


Abstract
6β-Naltrexol is the major metabolite of the opioid receptor antagonist, naltrexone, in humans. However, there are no functional studies of 6β-naltrexol in primates. The aim of this study was to compare the in vitro and in vivo potencies of naltrexone and 6β-naltrexol in rhesus monkeys. Affinity and potency were determined using radioligand displacement and stimulation of 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding in monkey brain membranes. In vivo apparent pA2 analysis was applied to compare the μ-opioid receptor (MOR) antagonist potency of both compounds in nondependent monkeys. In addition, the potencies of both compounds were determined in precipitating withdrawal manifested by increased respiratory parameters in acute morphine-dependent monkeys. In vitro assays revealed that naltrexone displayed 2-fold higher affinity and potency than 6β-naltrexol for the MOR binding site and for MOR agonist-stimulated [35S]GTPγS binding, respectively. 6β-Naltrexol (0.32-3.2 mg/kg) dose-dependently produced parallel rightward shifts of the dose-response curve of alfentanil-induced antinociception. Nevertheless, the apparent pA2 value of 6β-naltrexol (6.5) was 100-fold less potent than that of naltrexone (8.5) determined previously. 6β-Naltrexol was also less potent than naltrexone in antagonizing other MOR-mediated effects including respiratory depression and itch/scratching. Naltrexone (0.0032-0.032 mg/kg) and 6β-naltrexol (0.32-3.2 mg/kg) retained the same potency difference in precipitating withdrawal to a similar degree. Furthermore, 6β-naltrexol failed to block naltrexone-precipitated withdrawal in morphine-dependent monkeys. These results indicate that naltrexone and 6β-naltrexol display similar pharmacological actions with a large in vivo potency difference in monkeys such that 6β-naltrexol may play a minimal role in the therapeutic or antagonist effects of naltrexone in primates.
Opioid receptor antagonists have been used in the treatment of opioid addiction and overdose (Gonzalez et al., 2004). In addition, opioid antagonists, such as naltrexone, can be used to treat alcoholism and pruritus associated with liver diseases or opioid medications (Anton, 2001; Jones et al., 2002). Although opioid antagonists have wide therapeutic applications, withdrawal symptoms elicited by these antagonists have limited their pharmacotherapy in opioid-treated or -dependent subjects (O'Connor and Kosten, 1998; Gonzalez et al., 2004). Research and development of compounds that produce therapeutic effects but elicit reduced withdrawal symptoms have become important in the pharmacotherapy of opioid antagonists.
6β-Naltrexol is the major metabolite of naltrexone in humans, but not in rodents (Misra et al., 1976; Meyer et al., 1984). The plasma level of 6β-naltrexol is approximately 2- to 3-fold higher than that of naltrexone in humans (Meyer et al., 1984; Ferrari et al., 1998). However, there is no information on the characteristics of 6β-naltrexol in vivo in primates. More importantly, recent studies have found that pharmacological actions of 6β-naltrexol are different from those of naltrexone, and antagonist doses of 6β-naltrexol elicit less behavioral withdrawal signs in morphine-dependent mice (Wang et al., 2004; Raehal et al., 2005). These findings seem to indicate that 6β-naltrexol is a putative neutral antagonist in mice, and it may have greater therapeutic potential in treating opioid addiction and overdose (Raehal et al., 2005). Therefore, it is of interest to study the pharmacological actions of 6β-naltrexol in a primate species.
Although early studies of opioid dependence in monkeys relied on subjective observation of behaviors, monkeys during morphine withdrawal display several physiological responses closely resembling those in humans (Seevers, 1936; Holtzman and Villarreal, 1969; Heishman et al., 1989). For example, both morphine abstinence and antagonist administration have been reported to produce hyperpnea in morphine-dependent human subjects (Martin et al., 1968; Heishman et al., 1989). Increased respiratory frequency can be detected and be used as a sensitive, quantitative measure of precipitated opioid withdrawal in acute and chronic morphine-dependent monkeys (Paronis and Woods, 1997; Kishioka et al., 2000). Opioid antagonists have similar in vivo potency in precipitating withdrawal across different monkey behavioral assays (Valentino et al., 1983; France and Woods, 1989; Paronis and Woods, 1997). More importantly, opioid antagonists such as naltrexone retain the same potency in both producing μ-opioid receptor (MOR) antagonist effects and precipitating withdrawal in monkeys (France et al., 1990; Ko et al., 1998). These findings suggest that binding to opioid receptors is the mechanism by which antagonists precipitate withdrawal in opioid-dependent subjects (France et al., 1990). It is valuable to compare the potency of 6β-naltrexol under both experimental conditions in monkeys.
Therefore, the aim of this study was to characterize the pharmacological actions of 6β-naltrexol in a primate species. In the current study, the MOR binding affinities of both naltrexone and 6β-naltrexol were determined in the cortical and thalamic membranes of monkeys. In vitro antagonist potencies of both compounds were compared using a MOR agonist-induced [35S]GTPγS binding in the monkey thalamic membranes (Ko et al., 2003). Apparent pA2 analysis was used to determine the in vivo antagonist potency of 6β-naltrexol and to be compared with the pA2 value of naltrexone under similar conditions (Ko et al., 1998). Antagonist effects of 6β-naltrexol were also compared with naltrexone in different MOR-mediated behavioral endpoints including itch/scratching and respiratory depression (Ko et al., 2002, 2004). Furthermore, in vivo potencies of naltrexone and 6β-naltrexol in precipitating withdrawal were compared by measuring respiratory parameters in morphine-dependent monkeys (Paronis and Woods, 1997; Kishioka et al., 2000).
Materials and Methods
Subjects
Eighteen adult intact male and female rhesus monkeys (Macaca mulatta) with body weights ranging between 6.8 and 11.8 kg were used. They were housed individually with free access to water and were fed approximately 25 to 30 biscuits (Purina Monkey Chow; Purina, St. Louis, MO) and fresh fruit daily. No monkey had exposure to opioids for 1 month before the present study. The monkeys were housed in facilities accredited by the American Association for the Accreditation of Laboratory Animal Care. The studies were conducted in accordance with the University Committee on the Use and Care of Animals in the University of Michigan and the Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, DC, revised 1996).
Procedures
In Vitro Binding Assays: Ligand Binding Assay. Both cortical and thalamic membranes of monkeys were prepared according to the procedure described in a previous study (Ko et al., 2003). Cortical membranes (400 μg of protein) of monkeys were incubated for 75 min at 25°C with 1 nM [3H]DAMGO and varying concentrations of unlabeled 6β-naltrexol or naltrexone (0.03-100 nM) in 50 mM Tris buffer, pH 7.4. Thalamic membranes (100 μg of protein) were incubated with 0.2 nM [3H]diprenorphine and unlabeled 6β-naltrexol or naltrexone in 50 mM Tris buffer, pH 7.4, or in 50 mM Tris, pH 7.4, containing 100 mM NaCl and 10 μM GTPγS for studies of low-agonist affinity binding. Nonspecific binding was defined with 10 μM naloxone. Samples were rapidly filtered through glass fiber filters (32; Whatman Schleicher and Schuell, Keene, NH) mounted in a Brandel cell harvester (Brandel Inc., Gaithersburg, MD) and rinsed three times with Tris-HCl buffer. Filters were placed in polypropylene scintillation vials, and radioactivity retained on the filters was measured by liquid scintillation counting in 4 ml of EcoLume. Each experiment was carried out three times in duplicate.
In Vitro Binding Assays: [35S]GTPγS Binding Assay. Thalamic membranes (30 μg of protein) of monkeys were incubated for 60 min at 25°C in 50 mM Tris, pH 7.4, 1 mM EDTA, 5 mM MgCl2, 100 mM NaCl, 2.4 mM dithiothreitol (freshly prepared), 1 mU of adenosine deaminase, 100 μM GDP, 0.1 nM [35S]GTPγS, and varying concentrations of DAMGO (100 nM-100 μM) in the presence and absence of 20 nM 6β-naltrexol or 10 nM naltrexone. The reaction was terminated by rapidly filtering samples through glass microfiber filter mats, type GF/C (Whatman, Clifton, NJ) mounted in a Brandel harvester and rinsing three times with 50 mM Tris, pH 7.4, 5 mM MgCl2, and 100 mM NaCl. Filter mats were dried, and 0.1 ml of EcoLume was added to each sample. Filter mats were heat sealed in polyethylene bags, and radioactivity retained on the filters was measured by liquid scintillation counting in Wallac 1450 MicroBeta Liquid Scintillation and Luminescence Counter (PerkinElmer Life and Analytical Sciences, Boston, MA). Each experiment was carried out three times in duplicate.
Data Analysis. IC50 values were determined using GraphPad Prism version 4.0 (GraphPad Software Inc., San Diego, CA) and converted to Ki values according to Cheng and Prusoff (1973). The Kd values for [3H]DAMGO (0.7 nM) and [3H]diprenorphine (0.2 nM) are from Ko et al. (2003). [35S]GTPγS binding data were fit to a sigmoidal curve using GraphPad Prism to determine the EC50 value. Ke values for antagonist inhibition were calculated by the following equation: Ke = [nanomolar antagonist]/(dose ratio - 1), where dose ratio is the ratio of the EC50 for DAMGO in the presence and absence of the antagonist.
Behavioral Assays
Antinociception. The warm water (50°C) tail-withdrawal assay was used to evaluate thermal antinociceptive effects of the test compound. In brief, monkeys were seated in primate restraint chairs, and the lower part of their shaved tails (approximately 15 cm) were immersed in a thermal flask containing water maintained at 42, 46, or 50°C. Tail-withdrawal latencies were measured using a computerized timer by an experimenter who was blinded to experimental conditions. In each test session, monkeys were tested one to two times at three temperatures in a random order. If the monkeys did not remove their tails within 20 s (cutoff), the flask was removed, and a maximum time of 20 s was recorded. Test sessions began with control determinations at each temperature. Then, a MOR-selective agonist, alfentanil, was administered by either a cumulative or single dosing procedure. Subsequent tail-withdrawal latencies were determined starting 15 min after each injection.
Respiratory Function. The monkey was seated in a primate restraint chair, enclosed within a sound-attenuating chamber. A rectangular helmet (13.5 × 17.0 × 13.5 cm) was placed over the head of the monkey and sealed around its neck by two closely fitting latex shields. Gas (either air or a mixture of 5% CO2 in air) flowed into the helmet and was pumped out at a rate of 8 l/min. The monkeys' breathing produced changes in pressure inside the helmet that were measured with a pressure transducer connected to a polygraph (model 7; Grass Instruments, Quincy, MA); the data were recorded on a polygraph trace and in a microprocessor (IBM personal computer; IBM, White Plains, NY) via an analog-to-digital converter. The apparatus was calibrated routinely with known quantities of air. The polygraph integrator was connected to a computer, which analyzes the data collected over a 3-min period. The rate of breathing [respiratory frequency (f)] is determined directly. The minute volume (VE) is determined from the integration of the plethysmograph system. When the test compound was given in a cumulative dosing procedure, the test session contained six consecutive cycles. Each cycle was 30 min, which included the first 23-min exposure to air alone and remaining 7-min exposure to 5% CO2 mixed in air. Responses in the first two cycles were averaged as a baseline (control) value. Naltrexone, 6β-naltrexol, or vehicle solution was administered i.m. in the beginning of each cycle for the remaining dosing-injection cycles (i.e., from third to sixth cycles). The doses were increased by a 0.5 log unit throughout the test sessions. Each experimental condition was repeated at least once in all subjects.
Scratching Responses. Scratching responses, inferred as an itch sensation, were recorded on videotapes when monkeys were in their home cages. Each recording session was conducted for 15 min/test session (i.e., 16th-30th min after alfentanil administration). A scratch was defined as one short-duration (<1 s) episode of scraping contact of the forepaw or hind paw on the skin surface of other body parts. Scratches usually occurred repetitively at the same location. Scratching responses were scored by trained individuals who were blinded to experimental conditions.
Data Analysis. Individual tail withdrawal latencies were converted to percentage of maximum possible effect (%MPE) by the following formula: %MPE = [(test latency - control latency)/(cutoff latency 20 s - control latency)] × 100. In the respiration assay, data from the last 3 min of exposure to air and the second 3 min of exposure to CO2 mixed in air of each cycle were used for the data analysis. Individual respiratory parameters were averaged from the first two cycles and served as their own control values. Drug effects were determined from the remaining four dosing-injection cycles (i.e., from third to sixth cycles), and they were converted to percentage of their own control values. For data obtained from a cumulative dosing procedure, mean ED50 values were calculated after log transformation of individual ED50 values, which were calculated by leastsquares regression using the portion of the dose-effect curves spanning the 50% MPE or 150% control, and 95% confidence limits (CLs) were also determined (p < 0.05). In addition, dose ratios were calculated by dividing mean ED50 values in the presence of the antagonist by the baseline ED50 values. The significant shifts in dose-effect curves were defined when their 95% CL of ED50 values did not overlap. For in vivo apparent pA2 analysis, dose ratios were analyzed in a Schild plot, and individual pA2 values were obtained (Tallarida et al., 1979). Mean pA2 values were obtained from individual pA2 values.
Mean values (mean ± S.E.M.) were calculated from individual values for all behavioral endpoints. Comparisons were made for the same monkeys across all test sessions in the same experiment. All data obtained from a single dosing procedure were analyzed by one-way analysis of variance followed by the Newman-Keuls test for multiple (post hoc) comparisons. The criterion for significance was set at p < 0.05.
Experimental Designs
μ-Opioid Receptor Antagonist Potencies of Naltrexone and 6β-Naltrexol. A previous study has reported an in vivo apparent pA2 value of naltrexone against alfentanil-induced antinociception (Ko et al., 1998). For comparison, we used the same experimental conditions to determine the antagonist potency of 6β-naltrexol in nondependent (i.e., morphine-untreated) monkeys. Multiple doses of 6β-naltrexol (0, 0.1, 0.32, 1, and 3.2 mg/kg s.c.) were tested by using a single dosing procedure. The dose-response curve of s.c. alfentanil-induced antinociception was redetermined 30 min after pretreatment with 6β-naltrexol. Experiments were conducted once per week.
The second part of the antagonist study was to compare the potency of i.v. naltrexone and 6β-naltrexol against MOR-mediated behavioral effects. Although MOR agonists produce antinociception, respiratory depression, and itch/scratching mainly through central MOR activation in monkeys (Ko et al., 2002, 2004), it is unknown whether i.v. naltrexone and 6β-naltrexol have different in vivo potencies in blocking different behavioral endpoints. A single dose (0.056 mg/kg s.c.) of the MOR-selective agonist, alfentanil, was chosen because this dosing condition produced maximum behavioral responses measured in this study. Naltrexone (0.01 mg/kg), 6β-naltrexol (0.1 and 1 mg/kg), or vehicle solution (0.1 ml/kg) was administered intravenously through a saphenous vein 30 min before alfentanil administration. Experiments were conducted once per week.
Naltrexone- and 6β-Naltrexol-Precipitated Withdrawal. Previous studies have shown that naltrexone precipitated withdrawal measured by increased f and VE in acute and chronic morphine-dependent monkeys (Paronis and Woods, 1997; Kishioka et al., 2000). Therefore, we used the respiratory parameters to quantify the degree of acute dependence and to compare the potency of naltrexone and 6β-naltrexol in precipitating withdrawal. Our pilot study indicated that after 3 days of morphine treatment (6.4 mg/kg/day i.m.), naltrexone consistently produced a 2-fold increased f and VE of the baseline value when monkeys were exposed to air or 5% CO2 mixed in air. We further used this dosing regimen that acutely produced a minimum but significant degree of opioid dependence to compare the dose response curves of both compounds. Monkeys received two i.m. injections of 3.2 mg/kg morphine (i.e., at 9:00 AM and 3:00 PM) daily for 3 days. On the 4th day, effects of naltrexone (0.001-0.032 mg/kg i.m.) or 6β-naltrexol (0.1-3.2 mg/kg i.m.) were determined at 9:00 AM by using a cumulative dosing procedure. Effects of vehicle (0.1 ml/kg i.m.) were also studied under the same procedure (i.e., four dosing-injection cycles) in acute morphine-dependent monkeys. In addition, effects of naltrexone and 6β-naltrexol alone were tested in morphine-untreated monkeys. Experiments were normally conducted once per 2 to 3 weeks to assure that this acute morphine dependence had dissipated because our pilot studies, and previous studies have shown that dependence dissipated over the course of 5 to 7 days after cessation of acute or chronic morphine administration (Paronis and Woods, 1997; Kishioka et al., 2000).
Effect of 6β-Naltrexol on Naltrexone-Precipitated withdrawal. The third part of the behavioral study was to investigate whether 6β-naltrexol can act as a neutral antagonist by determining whether it can block naltrexone-precipitated withdrawal in acute morphine-dependent monkeys. Using the same dosing regimen for developing acute morphine dependence, effects of small doses of 6β-naltrexol that produced no or mild increments in f and VE on the dose-response curve of naltrexone-precipitated withdrawal were studied. Either vehicle (0.1 ml/kg) or 6β-naltrexol (0.1-0.32 mg/kg) was administered i.m. (i.e., at the beginning of the third cycle) 30 min before administration of naltrexone cumulative dosing in the remaining cycles (i.e., from fourth to sixth cycles). Likewise, experiments were conducted once per 2 to 3 weeks to assure that acute morphine dependence had dissipated before starting of the next experiment.
Drugs. [3H]DAMGO (51 Ci/mmol), [3H]diprenorphine (50 Ci/mmol), and [35S]GTPγS (1250 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences. Adenosine deaminase was from Calbiochem (San Diego, CA). EcoLume scintillation fluid was from MP Biomedicals (Irvine, CA). DAMGO, GDP, GTPγS, and all other biochemicals were from Sigma-Aldrich (St. Louis, MO). Alfentanil HCl, naltrexone HCl, 6β-naltrexol HCl, naloxone HCl (National Institute on Drug Abuse, Bethesda, MD), and morphine sulfate (Mallinckrodt, St. Louis, MO) were dissolved in sterile water. For systemic administration (i.e., s.c., i.m., i.v.), all compounds were administered at a volume of 0.1 ml/kg. Doses are presented in the compound forms listed above.
Results
In Vitro Binding Assays
Ligand Binding Assay. The ability of 6β-naltrexol and naltrexone to displace 1 nM [3H]DAMGO binding in cortical homogenates of monkey brain was measured over a range of concentrations. 6β-Naltrexol and naltrexone were able to dose-dependently displace the binding of [3H]DAMGO (Fig. 1) with Ki values of 0.74 ± 0.03 nM for 6β-naltrexol and 0.31 ± 0.04 nM for naltrexone. The differences in Ki values between 6β-naltrexol and naltrexone were statistically significant (p < 0.05).
The affinities of 6β-naltrexol and naltrexone were further examined for their abilities to displace the binding of the opioid [3H]diprenorphine (0.2 nM) (Fig. 2). Because [3H]diprenorphine is nonselective, these experiments were performed in homogenates of monkey thalamus, which expresses predominantly μ-opioid receptors (Ko et al., 2003). Experiments were performed in low-ionic strength 50 mM Tris buffer, pH 7.4, or in 50 mM Tris containing 100 mM NaCl and 10 μM GTPγS to promote a low-agonist affinity state of the receptor. Naltrexone had a higher affinity (Ki = 1.7 ± 0.3 nM) than 6β-naltrexol (Ki = 3.2 ± 0.3 nM) for displacing [3H]diprenorphine binding when performed in 50 mM Tris buffer (p < 0.05). When the assay was performed in the presence of NaCl and GTPγS, naltrexone (Ki = 1.6 ± 0.2 nM) retained its higher affinity compared with 6β-naltrexol (Ki = 2.6 ± 0.4 nM). Differences between the affinity of naltrexone and 6β-naltrexol were statistically significant (p < 0.05). Neither naltrexone nor 6β-naltrexol showed different binding properties in the different ionic conditions (p > 0.05).
[35S]GTPγS Binding Assay. Agonist-induced stimulation of G proteins was measured in homogenates of monkey thalamus (Fig. 3). DAMGO concentration-dependently increased the binding of [35S]GTPγS over basal by 52 ± 4.2% (EC50 = 0.76 ± 0.21 μM). Addition of 20 nM 6β-naltrexol resulted in an 18.3-fold rightward shift in the DAMGO concentration response curve (EC50 = 13.83 ± 4.0 μM). Likewise, addition of 10 nM naltrexone produced a 21.5-fold rightward shift in the DAMGO concentration response curve (EC50 = 17.54 ± 6.6 μM). The Ke value, the antagonist equilibrium affinity constant, for the degree of antagonist-induced shift is 1.17 ± 0.08 nM for 6β-naltrexol and 0.52 ± 0.09 for naltrexone (p < 0.05), suggesting that naltrexone has 2-fold higher affinity than 6β-naltrexol for the μ-opioid receptor.
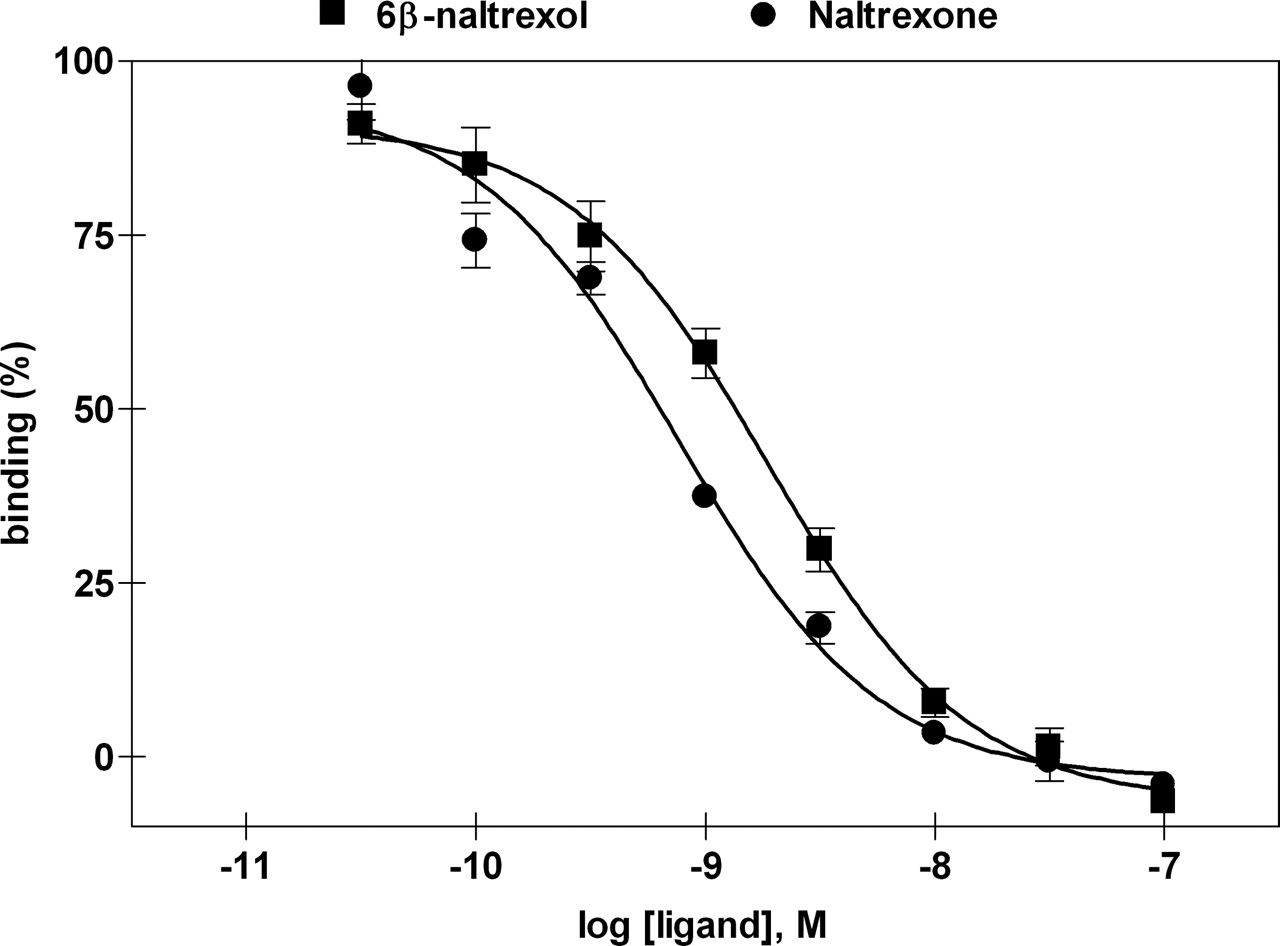
Displacement of [3H]DAMGO (1 nM) binding in cortical homogenates of monkey brain. Abscissa, log concentration (molar) of 6β-naltrexol or naltrexone. Ordinate, percentage of [3H]ligand bound. Data are expressed as mean ± S.E.M. of three experiments performed in duplicate. For details, see Materials and Methods.

Displacement of [3H]diprenorphine (0.2 nM) binding in thalamic homogenates of monkey brain. Experiments were performed in 50 mM Tris buffer, pH 7.4 (closed symbols, solid lines), or in 50 mM Tris, pH 7.4, containing 100 mM NaCl and 10 μM GTPγS (open symbols, dotted lines) for studies of low-agonist affinity binding. Abscissa, log concentration (molar) of 6β-naltrexol or naltrexone. Ordinate, percentage of [3H]ligand bound. Data are expressed as mean ± S.E.M for three experiments performed in duplicate. For details, see Materials and Methods.

Stimulation of [35S]GTPγS binding in thalamic homogenates of monkey brain. DAMGO concentration response curves were determined in the presence and absence of 20 nM 6β-naltrexol or 10 nM naltrexone as described in Materials and Methods. Abscissa, log concentration (molar) of DAMGO. Ordinate, percentage of [35S]GTPγS bound over basal. Data are expressed as mean ± S.E.M for three experiments performed in duplicate.
Behavioral Assays
μ-Opioid Receptor Antagonist Potencies of Naltrexone and 6β-Naltrexol.Figure 4 illustrates the antagonist effect of 6β-naltrexol against alfentanil-induced antinociception in 50°C water. Mean ED50 (95% CL) value of s.c. alfentanil-induced antinociception with vehicle pretreatment was 0.044 (0.039-0.048) mg/kg. Pretreatment with 6β-naltrexol dose-dependently produced rightward shifts of the dose-response curve of alfentanil-induced antinociception. Figure 4 also shows a Schild plot for 6β-naltrexol with values derived from individual dose ratios for each subject. The mean pA2 value of 6β-naltrexol was 6.50 (6.29-6.71) with a slope of -1.

In vivo antagonist potency of 6β-naltrexol against μ-opioid receptor agonist-induced antinociception in the monkey. Top panel, antagonist effect of s.c. 6β-naltrexol (30 min pretreatment) on the dose-response curve of alfentanil-induced antinociception in 50°C water. Abscissa, dose of s.c. alfentanil in milligrams per kilogram using a cumulative dosing procedure. Ordinate, percentage of maximum possible effect. Each data point represents the mean ± S.E.M. (n = 6). Bottom panel, Schild plot obtained for 6β-naltrexol. Abscissa, negative log unit of 6β-naltrexol in moles per kilogram. Ordinate, log of (dose ratio - 1). Each point was converted from individual dose ratios for each subject based on data in the top panel. Closed symbols represent different subjects (n = 6). The mean pA2 value and slope of 6β-naltrexol are shown with 95% confidence limits in parentheses.
The antagonist effects of i.v. naltrexone and 6β-naltrexol were determined against MOR-mediated behavioral effects (Fig. 5). Pretreatments with i.v. 1 mg/kg 6β-naltrexol and 0.01 mg/kg naltrexone significantly reversed s.c. alfentanil (0.056 mg/kg)-induced antinociception, scratching responses, and respiratory depression (p < 0.05). In contrast, pretreatment with i.v. 0.1 mg/kg 6β-naltrexol did not change the behavioral effects produced by alfentanil, compared with the vehicle pretreatment in the same monkeys (p > 0.05).

Comparison of antagonist effects of naltrexone and 6β-naltrexol against alfentanil-induced antinociception, scratching, and respiratory depression in monkeys. Abscissae, different pretreatment conditions. Naltrexone (0.01 mg/kg), 6β-naltrexol (0.1 and 1 mg/kg), or vehicle solution (0.1 ml/kg) was administered intravenously 30 min before s.c. administration of alfentanil (0.056 mg/kg). Each data point represents the mean ± S.E.M. (n = 6). The asterisk represents a significant difference from the vehicle pretreatment condition (*, p < 0.05). For other details, see Materials and Methods.
Naltrexone- and 6β-Naltrexol-Precipitated withdrawal. The potency difference between naltrexone and 6β-naltrexol in precipitating withdrawal was compared in acute morphine-dependent monkeys (Fig. 6). Both naltrexone- and 6β-naltrexol-induced increments in f and VE were similar in a dose-dependent manner whether dependent-monkeys were breathing air or air mixed with 5% CO2. For the sake of the simplicity of data presentation, only data during exposure to air are presented (Ko et al., 2002). Mean ED50 (95% CL) values of i.m. naltrexone-increased f and VE were 0.0042 (0.0027-0.0064) and 0.0034 (0.0022-0.0052) mg/kg, respectively. Mean ED50 values of i.m. 6β-naltrexol-increased f and VE were 0.34 (0.23-0.50) and 0.32 (0.20-0.49) mg/kg, respectively. Injection of vehicle solution every 30 min under the same dosing procedure did not significantly change both f and VE values in these acute morphine-dependent monkeys. There was no significant difference in the control values of monkeys under the different experimental conditions, nor did naltrexone (0.001-0.1 mg/kg) and 6β-naltrexol (0.1-10 mg/kg) change respiratory parameters f and VE in morphine-untreated (i.e., nondependent) monkeys (data not shown).
Effect of 6β-Naltrexol on Naltrexone-Precipitated Withdrawal.Figure 7 illustrates effect of 6β-naltrexol on naltrexone-precipitated withdrawal in acute morphine-dependent monkeys. Mean ED50 values of i.m. naltrexone-increased f and VE when morphine-dependent monkeys received vehicle pretreatment were 0.0041 (0.0031-0.0056) and 0.0030 (0.0018-0.0049) mg/kg, respectively. These ED50 values did not change when dependent monkeys received 0.1 mg/kg 6β-naltrexol pretreatment [ED50 values for f, 0.0040 (0.0026-0.0062); and for VE, 0.0032 (0.0018-0.0057) mg/kg]. In contrast, the potency of naltrexone in precipitating withdrawal seemed to be increased when dependent monkeys received 0.32 mg/kg 6β-naltrexol pretreatment [ED50 values for f, 0.0015 (0.0007-0.0031); and for VE, 0.0012 (0.0007-0.0020) mg/kg]. There was no rightward shift of the dose-response curve of naltrexone-increased respiratory parameters by pretreatment with 6β-naltrexol under these conditions.

Effects of naltrexone and 6β-naltrexol on respiration in the presence of air in acute morphine-dependent monkeys. Abscissae, dose of i.m. administration of the compound in milligrams per kilogram using a cumulative dosing procedure. Ordinates, percent change of the control value. Each data point represents the mean ± S.E.M. n = 6 in each panel. Open symbols represent the effect of vehicle in the last cycle of four-dosing injection/cycle regimen. For other details, see Materials and Methods.
Discussion
Radioligand binding assay showed that naltrexone exhibited approximately 2-fold higher affinity than 6β-naltrexol for sites labeled by [3H]DAMGO in the cortical membranes of the monkey (Fig. 1). Likewise, naltrexone displayed 2-fold higher affinity than 6β-naltrexol for sites labeled by [3H]diprenorphine in the monkey thalamic membranes. Both naltrexone and 6β-naltrexol retain similar binding properties under buffer conditions presenting high- and low-affinity states of the MOR (Fig. 2). These results are similar to those reported in another study showing that naltrexone displayed 5-fold higher affinity than 6β-naltrexol for sites labeled by [3H]DAMGO in guinea pig brain homogenate (Nelson et al., 1994). To compare the in vitro antagonist potency of both compounds, [35S]GTPγS binding was used to provide a functional correlate of ligand-binding experiments (Traynor and Nahorski, 1995; Ko et al., 2003). DAMGO concentration-dependently increased the binding of [35S]GTPγS over basal by 52% in the monkey thalamus, consistent with previous findings that the MOR is abundant and the maximum of G protein activation by a MOR agonist can be observed in the primate thalamic region (Sim-Selley et al., 1999; Ko et al., 2003). Furthermore, naltrexone was 2-fold more potent than 6β-naltrexol in antagonizing the concentration response curve of DAMGO-stimulated [35S]GTPγS binding (Fig. 3). These results indicate that both naltrexone and 6β-naltrexol are MOR antagonists with a slight potency difference in vitro in the monkey brain.

Effects of i.m. 6β-naltrexol (30-min pretreatment) on the dose-response curve of naltrexone-precipitated increased respiratory parameters in acute morphine-dependent monkeys. Abscissae, dose of i.m. naltrexone in milligrams per kilogram using a cumulative dosing procedure. Ordinates, percent change of the control value. Each data point represents the mean ± S.E.M. (n = 6) in each panel. For other details, see Fig. 6 and Materials and Methods.
To compare the in vivo antagonist potency of naltrexone and 6β-naltrexol, in vivo apparent pA2 analysis was used because this quantitative model provides a powerful approach for establishing receptor-mediated drug effects (Arunlakshana and Schild, 1959; Tallarida et al., 1979; Ward and Takemori, 1983). In the present study, 6β-naltrexol dose-dependently produced parallel rightward shifts of the dose-response curve of alfentanil-induced antinociception (Fig. 4), indicating that the agonist and antagonist compete for the same receptor site in a reversible manner (Arunlakshana and Schild, 1959; Tallarida et al., 1979). The pA2 value of 6β-naltrexol, 6.5, was approximately 100-fold less than the naltrexone pA2 value of 8.5 determined under the same behavioral assay conditions (Ko et al., 1998). It is expected that the pA2 values of the same antagonist are similar across different behavioral measures for effects mediated by a common receptor population (Ward and Takemori, 1983; Negus et al., 1993). Therefore, the in vivo antagonist potencies of naltrexone and 6β-naltrexol were further compared by using another administration route and other behavioral endpoints demonstrated to be mediated by central MOR (Ko et al., 2002, 2004). Again, i.v. 6β-naltrexol was much less potent than naltrexone, and 1 mg/kg 6β-naltrexol produced comparable blockade of alfentanil-induced antinociception, itch/scratching, and respiratory depression as 0.01 mg/kg naltrexone (Fig. 5). These results seem to indicate that both naltrexone and 6β-naltrexol produce MOR antagonist effects in vivo but with a consistent, large potency difference across different behavioral assays.
The in vitro binding assays may not be able to predict the in vivo apparent affinity for antagonists. For example, an opioid receptor antagonist, quadazocine, displayed 4-fold higher affinity than naltrexone for sites labeled by [3H]DAMGO in the monkey brain homogenate (Emmerson et al., 1994). However, the pA2 values of quadazocine (7.6-7.9) were 10-fold less potent than those of naltrexone (8.5-8.8) across different MOR-mediated behavioral endpoints (France et al., 1990; Negus et al., 1993; Ko et al., 1998). It is possible that 6β-naltrexol does not enter the brain as readily as naltrexone or that 6β-naltrexol is a better substrate than naltrexone for P-glycoprotein pumps removing ligands from the brain. Future studies measuring the concentrations of naltrexone and 6β-naltrexol in the monkey brain will be important to verify whether a large in vivo potency difference between ligands is due to the difference in the concentrations achieved in the vicinity of MOR receptors mediating the measured responses. Regardless, a large difference of in vivo potency between naltrexone and 6β-naltrexol has also been reported in rodent studies. 6β-Naltrexol was approximately 85- and 180-fold less potent than naltrexone in antagonizing MOR-mediated Straub tail and antinociception in mice, respectively (Chatterjie et al., 1975; Porter et al., 2002). In contrast, other studies showed that 6β-naltrexol was only 3- to 10-fold less potent than naltrexone in antagonizing morphine-induced antinociception and locomotor activity in mice (Wang et al., 2001; Raehal et al., 2005). Unfortunately, these mouse studies did not use the in vivo apparent pA2 analysis to systematically quantify and compare the antagonist potency of naltrexone and 6β-naltrexol across different behavioral endpoints.
In vivo potencies of naltrexone and 6β-naltrexol were further compared in acute morphine-dependent monkeys. In the present study, both antagonists dose-dependently precipitated withdrawal manifested by increased respiratory parameters (i.e., f and VE in Fig. 6). 6β-Naltrexol was approximately 80- to 95-fold less potent than naltrexone in precipitating withdrawal in morphine-treated monkeys. Both naltrexone and 6β-naltrexol at doses producing MOR antagonist effects also precipitated withdrawal in morphine-dependent monkeys. These results seem to agree with previous studies in monkeys indicating that opioid antagonists retain the same potency in producing MOR antagonist effects (France et al., 1990; Ko et al., 1998) and precipitating withdrawal (Valentino et al., 1983; France and Woods, 1989; France et al., 1990; Paronis and Woods, 1997) measured by different behavioral assays. More importantly, these findings are consistent with human studies showing that the doses of naloxone or naltrexone effective in blocking the effects of MOR agonists can also precipitate withdrawal in acute or chronic opioid-dependent subjects (Stitzer et al., 1991; Eissenberg et al., 1996; Walsh et al., 1996).
A recent study has shown that 6β-naltrexol may act as a neutral antagonist, whereas naltrexone displays an inverse agonist action, in acute morphine-dependent mice (Raehal et al., 2005). If a compound is a neutral antagonist, it is expected that this compound should block both agonist and inverse agonist actions (Kenakin, 2001). However, 6β-naltrexol failed to block naltrexone-precipitated withdrawal in acute morphine-dependent monkeys; instead, it potentiated the effects of naltrexone when the dose of 6β-naltrexol was increased (Fig. 7). There might be differences between primates and rodents in terms of the MOR basal signaling activity changed by MOR agonist administration. Naltrexone, but not 6β-naltrexol, slightly decreased [35S]GTPγS binding (i.e., 5-10% reduction from the basal level) in brain homogenates from morphine-treated mice (Wang et al., 2004). However, 6β-naltrexol at high doses can precipitate withdrawal in morphine-treated mice (Raehal et al., 2005). It will be valuable to explore whether a robust measure of inverse agonist action can be established in the morphine-treated monkey brain homogenates and to determine whether withdrawal precipitated by naltrexone and 6β-naltrexol can be correlated with their inverse agonist actions measured in vitro in morphine-treated monkeys. Nevertheless, except for a large difference of in vivo potency, the present results indicate that pharmacological actions of 6β-naltrexol are not significantly different from those of naltrexone in monkeys.
Withdrawal syndromes resulting from chronic dependence tend to be more intense and of longer duration than that from acute dependence; however, the observed signs and symptoms are qualitatively similar. The morphine dosing regimen used in this study produced a minimum but significant degree of physiological dependence on morphine to allow quantitative comparison of the potency of naltrexone and 6β-naltrexol. It is worth noting that precipitated withdrawal was measured at 18 h after the last injection of morphine, at which time point the amount of morphine was relatively low (Domino et al., 1987). Most withdrawal signs can be observed between 8 and 48 h postmorphine in both precipitated and abstinence-associated withdrawal in monkeys (Domino et al., 1987; France and Woods, 1989; Paronis and Woods, 1997; Kishioka et al., 2000). For comparison, 6β-naltrexol and naltrexone can precipitate withdrawal to a similar degree in mice implanted with morphine pellets, but they do not seem to do so when the degree of dependence is less severe (Fujimoto et al., 1975; Raehal et al., 2005). The only discrepancy among mouse studies is the difference of the MOR antagonist potency between naltrexone and 6β-naltrexol (i.e., 85-180-fold versus 3-10-fold) (Chatterjie et al., 1975; Wang et al., 2001; Porter et al., 2002; Raehal et al., 2005). Although in vivo pharmacological actions of the compound may depend on the physiological state of subjects under different antagonist doses and degrees of dependence, it is anticipated that a neutral antagonist would maintain its actions in a broader in vivo context.
In summary, this study demonstrates that naltrexone and 6β-naltrexol display similar pharmacological actions with different in vivo potencies in monkeys. Naltrexone is slightly more potent than 6β-naltrexol (i.e., 2-fold) in producing antagonist effects in vitro measured in the monkey brain membranes. However, naltrexone is much more potent than 6β-naltrexol (i.e., 100-fold) in producing in vivo MOR antagonist effects. More importantly, both antagonists retain the same relative potency in precipitating withdrawal in acute morphine-dependent monkeys. 6β-Naltrexol might not be a neutral antagonist because it failed to block the actions of naltrexone-precipitated withdrawal under these experimental conditions. Although 6β-naltrexol is 2- to 3-fold higher than naltrexone in the plasma level in humans (Meyer et al., 1984; Ferrari et al., 1998), 6β-naltrexol may play a minimal role in the therapeutic or antagonist effects of naltrexone in primates.
Acknowledgments
We thank Noreen Hughes, Tristan Edwards, and Wayne Yang for excellent technical assistance.
Footnotes
-
This work was supported by United States Public Health Service Grants DA-00254, DA-04087, DA-13685, and GM-07767.
-
doi:10.1124/jpet.105.094409.
-
ABBREVIATIONS: MOR, μ-opioid receptor; [35S]GTPγS, 5′-O-(3-[35S]thio)triphosphate; DAMGO, (d-Ala2,N-Me-Phe4,Gly5-ol)-enkephalin; %MPE, percentage of maximum possible effect; CL, confidence limit.
- Received August 19, 2005.
- Accepted October 27, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}