


Abstract
The involvement of multidrug resistance-associated protein 1 (Mrp1) and P-glycoprotein (mdr1) in the tissue distribution and excretion of grepafloxacin (GPFX), a fluoroquinolone antibiotic, was investigated using gene-deficient mice [mdr1a(–/–), mdr1a/1b(–/–), and mrp1(–/–)]. The plasma concentration-time profile of GPFX in mrp1(–/–) was nearly identical to that in mrp1(+/+), whereas that in mdr1a/1b(–/–) was higher than that in mdr1a/1b(+/+). The urinary clearance of GPFX in mdr1a/1b(–/–) was lower than that in mdr1a/1b(+/+), suggesting that the urinary excretion of GPFX is at least partially mediated by mdr1. The tissue-to-plasma concentration ratios during the β-phase (Kp β,) was significantly higher in the heart, trachea, kidney, spleen, and brown fat of mrp1(–/–) than those in mrp1(+/+). In MRP1-transfected LLC-PK1 cells, the efflux of GPFX after preloading into the cells was higher than that observed in the parent cell lines. These results suggest that GPFX is a substrate of MRP1 and that its distribution to these tissues might be limited by Mrp1. On the other hand, a higher Kp β, and of GPFX in mdr1a(–/–) mdr1a/1b(–/–) compared with mdr1a/1b(+/+) was observed only in the brain. GPFX was efficiently distributed to the lung parenchyma cells and pulmonary airspaces, including the epithelial lining fluid and macrophages that are the pharmacological target of GPFX, although the contribution of Mdr1 and Mrp1 to such distribution seems to be minor. Thus, the present findings reveal that the disposition of GPFX is at least in part governed by these two ABC transporters and that both Mrp1 and Mdr1 are involved in the limited distribution of GPFX to the distinct tissues, including pharmacological and/or toxicological targets by an active efflux mechanism.
GPFX is a new quinolone (NQ) antibiotic and has a high lipophilicity compared with other NQs. The distinct pharmacokinetic features of GPFX include its higher distribution to various tissues, especially lung (Akiyama et al., 1995a,b; Nakajima et al., 2000; Suzuki et al., 2002), although the exact mechanism for this has not yet been fully clarified. We have previously reported that GPFX specifically binds to phosphatidylserine among various phospholipids and that the tissue distribution of GPFX is well correlated with the tissue phosphatidylserine content (Suzuki et al., 2002). However, membrane transport processes such as an active influx and/or efflux also may contribute to the tissue distribution of GPFX. In fact, the distribution of GPFX and other NQs to the brain is restricted because of the limited permeability of these compounds through the blood-brain barrier by active efflux transporters such as P-glycoprotein (P-gp), an mdr1 gene product (Tsuji et al., 1988, Ooie et al., 1996, 1997, Murata et al., 1999, Tamai et al., 2000).
In mice, mdr1a is highly expressed in intestine, brain, testis, and lung, whereas mdr1b is highly expressed in adrenal, pregnant uterus, and ovary. Both genes are expressed in many other tissues (Croop et al., 1989; Schinkel et al., 1994, 1995). Transport studies with an MDR1-transfected cell line demonstrated that GPFX is a substrate of P-gp (Naruhashi et al., 2001). These findings suggest possible roles for P-gp in the secretion and/or distribution of GPFX in these organs. Indeed, several NQs, including GPFX, have been reported to be actively secreted across the apical membrane of human intestinal epithelial Caco-2 cells, and P-gp possibly limits the bioavailability of these drugs in vivo (Griffiths et al., 1993, 1994; Naruhashi et al., 2001; Yamaguchi et al., 2000, 2001, 2002). Transport studies with kidney epithelial cell line LLC-PK1 monolayers suggested that P-gp and/or some other transport system plays a role in the renal tubular secretion of NQs (Ito et al., 1997; Matsuo et al., 1998). In addition to these cell lines, mdr1 gene-deficient mice [mdr1a(–/–) and mdr1a/1b(–/–)] are efficient tools to elucidate the possible functions of P-gp in the whole body (Schinkel et al., 1994, 1997). The pharmacokinetics of many drugs such as digoxin, dexamethasone, cyclosporin A, taxol, and vinblastine has been examined in these deficient mice (Schinkel et al., 1994, 1995, 1997; Mayer et al., 1996; van Asperen et al., 1996; Sparreboom et al., 1997). Using these mice Yamaguchi et al. (2002) found that P-gp was involved in GFPX distribution to the brain, but not to the lung, heart, liver, small intestine, kidney, and testis although its contribution to other organs is still unknown.
In contrast to the many studies on P-gp described above, the involvement of multidrug resistance-associated proteins (Mrp) in the disposition of GPFX has not yet been fully characterized. Mrp1 is also expressed in ubiquitous organs, but its expression in the liver was minimal in normal condition. Using gene-deficient mice [mrp1(–/–)], Mrp1 was proposed to be involved in the efflux of etoposide from the cerebrospinal fluid space (Wijnholds et al., 2000). Mrp1 is highly expressed in the lung and also expressed in the epithelial lining bronchi and bronchioles (Stride et al., 1996). This suggests that the involvement of Mrp1 in the distribution of its substrate to the lung needs to be investigated. Mrp2 is, on the other hand, highly expressed in the liver, followed by the kidney and small intestine, and proposed to be mainly involved in the biliary excretion of various organic anions (Borst and Elferink, 2002; Payen et al., 2002; Suzuki and Sugiyama, 2002). Using Mrp2-deficient rats, we have previously reported that the biliary excretion of GPFX is partially mediated by Mrp2 (Sasabe et al., 1998). Such involvement of Mrp2 has also been reported for the biliary excretion of olamufloxacin (HSR-903), another NQ (Murata et al., 1998). In view of overlapping substrate specificity of Mrp1 with Mrp2 and the expression profile of Mrp1 in ubiquitous tissues, we have investigated here the possible contribution of Mrp1 to the tissue distribution and excretion of GPFX using mrp1(–/–) mice. We have examined the distribution of GPFX to many organs in mdr1a(–/–) and mdr1a/1b(–/–) mice to clarify the different roles of both Mrp1 and Mdr1 in the distribution of GPFX.
Materials and Methods
Materials. [14C]GPFX, (±)-1-cyclopropyl-6-fluoro-1,4-dihydro-5-methyl-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxulic acid monohydrochloride (specific activity 3.00 MBq/mg; radiochemical purity 97.1%), and [14C]levofloxacin, (–)-(S)-9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido[1,2,3-de][1,4] benzoxazine-6-carboxylic acid hemihydrate (LVFX; specific activity 1.96 GBq/mmol; radiochemical purity 98.5%) were obtained from Amersham Biosciences UK, Ltd. (Little Chalfont, Buckinghamshire, UK). [3H]Sucrose (specific activity 12.3 Ci/mmol; radiochemical purity >97%) was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). Unlabeled GPFX and LVFX were supplied by Otsuka Pharmaceutical Co. Ltd. (Tokyo, Japan). Geneticin solution, trypsin-EDTA solution, d-glucose, 1-chloro-2,4-dinitrobenzene (CDNB), 1-fluoro-2,4-dinitrobenzene, and the reduced form glutathione were obtained from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum (FBS) was purchased from Invitrogen (Tokyo, Japan). Scintillation cocktail (ACS-II) for the determination of radioactivity was obtained from Amersham Biosciences UK, Ltd. Hionic Fluor and Ultima Fluor AP were obtained from PerkinElmer Life and Analytical Sciences. Other chemicals of special grade or of analytical grade were obtained from Wako Pure Chemicals (Osaka, Japan) or Katayama Chemical Co. Ltd. (Osaka, Japan). 2,4-Dinitrophenyl-S-glutathione (DNP-SG) was synthesized for the HPLC quantification using the method reported by Hinchman et al. (1991).
Animals. Male FVB wild-type mice [mdr1a/1b(+/+)], mdr1a gene-deficient mice [mdr1a(–/–)], and mdr1a/1b gene-deficient mice [mdr1a/1b(–/–)] were purchased from Taconic Farms (Germantown, NY) and used at 7 to 14 weeks of age. Male FVB wild-type mice [mrp1(+/+)] and mrp1 gene-deficient mice [mrp1(–/–)] were purchased from Taconic Farms and used at 7 to 11 weeks of age.
Tissue Distribution Studies. [14C]GPFX was administered to mice through a tail vein at a dose of 5 mg/2 ml/kg. [14C]LVFX (5 mg/2 ml/kg) was also administered to mdr1a/1b(–/–) or mrp1(–/–) mice, respectively, in a control experiment. Blood samples were then collected from the tail vein at designated times with a heparinized syringe, and plasma was prepared by centrifuging the blood samples (1800g; 10 min). To determine the hematocrit value, blood was centrifuged at 12,000 rpm. The mice were killed by severing the inferior vena cava and aorta, and tissues were excised immediately. Cerebrospinal fluid (CSF) was collected at 6 h after the intravenous injection. The tissues were stored at –20°C until assay. The lung, liver, stomach, small intestine, large intestine, duodenum, jejunum, and ileum were homogenized with 1 ml of distilled water. Portions of the homogenate and other tissues were weighed and dissolved in 1 ml of Biomerit tissue-solubilizing solution (Nacalai Tesque, Kyoto, Japan) at 50°C for 30 min. The tissue lysate was neutralized with 1 ml of pH-adjusting solution (Nakalai Tesque). Then, after the addition of scintillation cocktail (Hionic-Fluor; PerkinElmer Life and Analytical Sciences), the radioactivity was determined using a liquid scintillation counter (LSC-1050; Aloka, Tokyo, Japan). The counting efficiency was corrected by the channels ratio method using an external standard. The radiochemical profiles of plasma and tissues were analyzed by gradient reversed phase HPLC with an automated gradient controller equipped with a type 510 pump (Waters, Milford, MA) connected to a flow scintillation analyzer type 500TR (PerkinElmer Life and Analytical Sciences). Two or 3 volumes of acetonitrile were added to the tissue homogenates, plasma, and blood cells to precipitate the protein. After centrifugation, the supernatant was evaporated to dryness. The residue was dissolved in 100 μl of methanol, and an aliquot was subjected to HPLC using a TSKgel ODS-80Ts column (4.6 mm i.d. × 15 cm; Tosoh, Tokyo, Japan). The mobile phase consisted of acetonitrile, water, and acetic acid [5:95:1 (A solution) and 45:55:1, (B solution), (v/v/v)]. Separation was achieved by running a linear gradient from 15 to 55% B solution for 20 min at a flow rate of 1 ml/min. The GPFX concentration in CSF was determined by HPLC using a mobile phase consisting of acetonitrile and 20 mM Na2SO4/0.1% H3PO4 [27:73 (v/v)].
Urinary and Biliary Excretions of GPFX. The bile duct and penis were connected to a polyethylene catheter under ether anesthesia. Bile and urine were collected over a period of up to 48 h after drug administration. The radioactivity in bile and urine was counted and unchanged GPFX and its metabolites were determined, after centrifugation, using the HPLC system described above.
Distribution of [14C]GPFX to the Pulmonary Airspace. The distribution of GPFX to lung compartments such as alveolar macrophages and epithelial lining fluid (ELF) was determined by a slight modification of the method described by Alblas and Furth (1979). Briefly, [14C]GPFX and [3H]sucrose were administered to mrp1(–/–), mrp1(+/+) mice, mdr1a/1b(–/–), and mdr1a/1b(+/+) mice through a tail vein at a dose of 5 mg/kg. Blood samples were collected from the inferior vena cava at designated times. The mice were killed by severing the inferior vena cava and aorta, and then the trachea was cannulated with a short length of intravenous catheter tubing (0.80 mm i.d.; Terumo, Tokyo, Japan). One milliliter of ice-cold Mg2+-free phosphate-buffered saline (PBS) containing 1 mM EDTA was gently instilled into the lung. After gently rubbing the lung, the fluid was slowly withdrawn. The alveolar lavage was repeated five times. The lavage fluids were pooled and centrifuged at 400g for 10 min to obtain macrophage cells. The cell pellet was washed twice with Mg2+-free PBS containing 1 mM EDTA and then the cells were resuspended in the same buffer. Viability counts were performed by trypan blue exclusion. After the lavage, the lung was excised immediately and homogenized with 1 ml of Mg2+-free PBS. To measure the radioactivity in the homogenate, cell suspension, and lavage fluid, each sample was solubilized with 1 ml of tissue-solubilizing solution. The lysate was neutralized with 1 ml of pH-adjusting solution and then scintillation cocktail was added to the vial. The unchanged GPFX in the lung compartments was determined by HPLC connected to a flow scintillation analyzer as described above. Protein concentrations in the lung compartments were determined by the method of Lowry et al. (1951), using a protein assay kit (Bio-Rad, Hercules, CA) with bovine serum albumin as a standard.
Determination of Blood Flow Rate. Under ether anesthesia, the abdomens were incised, and the blood flow rate at the portal vein was measured using an electromagnetic flowmeter (Transonic Systems Inc., Ithaca, NY).
Plasma Protein Binding of [14C]GPFX in Vitro. Plasma protein binding of GPFX was determined by ultrafiltration. Initially, a filtration tube (Centrifree YM30; Millipore, Billerica, MA), in which blank plasma had been placed, were centrifuged to obtain the filtrate (1800g; 20 min). Another filtration tube was filled with the filtrate obtained and then centrifuged. This pretreatment of the filtration tube reduced any nonspecific adsorption of GPFX to the membrane filter. Plasma samples with [14C]GPFX (final concentration 0.02 and 2 μg/ml) were placed in the pretreated filtration tubes, and then incubated at 37°C for 5 min. The plasma was centrifuged (1800g; 5 min) to yield a filtrate containing the unbound compound. The radioactivity in the plasma and filtrate was counted to determine the total and free concentration, respectively.
Efflux of GPFX and DNP-SG in LLC-MRP1 Cells. LLC-PK1 and LLC-MRP1 were kindly donated by Dr. P. Borst (The Netherlands Cancer Institute, Amsterdam, The Netherlands). LLC-PK1 and LLC-MRP1 were grown in M199 medium (Sigma-Aldrich) supplemented with 10% (v/v) fetal bovine serum and incubated at 37°C under 5% CO2 and 100% humidity. LLC-PK1 and LLC-MRP1 were seeded at a density of 1.5 × 106 cells/well on porous polyethylene terephthalate membrane filters (3-μm pore size, 4.2-cm2 filter area, Falcon culture insert; BD Biosciences, Franklin Lakes, NJ), and the cells were then used for the efflux studies on the third day after seeding. Efflux of DNP-SG was performed at room temperature (23°C). CDNB was applied to both the apical and basal compartments. At designated times after application, 500-μl aliquots of medium from both the apical and basal sides were taken to determine the DNP-SG concentration. At the last sampling point, each cell monolayer was washed twice with ice-cold PBS and dried in an air current. The cells were then lysed by the addition of 10% (v/v) perchloric acid to determine DNP-SG. After centrifugation, the supernatant was neutralized by adding 0.4 volumes of 3 M KOH/0.5 M 3-(N-morpholino) propanesulfonic acid and centrifuged again. The supernatant and the medium were extracted with an equal volume of ethyl acetate. After centrifugation, the organic layer was evaporated to dryness. The residue was dissolved in 100 μl of methanol, and an aliquot was subjected to HPLC with a TSKgel ODS-80Ts column (4.6 mm i.d. × 15 cm). Separation was achieved by a mobile phase of acetonitrile and 0.1% trifluoroacetic acid [1:3 (v/v)] at a flow rate of 1 ml/min. DNP-SG was detected at 365 nm and quantified from the peak area using a standard curve.
To determine the efflux of GPFX, the apical and basal medium were first replaced with Hanks' balanced salt solution/5% FBS with and without 5 to 6.25 μM [14C]GPFX, respectively. After preincubation for 1 h at 23°C, the cells were washed twice with ice-cold PBS, and sequential efflux was initiated by replacing both sides with Hanks' balanced salt solution/5% FBS. At designated times, 500-μl aliquots of the medium were taken from both sides to determine GPFX. At the last sampling point, the cell monolayer was washed twice with ice-cold PBS and dried up an air current. Then, 0.1 N NaOH was added to dissolve the cells, and the cell lysate obtained was neutralized with 1 N HCl. Determination of the unchanged GPFX was performed by gradient reversed phase HPLC with a flow scintillation analyzer. The mobile phase consisted of acetonitrile, water, and acetic acid [5:95:1 (A) and 45:55:1 (B), (v/v/v)]. Separation of GPFX in the sample was achieved by running a linear gradient of B from 15 to 55% for 20 min at a flow rate of 1 ml/min using a TSKgel ODS-80Ts column.
Data Analysis. To obtain pharmacokinetic parameters, the plasma concentration data for unchanged GPFX were fitted to a two-compartment model using a pharmacokinetic software package (WinNonlin, version 3.01; Pharsight, Mountain View, CA). The total clearance (CLtot,p) was obtained by dividing dose by area under the plasma concentration curve (AUC). The urinary and biliary clearance (CLbil;e,p and CLurine,p, respectively) was the amount of unchanged GPFX excreted into the urine and bile, respectively, for 48 h during which period more than 90% of dose was recovered in both urine and bile, divided by AUC. The metabolic clearance (CLmet,p) was the amount of metabolites excreted both into the urine and bile divided by AUC. The nonhepatic and nonrenal clearance (CLother,p) was obtained by the subtraction of CLurine,p, CLbil;e,p, and CLmet,p from CLtot,p. Differences between data in these mice strains were statistically analyzed using SAS, version 8.1. A P value less than 0.05 was regarded as significant difference (analysis of variance).
Results
Plasma Concentration Profile of Unchanged [14C]GPFX in Vivo. After intravenous administration of [14C]GPFX to mrp1(–/–), mdr1a(–/–), and mdr1a/1b(–/–), unchanged GPFX in plasma was determined and compared with that in control mice. GPFX was biexponentially eliminated from plasma (Fig. 1). The plasma concentration profile in mrp1(–/–) was very similar to that in mrp1(+/+), whereas that in mdr1a(–/–) and mdr1a/1b(–/–) was higher than that in mdr1a/1b(+/+) (Fig. 1).

Time profiles of the plasma concentration of GPFX after intravenous administration in mrp1(–/–) (A, open circle), mrp1(+/+) (A, closed circle), mdr1a(–/–) (B, open square), mdr1a/1b(–/–) (B, open triangle), and mdr1a/1b(+/+) mice (B, closed circle). Each plot and vertical bar represent mean ± S.E. of three to six mice.
Tissue Distributions of [14C]GPFX inmrp1(–/–)andmrp1(+/+)Mice. The tissue concentrations of unchanged GPFX after a single intravenous administration of [14C]GPFX to mrp1(–/–) and mrp1(+/+) were examined. In preliminary studies, the concentrations of radioactivity in each tissue had almost reached equilibrium with that in plasma within 4 h of intravenous administration, and therefore, the tissue-to-plasma concentration ratio during the β-phase (Kp,β) was determined as the mean value of Kp at 4 and 6 h (Table 1). In mrp1(+/+), a higher Kp,β was observed in lung, liver, kidney, pancreas, small intestine, and large intestine, whereas the Kp,β value in brain and fat was relatively small (Table 1). The Kp,β in trachea, heart, kidney, spleen, and brown fat of mrp1(–/–) was significantly higher than that in mrp1(+/+), whereas that in other tissues was fairly similar for each strain (Table 1).
Tissue-to-plasma concentration ratio in β-phase (Kp,β) of [14C]GPFX in mrp1(–/–) and mrp1(+/+) mice
Tissue Distributions and Excretion of GPFX inmdr1a(–/–),mdr1a/1b(–/–), andmdr1a/1b(+/+)Mice. The Kp,β values in the brain of mdr1a(–/–) and mdr1a/1b(–/–) were 2.4 times higher than that in mdr1a/1b(+/+) (Table 2). This result is compatible with a previous report (Tamai et al., 2000; Yamaguchi et al., 2002) and supports the hypothesis that Mdr1a might contribute to the limited distribution of GPFX to the brain. In other tissues except the large intestine, the Kp,β values in mdr1a(–/–) and mdr1a/1b(–/–) were comparable with that in mdr1a/1b(+/+) (Table 2). The distribution of LVFX, another new quinolone analog, was also examined in these mouse strains to examine whether the observed difference in tissue distribution is specific for GPFX. The Kp,β of [14C]LVFX in all tissues was markedly lower than that of [14C]GPFX (Table 2). No significant difference among mdr1a(–/–), mdr1a/1b(–/–) and mdr1a/1b(+/+) was observed for the Kp,β of LVFX in the brain (Table 2), confirming the observation by Tamai et al. (2000). In addition, no difference among these strains was found for all the other tissues examined (Table 2), although both GPFX and LVFX have also been reported to be substrates of MDR1 (Naruhashi et al., 2001). Thus, limited distribution to the brain by Mdr1 is observed only for GPFX, and distribution of both GPFX and LVFX to other tissues is not affected by Mdr1.
Tissue-to-plasma concentration ratio in β-phase (Kp,β) of GPFX and LVFX in mdr1a(–/–), mdr1a/1b(–/–), and mdr1a/1b(+/+) mice
The CLtot,p of GPFX in mdr1a(–/–) and mdr1a/1b(–/–) was reduced by about 30 and 20%, respectively, compared with that in mdr1a/1b(+/+) (Table 3). The CLurine,p of GPFX in mdr1a(–/–) was significantly smaller than that in mdr1a/1b(+/+) (Table 3). The CLurine,p of GPFX in mdr1a/1b(–/–), compared with that in mdr1a/1b(+/+), was reduced by about 30%, although this difference was not statistically significant (Table 3). The CLother,p of GPFX was also lower in mdr1a(–/–) and mdr1a/1b(–/–) compared with the control mice (Table 3). Because the hepatic clearance (sum of CLbile,p and CLmet,p) was not very different from the hepatic plasma flow rate, we also measured the blood flow rate in the portal vein of each strain and found that the hepatic blood flow in mdr1a /1b(–/–) was slightly lower than that in mdr1a/1b(+/+) (Table 3). The plasma protein binding of [14C]GPFX at 2 μg/ml was measured in each strain (Table 3). Although there was a statistically significant difference between mdr1a/1b(+/+) and mdr1a/1b(–/–) at 2 μg/ml, this difference was small (Table 3). In addition, the plasma protein binding of GPFX was low, at most 35 to 40% (Table 3).
Comparison of GPFX clearance, plasma protein binding, and portal vein blood flow rate among mdr1a(–/–), mdr1a/1b(–/–), and mdr1a/1b(+/+)
Distribution of [14C]GPFX to Pulmonary Airspace. To characterize the distribution mechanism of GPFX to its pharmacological target site, unchanged GPFX was determined in lung parenchyma cells and pulmonary airspaces, including ELF and macrophages after intravenous administration (Fig. 2). At 4 to 8 h after administration to both mdr1a/1b(–/–) and mdr1a/1b(+/+), the radioactivity in the pulmonary airspaces, including lavage fluid, accounted for 11 to 13% of the total radioactivity distributed to the whole lung. No difference between mdr1a/1b(–/–) and mdr1a/1b(+/+) was observed in the distribution of GPFX to the pulmonary airspaces. The alveolar macrophage-to-plasma concentration ratio of unchanged GPFX in mdr1a/1b(–/–) was significantly lower than that in mdr1a/1b(+/+), although this difference was not very marked (Fig. 2). The ELF-to-plasma concentration ratio in mdr1a/1b(–/–) was not statistically different, and almost identical to that in mdr1a/1b(+/+) (Fig. 2). The distribution of GPFX to the pulmonary airspace was also examined in mrp1(–/–) at 4 h after intravenous administration. No statistically significant difference was observed in the distribution ratio to the pulmonary airspaces, alveolar macrophages, and ELF between mrp1(–/–) and mrp1(+/+) (Fig. 2A).

Distribution of GPFX to pulmonary airspaces in mdr1a/1b(–/–), mdr1a/1b(+/+), mrp1(–/–), and mrp1(+/+) mice, after a single intravenous administration of [14C]GPFX. After administration, the amount of GPFX in alveolar macrophages of mdr1a/1b(–/–) (A, closed circle), mdr1a/1b(+/+) (A, open circle), mrp1(–/–) (B, closed diamond), and mrp1(+/+) (B, open diamond), and ELF of mdr1a/1b(–/–) (A, closed triangle), mdr1a/1b(+/+) (A, open triangle) mrp1(–/–) (B, closed square), and mrp1(+/+) (B, open square) was determined and normalized by the plasma concentration. Each point and vertical bar represent the mean ± S.E. of three to four mice. *, P < 0.05 significantly different from mdr1a/1b(+/+).
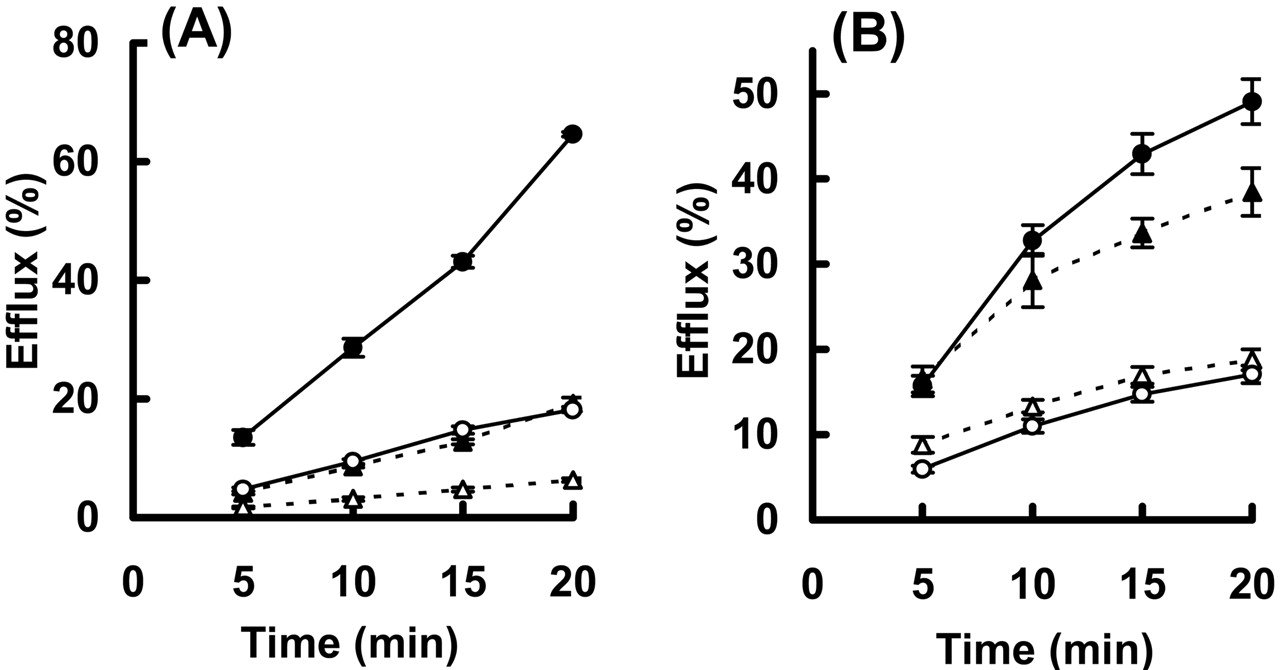
Efflux of GPFX and DNP-SG by LLC-MRP1 Cells. Although GPFX is reported to be a substrate of P-gp (Naruhashi et al., 2001), no information is available on whether GPFX is transported by MRP1. To support the hypothesis that Mrp1 is involved in GPFX distribution to several tissues, resulting in the increase in the Kp,β of the corresponding tissues in mrp1(–/–), a transport study using LLC-MRP1 was performed to examine whether GPFX is a substrate of MRP1. First, to exclude the possibility of depolarization in LLC-PK1 cells during the culture period, efflux of DNP-SG, a typical substrate of MRP1, to the basal and apical sides was examined in the present study (Fig. 3A). The efflux of DNP-SG to the basal side in LLC-MRP1 during the incubation with CNDB was 2.3-fold higher than that in LLC-PK1 (Fig. 3A). On the other hand, the efflux to the apical side in LLC-MRP1 was close to that in LLC-PK1 (Fig. 3A). The total amount of DNP-SG recovered both in the cells and the medium after the 20-min efflux study, representing the conjugating activity of CDNB to DNP-SG, was 1.4-fold higher in LLC-PK1 than that in LLC-MRP1 (Fig. 3, legend). The efflux of DNP-SG specifically to the basal side is similar to that obtained in LLC-PK1 cells at 3 days after seeding as described by Evers et al. (1996). Then, the efflux of GPFX in LLC-MRP1 cells was examined (Fig. 3B). The intracellular accumulation of unchanged GPFX after 1-h loading in LLC-MRP1 were almost equal to that in LLC-PK1 (Fig. 3, legend). The efflux of GPFX to the basal side in LLC-MRP1 was higher than that in LLC-PK1, and the efflux to the apical side was almost identical in between LLC-MRP1 and LLC-PK1 (Fig. 3B). Considering that MRP1 is expressed in the basal membranes of LLC-MRP1 cells (Evers et al., 1996), this result suggests that GPFX is a substrate of MRP1. However, the difference in GPFX efflux to the basal side between LLC-MRP1 and LLC-PK1 was not very marked, compared with that for DNP-SG (Fig. 3). This is probably because of the high background in the membrane penetration of GPFX: efflux of GPFX observed in LLC-PK1 cells was still high, the recovery (sum of the amount effluxed to both basal and apical sides) after 20 min being 57%, which is close to that in LLC-MRP1 cells (66%; Fig. 3B). On the other hand, the recovery of DNP-SG in LLC-PK1 cells was only 26%, which was much lower than that found in LLC-MRP1 cells (83%; Fig. 3A). GPFX has been developed to exhibit a high degree of tissue distribution (Akiyama et al., 1995a,b; Nakajima et al., 2000; Suzuki et al., 2002), and therefore its membrane permeability could be high. This may hinder the detection of MRP1-mediated transport in LLC-PK1 cell lines.

Efflux of DNP-SG (A) and GPFX (B) in LLC-PK1 and LLC-MRP1 monolayers. A, CDNB was applied to both basal and apical sides, and the amount of DNP-SG recovered from each side was determined at room temperature. The efflux was normalized by the amounts of DNP-SG recovered from both medium and intracellular space after the efflux study for 20 min (4.69 ± 0.18 and 3.24 ± 0.14 nmol/well in LLC-PK1 and LLC-MRP1, respectively). B, GPFX applied to the apical side and preincubated for 1 h at room temperature, followed by replacement with fresh medium and determination of the efflux of GPFX on each side. The efflux was normalized by the intracellular accumulation of [14C]GPFX after the 1-h preincubation period (405 ± 21 and 379 ± 19 pmol/well in LLC-PK1 and LLC-MRP1, respectively). Open and closed symbols represent apical and basal medium in the LLC-PK1 (triangles) and LLC-MRP1 (circles). Each plot and bar represent the mean ± S.E. of six samples in the two experiments.
Discussion
Because the antibacterial activity of an antibiotic depends on its exposure to the pharmacological target sites, it is important to characterize the molecular mechanism(s) that affects the tissue distribution of NQs. GPFX is highly distributed to many tissues, compared with other NQs, which is a pharmacokinetic advantage in terms of its antibacterial activity. Nevertheless, the exact mechanism for the delivery of GPFX has not yet been fully clarified. In the present, study the possible involvement of two ATP-binding cassette transporters, Mrp1 and Mdr1, in tissue distribution of GPFX was examined, and an increase in Kp,β was observed in the trachea, heart, kidney, spleen, and brown fat of mrp1(–/–), whereas a similar increase was observed only in the brain of mdr1a(–/–) and mdr1a/1b(–/–) (Tables 1 and 2). Considering that GPFX is a substrate of MDR1 (Naruhashi et al., 2001) and MRP1 (Fig. 3), this result suggests that both transporters are involved in the efflux of GPFX in these tissues. Although MDR1 has been suggested to play an important role in the tissue distribution of various types of therapeutic agents, especially to the brain (Schinkel, 1998; Tanigawara, 2000), information on the possible involvement of MRP1 in the tissue distribution of therapeutic compounds is limited. The present findings thus suggest MRP1 makes a significant contribution to the distribution of its substrates to several tissues.
The plasma concentration profile of GPFX after its intravenous injection was slightly higher in mdr1a(–/–) and mdr1a/1b(–/–) than in wild-type mice (Fig. 1). Therefore, Mdr1a seems to be partially involved in the overall elimination of GPFX from the body. The lower CLurine,p in those knockout mice suggests that urinary excretion is at least partly mediated by Mdr1. However, the reduction in CLurine,p was not enough to explain that in the CLtot,p of mdr1a(–/–) and mdr1a/1b(–/–) (Table 3). A reduction in CLother,p was also observed in mdr1a(–/–) and mdr1a/1b(–/–), suggesting that the elimination of GPFX other than by urinary excretion, biliary excretion, and metabolism is reduced in those knockout mice. Yamaguchi et al. (2000, 2001) reported a similar reduction in the intestinal secretion of GPFX in mdr1a(–/–) and mdr1a/1b(–/–). Naruhashi et al. (2001, 2002) have proposed that GPFX is secreted in the small intestine and Caco-2 monolayers via Mdr1. Therefore, the reduction in CLother,p in the mice used in present study might be due to a deficiency of P-gp transport in the gut. The plasma protein binding of GPFX was relatively similar for each strain, whereas hepatic blood flow in mdr1a(–/–) and mdr1a /1b(–/–) was 75 to 85% that in mdr1a/1b(+/+) mice (Table 2). Considering that the hepatic clearance (the sum of CLbile,p and CLmet,p) was close to the blood flow rate, the change in portal vein blood flow rate exhibited by those knockout mice might account for the difference in metabolic clearance among the strains. On the other hand, the plasma concentration of GPFX in mrp1(–/–) was similar to that in mrp1(+/+) (Fig. 1). Therefore, the contribution of Mrp1 to the excretion of GPFX in mice might be minor. We have previously reported that the biliary excretion of GPFX is partially mediated by Mrp2. Therefore, the contribution of Mrp1 to the excretion process of GPFX in mice might be minor, although the biliary excretion of GPFX is partially mediated by Mrp2 (Sasabe et al., 1998).
Wijnholds et al. (2000) demonstrated a significant increase in radioactivity in mrp1(–/–) after the administration of [3H]etoposide, an MRP1 substrate, in heart and brown adipose tissue, compared with mrp1(+/+). This finding could be compatible with the possible involvement of Mrp1 in the distribution of GPFX to these two tissues (Table 1). However, we observed no difference in the distribution of estradiol 17β-glucuronide, another typical substrate of MRP1, to these tissues as well as kidney and spleen between the two strains when we determined the parent compound by HPLC at 10 min after its injection (data not shown). Thus, the involvement of transporters in tissue distribution is highly substrate-dependent, and a number of other transporters should also be considered to appreciate the whole picture as far as the distribution mechanism of GPFX is concerned. Wijnholds et al. (2000) have also reported a higher etoposide concentration in the CSF of triple-knockout mice of mrp1/mdr1a/mdr1b(–/–) than in that of double-knockout mice mdr1a/mdr1b(–/–), suggesting that Mrp1 is involved in the efflux of etoposide into plasma in the choroid plexus. On the other hand, we observed no significant difference in the Kp,β of GPFX in the CSF between mrp1(–/–) and mrp1(+/+). Although the exact reason for such a discrepancy is not clear, the similarity in the Kp,β of GPFX between mrp1(–/–) and mrp1(+/+) can be explained if transporters other than Mrp1 are mainly involved in the efflux of GPFX from the CSF, even if Mrp1 also acts as an efflux transporter in the choroid plexus.
Both Mdr1 and Mrp1 are reported to be expressed in lung (Croop et al., 1989; Schinkel et al., 1994, 1995; Stride et al., 1996). To understand the possible functions of these transporters for the disposition of GPFX in its target organs, GPFX concentrations in alveolar macrophages and ELF were compared among mdr1a/1b(–/–), mrp1(–/–), and wild-type mice. The distribution to alveolar macrophages, the lung parenchyma cells, and ELF was relatively similar for each strain (Fig. 2). A significant difference was observed in the alveolar macrophage-to-plasma concentration ratio between mdr1a/1b(–/–) and mdr1a/1b(+/+) (Fig. 2B). Considering that this difference was significant only at 4 h after injection (Fig. 2B), further studies are needed to prove any involvement of mdr1 in GPFX distribution. Thus, although GPFX is a substrate of MDR1 and MRP1, these transporters do not play a major role in the higher distribution of GPFX to the lung. On the other hand, the possible contribution of MDR1 and MRP1 to GPFX distribution to the brain and heart, respectively, should also be noted because NQs exhibit an unexpected side effect in these organs. Various drugs can sometimes cause cardiac adverse effects, for example, prolongation of the QT intervals, which is commonly considered to be associated with the distribution to myocardial tissues. The present findings should promote the further investigation of the involvement of the Mrp family in drug distribution to the heart and its relevance to this potential side effect.
In conclusion, GPFX is a substrate of MRP1 and its distribution several tissues, including heart, trachea, kidney, and spleen, is restricted partially by the efflux by GPFX, whereas MDR1 is involved in the efflux of GPFX from the brain and excretion into the urine. Thus, multiple ATP-binding cassette transporters, MDR1, MRP1, and MRP2, are involved in the distribution of GPFX, thereby restricting its exposure both to pharmacological and toxicological targets such as the bile, brain, and heart.
Acknowledgments
We thank Dr. P. Borst for the donation of LLC-MDR1 and LLC-MRP1 and for suggestions about supply and use of gene-deficient mice [mdr1a(–/–), mdr1a/1b(–/–), and mrp1(–/–)]. We also thank Dr. A. Tsuji (Kanazawa University) and Dr. I. Tamai (Tokyo University of Sciences) for many helpful suggestions.
Footnotes
-
DOI: 10.1124/jpet.104.065201.
-
ABBREVIATIONS: NQ, new quinolone; GPFX, grepafloxacin; P-gp, P-glycoprotein; Mrp, multidrug resistance-associated protein; Mdr, multidrug resistance; LVFX, levofloxacin; CDNB, 1-chloro-2,4-dinitrobenzene; FBS, fetal bovine serum; DNP-SG, 2,4-dinitrophenyl-S-glutathione; HPLC, high-performance liquid chromatography; CSF, cerebrospinal fluid; ELF, epithelial lining fluid; PBS, phosphate-buffered saline; AUC, area under the curve.
- Received January 14, 2004.
- Accepted April 30, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}