Van Dyke et al., 1982).
secretion into bile (Meier et al., 1985) and also contributes to BSIF formation (Hardison and Wood, 1978; Van Dyke et al., 1982).
Intrahepatic cholestasis may be induced by a variety of dissimilar chemical compounds, including steroid hormones. A structure-activity relationship has been established for steroid-glucuronide-induced cholestasis (Vore and Slikker, 1985). The glucuronide conjugates of the steroid D-ring of estradiol, estriol, and ethynylestradiol exert potent cholestatic properties in the rat and induce an acute but reversible decrease in bile flow after an intravenous bolus dose. Both BSDF and BSIF fractions of bile flow are similarly decreased immediately after a dose of estradiol-17β-d-glucuronide (E2-17G), followed by a compensatory increase of BSDF, which leads to bile flow normalization (Meyers et al., 1980). We recently demonstrated a marked endocytic internalization of Mrp2 from the canalicular membrane into intracellular membranes at 20 and 75 min after administration of E2-17G to rats in parallel with a decrease in Mrp2 transport activity (Mottino et al., 2002). Bile flow was partially restored (40% of control) 75 min after E2-17G, even though the endocytic retrieval of Mrp2 from the canalicular membrane into intracellular compartments was indistinguishable from that observed at 20 min, when bile flow was 15% of control. This agrees well with a compensatory role of BSDF in bile flow restoration, which in turn may result from a different balance between insertion/retrieval of Mrp2 and Bsep during the phase of recovery from cholestasis. Whereas factors leading to insertion of ABC transporters to the canalicular membrane such as cAMP and Ca2+/protein kinase C activation are better understood (for review, see Haussinger et al., 2000; Kipp and Arias, 2002), far less is known about the events governing internalization of transporters (e.g., in response to E2-17G) from the apical domain to intracellular vesicles. The latter phenomenon may be crucial in determining impairment in transport function under conditions of acute estrogen-induced cholestasis. In preliminary studies, we observed that the biliary excretion of glutathione was markedly impaired after E2-17G and did not recover in parallel with either bile flow or the biliary excretion of a classic Mrp2 substrate, dinitrophenyl-S-glutathione (DNP-SG). In the current study, we therefore evaluated the biliary excretion and hepatic content of total glutathione in E2-17G-induced cholestasis in vivo and in the isolated perfused rat liver. We also examined the bile secretory rate of  and the potential interference of bilirubin conjugates with glutathione secretion into bile. The data show that E2-17G severely impaired the biliary secretion of both GSH and GSSG in vivo and in the perfused liver without influencing their liver content, thus indicating selective inhibition of canalicular transport of these glutathione species.
and the potential interference of bilirubin conjugates with glutathione secretion into bile. The data show that E2-17G severely impaired the biliary secretion of both GSH and GSSG in vivo and in the perfused liver without influencing their liver content, thus indicating selective inhibition of canalicular transport of these glutathione species.
Materials and Methods
Chemicals. E2-17G, estradiol-3-d-glucuronide (E2-3G), NADPH, GSH, glutathione reductase, and acivicin were obtained from Sigma-Aldrich (St. Louis, MO). Radioactive E2-17G (6,7-3H, 40.50 Ci/mmol) and E2-3G (6,7-3H, 57.00 Ci/mmol) were purchased from PerkinElmer Life Sciences (Boston, MA). All other chemicals were of analytical grade purity and used as supplied.
Animals and Experimental Protocols. Female, Sprague-Dawley rats (190–210 g) (Harlan, Indianapolis, IN) were used throughout. The rats had free access to food and water and were maintained on a 12-h automatically timed light and dark cycle. All procedures involving animals were conducted in accordance with National Institutes of Health guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Kentucky.
In Vivo Studies. The rats were anesthetized with urethane (1,000 mg/kg i.p.) and thus maintained throughout. Body temperature was measured with a rectal probe and maintained at 37°C with a heating pad connected to a temperature regulator (model 73A; YSI Inc., Yellow Springs, OH). The femoral vein and the common bile duct were cannulated with polyethylene tubing (PE50 and PE10, respectively). Saline was administered intravenously throughout the experiment to replenish body fluids. E2-17G (15 μmol/kg in saline/propylene glycol/ethanol, 10:4:1; 4.25 mM) or solvent was administered via the femoral vein 30 min after bile duct cannulation (Meyers et al., 1980). Bile was collected at 10-min intervals for 120 min in preweighed tubes containing 0.1 ml of 10% sulfosalicylic acid to minimize oxidation of GSH (Griffith, 1980) and immediately used in total glutathione and GSSG assays. Bile flow was determined gravimetrically, assuming a density of 1 g/ml. To determine intrahepatic content of glutathione species, livers were perfused for 30 s with cold saline 20, 80, or 120 min after E2-17G, and the median lobe gently removed and homogenized [20% (w/v) in saline]. Two volumes of the homogenate were mixed with 1 volume of 10% sulfosalicylic acid, centrifuged at 5,000g for 5 min, and the supernatant immediately used in total glutathione and GSSG assays. In additional experiments, bile was either collected under liquid petroleum jelly and immediately used in  assay or collected under dim light and kept at –20°C until used in the total bilirubin assay.
assay or collected under dim light and kept at –20°C until used in the total bilirubin assay.
In a separate set of experiments, the effects of E2-17G or solvent on biliary excretion of total glutathione and GSSG were evaluated in animals after retrograde administration of 20 μmol/kg acivicin over 1 min into the common bile duct. Acivicin irreversibly inhibits intrabiliary γ-glutamyl transpeptidase (GGT)-mediated hydrolysis of glutathione (Ballatori et al., 1986). Basal bile collection started 10 min later. The protocol for injection of E2-17G or solvent was the same as described above.
Isolated Perfused Rat Liver (IPRL) Studies. Rats were anesthetized with urethane (1,000 mg/kg i.p.) and the bile duct cannulated with PE10 tubing. Livers were perfused via the portal vein as described previously (Vore et al., 1978) with 200 ml of Krebs-Ringer bicarbonate buffer at a constant flow rate of 20 ml/min in a recirculating design. The perfusate was continuously oxygenated with 95% O2, 5% CO2. After a 20-min equilibration, basal bile flow was collected for 10 min, followed by addition of solvent, E2-17G, or E2-3G (15 μmol/kg; 4.25 mM in saline/propylene glycol/ethanol, 10:4:1) to the reservoir at zero time. Bile was collected at 10-min intervals for 60 min for determination of bile flow and glutathione species as described above. Samples (200 μl) from the reservoir were also taken at 10-min intervals for glutathione and lactate dehydrogenase (LDH) activity determinations. At the end of the collecting period, the median lobe was removed and used in homogenate preparation as described above. Experiments were considered valid only if basal bile flow was greater than 15 μl/min/kg (∼0.6 μl/min/g liver). Viability of preparations was constantly checked by determining LDH activity in the reservoir; experiments exhibiting activities over 20 U/l were not used.
In a separate set of experiments, we evaluated the biliary excretion of E2-17G or E2-3G in the IPRL. Perfusions were conducted in a recirculating manner as described above using [3H]E2-17G or [3H]E2-3G (3.0 μCi; 15 μmol/kg) added to the reservoir at zero time. Bile and perfusate were collected at 10-min intervals for 60 min for determination of bile flow, LDH activity and radioactivity. At the end of the collecting period, the median lobe was removed and homogenized [20% (w/v) in saline] and an aliquot assayed for radioactivity. Criteria for viability of perfusions were as described above.
Analytical Methods. Total glutathione (GSH + GSSG) and GSSG in bile and liver homogenate from both in vivo and IPRL studies as well as in the perfusate of the IPRL model were determined spectrophotometrically by using the recycling method of Tietze (1969), as modified by Griffith (1980). Total glutathione was expressed as equivalents of GSH, and GSSG as equivalents of GSH relative (percentage) to total glutathione. LDH activity in the perfusate was determined using a commercial kit (catalog no. 340-LD; Sigma-Aldrich). Total bilirubin (conjugated + unconjugated) was determined in bile using a commercial kit (catalog no. 550-A; Sigma-Aldrich). Biliary  concentrations were calculated from pH and pCO2 data using the Henderson-Hasselbalch equilibrium equation; pH and pCO2 were measured immediately after bile collection in an automated blood-gas analyzer (Compact 1; AVL Medical Instruments AG, Schaffhausen, Switzerland). In experiments using [3H]E2-17G and [3H]E2-3G, total radioactivity was quantitated in aliquots of bile, perfusate, and liver homogenates after addition of 5 ml of Bio-Safe II cocktail (Research Products International, Mount Prospect, IL) and counting in a 1500 series Tri-Carb liquid scintillation analyzer (PerkinElmer Life Sciences, Boston, MA).
concentrations were calculated from pH and pCO2 data using the Henderson-Hasselbalch equilibrium equation; pH and pCO2 were measured immediately after bile collection in an automated blood-gas analyzer (Compact 1; AVL Medical Instruments AG, Schaffhausen, Switzerland). In experiments using [3H]E2-17G and [3H]E2-3G, total radioactivity was quantitated in aliquots of bile, perfusate, and liver homogenates after addition of 5 ml of Bio-Safe II cocktail (Research Products International, Mount Prospect, IL) and counting in a 1500 series Tri-Carb liquid scintillation analyzer (PerkinElmer Life Sciences, Boston, MA).
Statistical Analysis. Data are presented as the mean ± S.D. Comparison between solvent and E2-17G groups in in vivo experiments was performed using the Student's t test. In the IPRL, a one-way analysis of variance followed by the Bonferroni test was used to compare the effects of solvent, E2-17G, or E2-3G. Time-related changes in biliary total bilirubin secretion and in liver glutathione content in in vivo experiments were analyzed by this same test. Values of p < 0.05 were considered to be statistically significant.
Results
Effect of E2-17G on Biliary Secretion and Liver Content of Glutathione in Vivo. Bile flow decreased substantially immediately after E2-17G injection, partially recovered at 70 to 80 min, and reached solvent values at 120 min (Fig. 1A), as observed previously for doses ranging from 11 to 17 μmol/kg (Meyers et al., 1980; Mottino et al., 2002). Whereas the concentration of total glutathione in bile decreased slightly over time in animals from the solvent group, the decrease was marked in E2-17G-treated animals relative to solvent values from 40 min onwards (Fig. 1B). Despite normalized bile flow in E2-17G-treated rats at 120 min, glutathione concentrations remained significantly impaired by 75% with respect to the solvent group. Similarly, the biliary excretion of total glutathione was severely inhibited in response to E2-17G throughout the experimental period (Fig. 1C). GSSG represented 26 ± 7% of total glutathione in basal bile (N = 6), and was not substantially affected by the solvent or by E2-17G at any time (data not shown). This clearly indicates that E2-17G affected excretion of GSSG and the remaining component of total glutathione, GSH, to a similar degree. To determine whether the decreased biliary excretion of glutathione led to its retention in liver, or whether E2-17G also decreased hepatic glutathione, we examined the liver content of total glutathione under basal conditions and after injection of solvent or E2-17G at 20, 80, and 120 min (Fig. 1D). The glutathione levels in liver from both solvent and E2-17G-treated animals decreased somewhat with time, but this did not reach statistical significance. The relative content of GSSG in liver under basal conditions was 4.8 ± 0.5% of total glutathione (N = 4), and was not substantially affected by solvent or E2-17G at any time analyzed (data not shown). Calculation of the bile/liver concentration ratios for glutathione further illustrated the decreased ability of the liver to secrete glutathione in bile after E2-17G (Fig. 1E).

Biliary excretion and hepatic concentration of total glutathione in vivo. Rats were treated with solvent (S) or E2-17G (15 μmol/kg), and bile flow (A), total glutathione concentration (B) and excretion rate (C) were evaluated in bile samples. Liver content of total glutathione (D) was determined at the indicated time after solvent or E2-17G administration. The bile/liver total glutathione concentration ratios (E) were calculated from the data in B and D. Data represent means ± S.D. of three to four rats per group. *, p < 0.05, significantly different from E2-17G.
To ascertain whether the decreased biliary concentration and excretion rate of glutathione could be an artifact resulting from its increased metabolism by GGT under cholestatic conditions, we administered acivicin by retrograde infusion into the bile duct before injecting E2-17G. Although biliary glutathione concentrations were slightly higher (∼20%) in acivicin-treated animals, both under basal conditions and after solvent or E2-17G administration, the changes in bile flow and biliary glutathione concentration in response to E2-17G were not affected (data not shown).
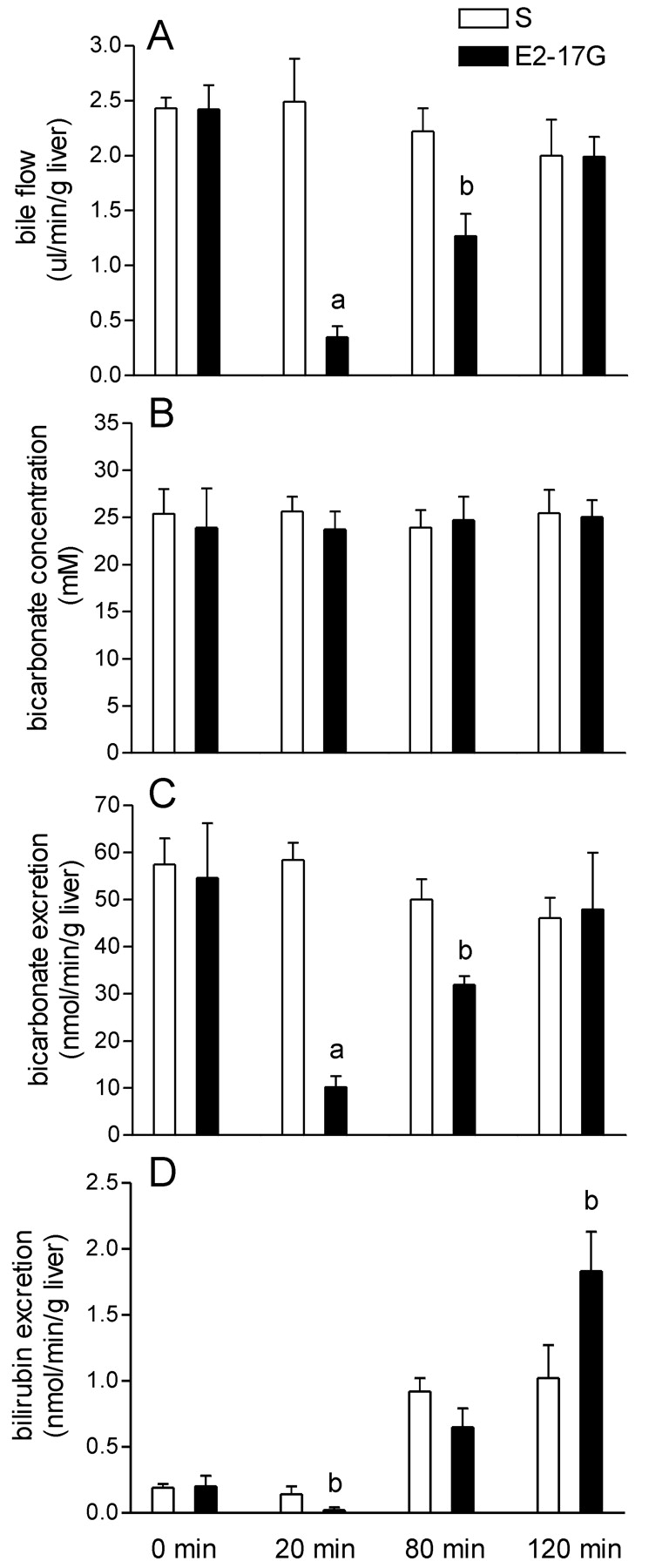
Because  also contributes to BSIF, we evaluated its biliary secretory rate. The concentration of
also contributes to BSIF, we evaluated its biliary secretory rate. The concentration of  did not vary in response to E2-17G administration (Fig. 2B), and therefore its biliary secretion (Fig. 2C) paralleled bile flow (Fig. 2A).
did not vary in response to E2-17G administration (Fig. 2B), and therefore its biliary secretion (Fig. 2C) paralleled bile flow (Fig. 2A).

Biliary excretion of  and total bilirubin in vivo. Rats were treated with solvent (S) or E2-17G (15 μmol/kg), and bile flow (A),
and total bilirubin in vivo. Rats were treated with solvent (S) or E2-17G (15 μmol/kg), and bile flow (A),  concentration (B) and excretion rate (C) as well as total bilirubin excretion rate (D) were evaluated in bile samples. Data represent means ± S.D. of three rats per group. a, significantly different from S (p < 0.005); b, significantly different from S (p < 0.05).
concentration (B) and excretion rate (C) as well as total bilirubin excretion rate (D) were evaluated in bile samples. Data represent means ± S.D. of three rats per group. a, significantly different from S (p < 0.005); b, significantly different from S (p < 0.05).
Glutathione secretion into bile may be competitively inhibited by exogenous or endogenous organic anions, particularly if they accumulate in the liver as a consequence of decreased canalicular transport. We therefore examined the biliary excretion of total (unconjugated + conjugated) bilirubin. The major pigments in bile, monoglucuronosyl and bisglucuronosyl bilirubin conjugates, are prototypical substrates of Mrp2 and may thus inhibit glutathione secretion into bile. Figure 2D shows a 5-fold increase in the total bilirubin secretion rate at 80 and 120 min (p < 0.05) in solvent-treated rats, indicating increased production of the pigment, most probably as a consequence of solvent-induced intravascular hemolysis. In E2-17G-treated rats, total bilirubin excretion was decreased at 20 min, reached the same level at 80 min, and was higher at 120 min with respect to solvent controls.
Effect of E2-17G and E2-3G on Biliary Secretion, Efflux into the Perfusate, and Liver Content of Glutathione in IPRL. Although increased biliary excretion of bilirubin conjugates did not affect glutathione excretion in vivo in solvent-treated rats, it was possible that these pigments might inhibit transport of glutathione species under conditions of reduced transport activity after E2-17G. We therefore used the IPRL perfused with Krebs-Ringer bicarbonate buffer without addition of red blood cells to avoid the potential interference of bilirubin derivatives. Similarly, E2-17G, an Mrp2 substrate and organic anion, might itself inhibit glutathione transport. To test this possibility, we compared the potential inhibitory effects of the cholestatic E2-17G and noncholestatic E2-3G on the biliary excretion of glutathione in the IPRL model.
Figure 3 shows the effects of E2-17G, E2-3G, and solvent control on bile flow and biliary concentration and excretion rate of total glutathione in the perfused liver. E2-17G substantially decreased bile flow relative to both E2-3G and solvent throughout the experimental period (Fig. 3A). Interestingly, both steroid glucuronide conjugates decreased the biliary concentration of glutathione, which was slightly more pronounced in the E2-17G group at later times (Fig. 3B). As expected from the decrease in both bile flow and glutathione concentration in bile, glutathione excretion was substantially decreased by E2-17G (Fig. 3C). The biliary excretion rate of glutathione was similar between the solvent and E2-3G groups except for the last two collection periods, where both E2-17G and E2-3G significantly decreased glutathione excretion. The cumulative excretion of total glutathione at 60 min was similar for solvent and E2-3G groups (72.4 ± 21.2 and 59.7 ± 11.4 nmol/g liver, respectively) and was significantly decreased by E2-17G treatment (29.0 ± 9.3 nmol/g liver, p < 0.05). The proportion of GSSG in basal bile in the IPRL was 36.9 ± 6.5% (N = 9) of total glutathione and was not affected by treatment with solvent, E2-17G, or E2-3G (data not shown). The amount of total glutathione that accumulated in the perfusate during the 60-min period was significantly higher for E2-17G (43 ± 5 nmol/g liver, N = 3, p < 0.05) than for E2–3G (26 ± 6 nmol/g liver, N = 3), whereas it did not differ from the solvent group (34 ± 7 nmol/g liver, N = 3). This finding could reflect differential transport of glutathione across the basolateral membrane after E2-17G versus E2-3G administration and/or increased paracellular reflux due to alteration of tight junction permeability under E2-17G-induced cholestatic conditions (Kan et al., 1989). The proportion of GSSG in the perfusate was 8 ± 0.4% (N = 9) under basal conditions and was not affected by any treatment. As shown for E2–17G in vivo, the total hepatic glutathione level did not vary in response to E2-17G or E2-3G administration (Fig. 3D), indicating that the impairment in biliary excretion induced by these agents is not a consequence of changes in the hepatic level of glutathione. Liver GSSG represented 5.0 ± 0.7% of total glutathione and was not modified by addition of solvent, E2-17G or E2-3G to the perfusate (data not shown). Finally, calculation of the bile/liver concentration ratios for glutathione in Fig. 3E demonstrated that both E2-17G and E2-3G significantly inhibited the ability of the liver to secrete glutathione in bile.

Biliary excretion and hepatic content of total glutathione in the perfused liver. Solvent (S), E2-17G, or E2-3G (15 μmol/kg) were added to the perfusate, and bile flow (A), total glutathione concentration (B), and excretion rate (C) were evaluated in bile samples. Liver content of total glutathione (D) was determined at the indicated time after solvent or steroid administration. The bile/liver total glutathione concentration ratios (E) were calculated from the data in B and D. Data represent means ± S.D. of three rats per group. a, S significantly different from E2-17G (p < 0.05); b, E2-3G significantly different from E2-17G (p < 0.05); c, S significantly different from E2-3G (p < 0.05).
Disposition of [3H]E2-17G and [3H]E2-3G in the IPRL. We examined the disposition of [3H]E2-17G and [3H]E2-3G in the IPRL after their addition (15 μmol/kg) to the perfusate. Disappearance of radiolabel from the perfusate was similar for both [3H]E2-17G and [3H]E2-3G (Fig. 4A), even though the biliary excretion of [3H]E2-17G-derived radioactivity was significantly impaired as a result of the cholestasis (Fig. 4B). Consequently, the hepatic content of radioactivity derived from [3H]E2-17G at 60 min was significantly higher than for [3H]E2-3G (Fig. 4C) and was calculated as ∼370 and 215 nmol/g liver, respectively.

Disposition of [3H]E2-17G and [3H]E2-3G in the perfused liver. [3H]E2-17G and [3H]E2-3G (3 μCi; 15 μmol/kg) were added to the perfusate at zero time. Radioactivity in the perfusate (A), bile (B), and liver homogenate (C) were determined as described under Materials and Methods and expressed as a percentage of the initial dose. Total radioactivity in liver was determined at the end of the experiment. Data represent means ± S.D. of three rats per group. *, p < 0.05 significantly different from E2-17G.
Discussion
The present studies demonstrate a severe impairment in biliary excretion of glutathione during E2-17G-induced cholestasis in vivo that persisted despite the recovery of bile flow. The sustained decrease in excretion of glutathione was also in marked contrast to the relatively rapid recovery in the biliary excretion of DNP-SG. The biliary excretion of DNP-SG was significantly inhibited at 20 and 75 min after E2-17G administration, but was completely restored at 180 min (Mottino et al., 2002). This time course of inhibition of Mrp2 activity is in excellent agreement with confocal microscopy analyses of the liver, demonstrating that Mrp2 localizes within the canalicular space in control liver, is redistributed into intracellular structures at 20 and 75 min after E2-17G administration, but returns to its canalicular localization by 180 min (Mottino et al., 2002). In these initial studies, we found that the concentration of total glutathione in bile had not recovered at 3 h. The marked discrepancy between the recoveries of Mrp2 localization and biliary excretion of DNP-SG versus glutathione indicated that factors other than retrieval of Mrp2 contributed to the decreased biliary excretion of glutathione. In the present studies, we examined several factors (see below) that might mediate the persistent inhibition of glutathione transport into bile.
Decreased Hepatic Content and Proportion of GSH versus GSSG. E2-17G-induced depletion of hepatic glutathione could readily explain a decrease in its biliary excretion. Although the hepatic content of GSSG and GSH declined somewhat over time after E2-17G (about 40% over 2 h), this was not significantly different from the 25% decline in hepatic total glutathione in control livers. The reason for the decline in hepatic total glutathione in control liver is not known but may be due to interruption of the enterohepatic circulation and irretrievable loss of glutathione and derived amino acids. The declining hepatic glutathione in control animals provided the rationale for focusing the current studies on the acute effects of E2-17G-induced cholestasis on glutathione excretion. The observation that treatment with E2-17G did not further affect liver GSH or GSSG content over time indicated that the profound decrease in biliary excretion resulted from factors other than an impaired hepatic level. The decreased biliary excretion of total glutathione after E2-17G treatment would be expected to lead to its retention and increased hepatic content. However, because the difference in biliary excretion between control and E2-17G-treated rats represented less than 15% of the total liver content, any increase was likely masked by interindividual variations. Alternatively, increased GSH efflux across the basolateral membrane via organic anion transporting protein/Mrp transporters (Li et al., 2000; Mao et al., 2000), as suggested by IPRL experiments, could contribute to the lack of increased hepatic GSH. The ratios of oxidized to reduced glutathione in liver, bile, and perfusate were also unaffected by E2-17G, indicating that E2-17G did not induce oxidative stress. Finally, retrograde administration of acivicin into the bile duct to inhibit GGT-mediated metabolism of glutathione did not restore biliary excretion of GSH or GSSG after E2-17G, thus ruling out hydrolysis and re-uptake of the derived amino acids along the biliary tree.
Competitive Inhibition of GSH and GSSG Transport by Other Organic Anions. Several lines of evidence indicate that GSH is a relatively low-affinity Mrp2 substrate with Km values in the millimolar range (Ballatori and Rebbeor, 1998; Rebbeor et al., 2002), whereas GSSG is transported by MRP1 with a Km of ∼100 μM (Leier et al., 1996). This difference in Km value likely explains the observation that the proportion of total glutathione in bile as GSSG is 26 to 36%, whereas that in liver is only 5%. Ballatori and Clarkson (1985) demonstrated that bromosulfophthalein and phenol-3,6-dibromophthalein disulfonate (a nonmetabolizable analog of bromosulfophthalein) similarly inhibited the biliary excretion of GSH and GSSG in a dose-dependent manner. Thus, accumulation of alternative Mrp2 substrates in liver during cholestasis could also lead to competitive inhibition of GSH and GSSG transport across the canalicular membrane. Biliary excretion of total bilirubin in vivo was decreased at 20 min after E2-17G, but subsequently increased significantly at 80 and 120 min. Impairment of canalicular secretion of total pigments during the acute phase of cholestasis agrees well with the previously described endocytic retrieval and decreased activity of Mrp2 (Mottino et al., 2002). The fact that the excretion of these pigments at 80 min was the same as in solvent rats, despite the decreased activity of Mrp2 observed 75 min after E2-17G administration, could be due to its accumulation in liver and generation of a larger gradient; this is also the likely basis for its increased secretory rate in the E2-17G group observed at 120 min. Because the monoglucuronosyl and bisglucuronosyl bilirubin conjugates are high-affinity Mrp2 substrates, with Km values of 0.8 and 0.5 μM, respectively (Kamisako et al., 1999), their intrahepatic accumulation could readily compete for transport of the relatively low-affinity GSSG and GSH. To assess the role of bilirubin conjugates, we carried out further studies in the liver perfused with buffer, where we could exclude red blood cells. We also assessed the role that E2-17G and its noncholestatic regio-isomer E2-3G might play in competing with glutathione for canalicular transport. Importantly, the concentration of glutathione in bile decreased significantly following addition of E2-17G, indicating that intracellular accumulation of bilirubin products is not an essential component of the E2-17G-induced inhibition of canalicular glutathione transport in vivo. Interestingly, E2-3G also decreased the biliary concentration of glutathione, even though it did not inhibit bile flow. These data indicate that the steroid glucuronides themselves inhibit the biliary excretion of glutathione. E2-17G is a high-affinity Mrp2 substrate (Km value of 7 μM; Ito et al., 2001), and recent studies in our laboratory have also identified E2-3G as an Mrp2 substrate, with Km values of ∼19 μM (P. M. Gerk and M. Vore, manuscript submitted for publication). E2-17G-3-sulfate is a major metabolite of E2-17G (Meyers et al., 1980), and preliminary studies show that it is also a potent inhibitor of Mrp2-mediated transport (Gerk et al., 2003). Whereas E2-3G was readily excreted in bile, the cholestatic activity of E2-17G resulted in significantly greater retention of E2-17G-derived radioactivity in liver. The approximate concentrations of [3H]E2-3G and [3H]E2-17G derived radioactivity in liver at 60 min were 0.2 and 0.37 mM, respectively, concentrations much greater than their Km values for Mrp2-mediated transport. In contrast, the concentrations of GSSG (∼0.1 mM) and GSH (∼3.2 mM) in liver are similar to their Km values for transport. Together, these data provide strong evidence that E2-17G retained in liver cis-inhibits the biliary excretion of GSH and GSSG.
Bile flow is dependent on the secretion of osmotically active solutes that bring water with them passively until osmotic equilibrium is achieved. A similar and parallel inhibition of the transporters mediating biliary excretion of osmotically active solutes should therefore lead to a decrease in bile flow, with little change in the concentration of the solutes, as was postulated to occur in phalloidin-induced cholestasis (Rost et al., 1999). The concentration of glutathione in bile did not change during the onset of cholestasis in vivo (i.e., at 10 min; Fig. 1B) nor does the concentration of bile acids (Meyers et al., 1980). Bicarbonate is also an important osmolyte in bile that contributes to BSIF, and its concentration in bile did not change throughout E2-17G cholestasis. These data imply that each of the transporters that contribute to bile flow, e.g., Bsep, Mrp2, and AE2, are equally inhibited at early times. We recently showed that Bsep also undergoes rapid endocytic retrieval after E2-17G (Crocenzi et al., 2003), resulting in loss of canalicular bile acid transport. The rapid recovery of BSDF in vivo probably reflects the reinsertion of Bsep into the canalicular membrane and transport of accumulated bile acids into bile. Although Mrp2 is also restored to its place in the canalicular membrane at the same time as is Bsep, the accumulated Mrp2 substrates in the hepatocyte cis-inhibit GSH and GSSG transport into bile, resulting in a sustained inhibition of BSIF. The decreased glutathione concentration in bile provides an explanation for the observed decrease in BSIF in E2-17G cholestasis (Meyers et al., 1980) and for the lack of recovery of bile flow in the perfused liver. In vivo, a compensatory increase in BSDF contributes significantly to the spontaneous recovery of bile flow after E2-17G cholestasis (Meyers et al., 1980). Because the bile salt pool is severely depleted in the IPRL, BSDF does not contribute significantly to bile formation, thus providing a likely explanation for the differential recoveries of bile flow. Together, the data suggest that early cholestasis after E2-17G is due to simultaneous retrieval of Bsep, Mrp2, and probably AE2, whereas cholestasis at later times reflects primarily cis-inhibition of glutathione transport into bile.
In summary, we have demonstrated a severe impairment in the biliary secretion of GSH and GSSG in vivo and in the perfused liver during E2-17G cholestasis, whereas there is no change in the hepatic content of glutathione. We conclude that E2-17G-induced endocytic retrieval of Mrp2 and Bsep, followed by cis-inhibition of canalicular glutathione transport by E2-17G itself, mediates the potent cholestatic activity of E2-17G.
Acknowledgments
We express our gratitude to Drs. Daret St. Clair, Phil Gerk, Marcelo G. Roma, and Fernando A. Crocenzi for valuable suggestions.
Footnotes
-
This study was supported by U.S. Public Health Service Grant GM-55343 (to M.V.), and Consejo Nacional de Investigaciones Científicas y Técnicas, Agencia Nacional de Promoción Científica y Tecnológica, and Fundación Antorchas (to A.D.M.).
-
ABBREVIATIONS: Bsep, bile salt export pump; BSDF, bile salt-dependent bile flow; BSIF, bile salt-independent bile flow; Mrp2, multidrug resistance-associated protein 2; GSSG, oxidized glutathione; GSH, reduced glutathione; AE2, anion exchanger 2; E2-17G, estradiol-17β-d-glucuronide; DNP-SG, dinitrophenyl-S-glutathione; E2-3G, estradiol-3-d-glucuronide; PE, polyethylene; GGT, γ-glutamyl transpeptidase; LDH, lactate dehydrogenase; IPRL, isolated perfused rat liver.
-
DOI: 10.1124/jpet.103.054544.
- Received May 14, 2003.
- Accepted June 12, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}