


Abstract
Many studies have reported greater drug uptake into brain than that predicted based upon existing models using the free fraction (fu) of drug in arterial serum. To explain this difference, circulating plasma proteins have been suggested to interact with capillary membrane in vivo to produce a conformational change that favors net drug dissociation and elevation of fu. Albumin, the principal binding protein in plasma, has two main drug binding sites, Sudlow I and II. We tested this hypothesis using drugs that bind selectively to either site I (warfarin) or site II (ibuprofen), as well as mixed ligands that have affinity for both sites (tolbutamide and valproate). Brain uptake was determined in the presence and absence of albumin using the in situ rat brain perfusion technique. Unidirectional brain uptake transfer constants (Kin) were measured and compared with those predicted using the modified Kety-Crone-Renkin model: Kin = F(1 – e–fu × PSu/F), where F is perfusion flow and PSu is the permeability-surface area product to free drug of brain capillaries. The results demonstrated good agreement between measured and predicted Kin over a 100-fold range in perfusion fluid albumin concentration using albumin from three different species (i.e., human, bovine, and rat), as well as whole-rat serum. Kin decreased in the presence of albumin in direct proportion to perfusion fluid fu with constant PSu. The results show that brain uptake of selected Sudlow site I and II ligands matches that predicted by the modified Kety-Crone-Renkin model with no evidence for enhanced dissociation.
Central nervous system drug development is complicated by the presence of blood-brain barrier, which is formed by the brain capillary endothelium (Hawkins and Davis, 2005). Most drugs gain access to the brain from the circulation either by dissolving in and diffusing across the lipophilic brain capillary cell membranes or by being transported across by one or more carrier- or receptor-mediated mechanisms (Begley and Brightman, 2003). Many parameters influence the ability of a drug to distribute to brain. One of these is plasma protein binding.
Drug distribution within the body is generally held to be driven by the free unbound concentration (Cu) of drug in circulating plasma (free drug hypothesis) (Buxton, 2005). Most drugs rapidly exchange between free and bound forms within the circulation (Talbert et al., 2002). One of the more commonly utilized models to analyze capillary transport is the modified Kety-Crone-Renkin (parallel tube) model that incorporates rapid mass exchange between the free and plasma protein-bound pools as blood transits the capillary bed (Pardridge and Landaw, 1984; Robinson, 1990; Morgan and Huang, 1993; Smith, 2003). This model is based upon the capillary permeability-surface area product to free drug (PSu), tissue blood flow (F), and the free fraction (fu) of drug in arterial plasma or serum. However, a large number of studies have reported that arterial fu is not adequate to account for measured brain uptake in vivo even when mass action exchange between the bound and free pool in serum is incorporated (Tanaka and Mizojiri, 1999; Videbaek et al., 1999; Pardridge, 2001). Several hypotheses have been put forward to explain this discrepancy. The most commonly referenced is the enhanced-dissociation hypothesis, which posits that an interaction between plasma proteins and brain capillary membranes in vivo induces a conformational change in plasma protein structure that reduces binding affinity and raises capillary drug fu during blood passage through the capillary bed. According to this hypothesis, plasma fu and Cu measured in vitro substantially underestimate in vivo values in cerebral capillaries. The magnitude of the discrepancy has been reported to be 5–50-fold (Pardridge, 2001). Not all studies support the enhanced-dissociation hypothesis, and even within studies reporting enhanced dissociation, some compounds do not exhibit the effect (Pardridge and Landaw, 1984). As a result of this variability, it has been questioned whether brain uptake can be predicted from in vitro parameters in the absence of in vivo measurements.
Of the many plasma proteins that bind drugs and hormones, albumin is one of the most important. It is the most abundant protein in blood plasma with concentrations of 3.5 to 5.0 g/dl in humans and 2.7 to 3.3 g/dl in rodents (Peters, 1996). It has been shown to shuttle a broad range of endogenous and exogenous ligands, including >70% of drugs (Kratochwil et al., 2002). Albumin has two main drug binding sites characterized as Sudlow site I and Sudlow site II (Sudlow et al., 1975), which bind drugs selectively. Site I, also known as the warfarin binding site, is formed by a pocket in subdomain IIA of human serum albumin (Kragh-Hansen et al., 2002). Warfarin is the selective probe drug for this site (Petitpas et al., 2001). Site II is located in subdomain IIIA and is known as the benzodiazepine binding site. Ibuprofen and diazepam are selective drug probes for site II (Kragh-Hansen et al., 2002).
We believed that if enhanced dissociation occurs in the brain capillary circulation, some of the variability in the literature may relate to the fact that the capillary-mediated conformational change in plasma protein structure may alter the binding affinity of only certain sites on plasma proteins. Therefore, to investigate this, we chose a series of agents that bind selectively to Sudlow site I, site II, or both and determined the effects of plasma albumin on brain uptake using in situ rat brain perfusion technique. Given that albumin differs among species in binding site selectivity and structure, albumin from three separate species (i.e., human, bovine, and rat) was examined. Warfarin was used as the Sudlow site I-specific ligand, whereas ibuprofen was used for site II. Tolbutamide and valproic acid, which bind to a limited extent at both sites, were also tested. All experiments were performed at tracer concentrations relative to binding affinity to maximize binding selectivity to specific sites. The four agents chosen for study are moderate in blood-brain barrier permeability and are not predicted to show substantial flow dependence of uptake at physiologic flow rates. A preliminary abstract of this study has been published (Mandula et al., 2005).
Materials and Methods
Materials
[4-Phenyl-3H]-(R,S)-warfarin (13.7 Ci/mmol) was purchased from Moravek Biochemicals Inc. (Brea, CA). [2-14C]Diazepam (56 mCi/mmol) was obtained from Amersham Biosciences (Piscataway, NJ). [Carboxyl-14C]-(R,S)-ibuprofen (55 mCi/mmol), [carbonyl-14C]tolbutamide (54 mCi/mmol), [carboxyl-14C]valproic acid (55 mCi/mmol), [N-methyl-3H]diazepam (85 Ci/mmol), [methoxy-14C]inulin (8.6 mCi/g), and [methoxy-3H]inulin (225 mCi/g) were procured from American Radiolabeled Chemicals, Inc. (St. Louis, MO). Bovine serum albumin (fraction V, A3059), human serum albumin (fraction V, A3059), and rat serum albumin (fraction V) were purchased from Sigma-Aldrich (St. Louis, MO). All tracers had a radiochemical purity of >99%, as reported by the manufacturer.
Animals
The study was approved by the Institutional Animal Care and Use Committee. All of the experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Adult Sprague-Dawley male rats (250–350 g) were purchased from Charles River Laboratories (Wilmington, MA) and were allowed free access to food and water until the morning of the experiment.
Perfusion Fluid
Perfusion fluid consisted of bicarbonate-buffered perfusion saline (128 mM NaCl, 24 mM NaHCO3, 4.2 mM KCl, 2.4 mM NaH2PO4, 1.5 mM CaCl2, 0.9 mM MgSO4, and 9 mM d-glucose; pH 7.40 ± 0.05) with or without varying concentrations of added serum albumin or freshly collected rat whole serum (collected on the morning of the day of the experiment and used within 1 h). To each perfusion, fluid was added with 3H or 14C drug (0.04–1.0 μCi/ml) and correspondingly labeled (14C or 3H) with inulin or diazepam to measure brain vascular volume (Vv) or cerebral perfusion fluid flow (F), respectively, by dual-label liquid scintillation counting (LSC). All unlabeled drug agents were added to perfusion fluid from buffered (pH 7.4) stock solution to ensure control of perfusion fluid pH.
Surgical Procedures
Radiolabeled drug uptake into brain across the blood-brain barrier was measured using the in situ rat brain perfusion technique (Smith, 1996). Rats were anesthetized using sodium pentobarbital (40 mg/kg i.p. Nembutal; Abbott Laboratories, North Chicago, IL). The left external carotid artery was ligated with surgical silk, and the left common carotid artery was catheterized with PE-60 tubing filled with heparinized 0.9% NaCl (100 IU/ml). The left pterygopalatine artery was not occluded. The common carotid artery catheter was connected via a four-way valve to dual-thermostated (37°C) glass syringes mounted in a Harvard infusion pump (Harvard Biosciences, South Natick, MA) (Smith, 1996). A heating pad connected to a feedback device (YSI model 73A; Yellow Springs, OH) kept rat body temperature at 37°C as measured using a rectal probe.
To begin the perfusion, the heart was severed to stop blood flow to the brain, and then perfusion fluid was infused into the carotid artery catheter at a rate (5 or 20 ml/min) to maintain perfusion pressure at 80 to 120 mm Hg. An initial tracer-free perfusion (∼30 s) was performed to wash out circulating blood from the vasculature, and then the fluid was switched to matching perfusion fluid containing radiotracers. Concentrations were chosen to obtain a brain [3H]/[14C] ratio of 3 to 5:1 and counts >5 to 10 times background. After 10 to 30 s of tracer uptake, the animal was decapitated, and the perfusion pump was turned off. The perfused left cerebral hemisphere was dissected into regions, as described by Takasato et al. (1984), which were weighed in glass vials. In addition, duplicate specimens of perfusion fluid were collected for determination of tracer concentration and drug-free fraction.
Sample Preparation
Brain samples were digested overnight at 55°C in 1 ml of Solvable tissue solubilizer (Packard, Meriden, CT). The next morning, 10 ml of Scintisafe 30% scintillation cocktail (Fischer Scientific, Pittsburgh, PA) was added to each vial, and then the vial was counted for [3H] and [14C] activity by LSC. Disintegrations per minute were obtained from counts per minute with correction for background, quench, and counting efficiency (Beckman LS 6500; Beckman Coulter, Fullerton, CA).
Plasma Protein Binding
Ultrafiltration. Drug fu in perfusion fluid was measured by ultrafiltration using Microcon centrifugal filter devices (molecular mass cutoff 10 kDa) purchased from Amicon Bioseperations (Bedford, MA). Perfusion fluid was collected immediately at the end of perfusion (volume 500 μl) and spun at 4000 rpm at 37°C for 8 min under 95% O2 and 5% CO2. Centrifugation conditions were set to maintain temperature and pH at 37°C and 7.4, respectively. Drug concentration in the initial fluid, as well as filtrate and retentate, was determined by LSC. Protein-free fluid was also filtered to determine drug binding to the membrane (Barre et al., 1985; March and Blanke, 1985).
Equilibrium Dialysis. Acrylic equilibrium dialysis cells (1 ml) (Bel-Art Products, Pequannock, NJ) were used to confirm fu by ultrafiltration. The two halves of the cells are separated by a 6-kDa molecular mass cutoff dialysis membrane (Bel-Art Products). One of the cells contained protein-free buffer, and the other cell contained drug in protein-containing perfusion fluid. The equilibrium time was determined for each drug of interest. The cells were suspended in a shaker maintained at 37°C until equilibrium was achieved, and then the fluid from each chamber was sampled to determine tracer concentrations. Drug concentration was determined by LSC. Control experiments in the absence of plasma protein confirmed attainment of equal concentrations in the two compartments at equilibrium. Experiments with warfarin and ibuprofen showed that fu by the two methods matched and did not differ significantly.
Distribution Coefficient
The octanol/saline distribution coefficient (log Doct 7.4) was measured using radiolabeled drug in phosphate buffer (0.1 M) and an equal volume of water-saturated n-octanol. After vortexing for 1 min, the mixture was centrifuged, and the two phases were allowed to separate. Aliquots of the separated phases were assayed for tracer activity by LSC. Samples were analyzed in duplicate and re-extracted three times to ensure stability of the log Doct 7.4 value.
Data Analysis and Calculations
The initial time course (10–30 s) of drug uptake into brain was measured to determine the time over which brain uptake was linear and unidirectional (Smith, 2003). From this data, transfer constants (Kin) for unidirectional brain uptake were calculated using the linear sloping method, as described previously (Smith, 2003),  where Qbr is the vascularly corrected quantity of drug tracer in brain (disintegrations per minute/gram) at the end of the perfusion, Vi is the cerebral vascular volume separate from that of inulin (milliliter/gram), Ctot is the perfusion fluid total concentration of labeled drug tracer (disintegrations per minute/milliliter), and T is the net tracer perfusion time (seconds). Qbr was calculated from the total measured drug content in brain (Qtot, disintegrations per minute/gram) as Qbr = Qtot – VvCpf, where Vv is the measured brain inulin volume [(brain inulin disintegrations per minute/gram)/(perfusion fluid inulin disintegrations per minute/milliliter)] (Takasato et al., 1984). Data were expressed as a brain uptake space (milliliter/gram), defined as Qbr/Ctot. The initial time course was measured in the absence of plasma protein. Once the linear range of initial uptake was determined, subsequent experiments determined Kin using the single time point method (Smith, 2003),
where Qbr is the vascularly corrected quantity of drug tracer in brain (disintegrations per minute/gram) at the end of the perfusion, Vi is the cerebral vascular volume separate from that of inulin (milliliter/gram), Ctot is the perfusion fluid total concentration of labeled drug tracer (disintegrations per minute/milliliter), and T is the net tracer perfusion time (seconds). Qbr was calculated from the total measured drug content in brain (Qtot, disintegrations per minute/gram) as Qbr = Qtot – VvCpf, where Vv is the measured brain inulin volume [(brain inulin disintegrations per minute/gram)/(perfusion fluid inulin disintegrations per minute/milliliter)] (Takasato et al., 1984). Data were expressed as a brain uptake space (milliliter/gram), defined as Qbr/Ctot. The initial time course was measured in the absence of plasma protein. Once the linear range of initial uptake was determined, subsequent experiments determined Kin using the single time point method (Smith, 2003), 
Drug uptake Kin in the absence of plasma protein (fu = 1.0) was analyzed using the Kety-Crone-Renkin equation (Crone and Levitt, 1984; Robinson, 1990; Smith, 2003),  where F is the tissue blood flow or perfusion flow rate. For drugs that bind rapidly and reversibly to plasma proteins, the modified Kety-Crone-Renkin equation was used (Robinson, 1990; Morgan and Huang, 1993),
where F is the tissue blood flow or perfusion flow rate. For drugs that bind rapidly and reversibly to plasma proteins, the modified Kety-Crone-Renkin equation was used (Robinson, 1990; Morgan and Huang, 1993),  where fu is the unbound fraction of labeled drug in perfusion fluid. Equation 4 assumes that 1) drug binding and dissociation from plasma protein is sufficiently rapid that bound and free drug are essentially in equilibrium at all points in the capillary bed, 2) only free (unbound) drug contributes to brain uptake, and 3) all brain capillaries are equal (no capillary heterogeneity). Equation 4 can be rearranged and expressed in terms of either in vivo PSu or fu as,
where fu is the unbound fraction of labeled drug in perfusion fluid. Equation 4 assumes that 1) drug binding and dissociation from plasma protein is sufficiently rapid that bound and free drug are essentially in equilibrium at all points in the capillary bed, 2) only free (unbound) drug contributes to brain uptake, and 3) all brain capillaries are equal (no capillary heterogeneity). Equation 4 can be rearranged and expressed in terms of either in vivo PSu or fu as, 

Cerebral perfusion fluid flow was determined in the absence of protein from brain uptake of radiolabeled diazepam (Smith, 1996, 2003). Calculated in vivo fu from brain perfusion studies was compared with fu measured in vitro using ultrafiltration or equilibrium dialysis. Brain capillary unidirectional extraction (Eu) for free drug was calculated as E = (1 – e–PSu/F) (Crone and Levitt, 1984).
Statistical Analysis
Data are presented as mean ± S.E.M., unless otherwise noted. Prism version 4.0 (GraphPad Software, Inc., San Diego, CA) was used for performing statistical analyses. Means were compared for statistical differences using one-way analysis of variance and Dunnett's test for multiple comparisons with a control group. Linear regression analysis was performed with Prism. A p value of <0.05 was considered statistically significant.
Results
Linear Brain Uptake.Figure 1 illustrates the time course (10–30 s) for brain uptake of the four radiolabeled ligands used in this study during perfusion at tracer concentration in the absence of protein (fu = 1.0). Brain uptake for each compound was corrected for residual intravascular tracer using inulin. All time courses exhibited linear uptake over 30 s and extrapolated to ∼0 ml/g at T = 0 s. The magnitude of uptake varied ∼8-fold among the solutes, with ibuprofen being the fastest and tolbutamide being the slowest. From these data, blood-brain barrier PSu values were calculated using the Kety-Crone-Renkin equation (eq. 3). The values are summarized in Table 1. Brain-free drug extraction for most compounds was <25%.
Calculated cerebrovascular Kin, PSu, F, Vv, and Eu for the four drug ligands studied in this article Data represent mean ± S.E.M. (n = 3-9). Values are for the 5 ml/min infusion rate.

Unidirectional linear uptake of site-specific albumin ligands for a period of 10 to 30 s. Data represent mean ± S.E.M. (n = 3–5) values of brain space (Qbr/Ctot) for left cerebral hemisphere after brain perfusion. Tracer concentrations were [3H]warfarin (0.01 μM), [14C]ibuprofen (1 μM), [14C]valproate (1.5 μM), and [14C]tolbutamide (3 μM). The pooled cerebral hemisphere includes the cerebral cortex, hippocampus, striatum, thalamus/hypothalamus, and midbrain/colliculi.
To ensure that the blood-brain barrier PSu values were accurate, uptake was determined at two infusion rates, 5 and 20 ml/min. The 5 ml/min infusion rate yielded a cerebral F of 0.024 ± 0.006 ml/s/g, which matches normal cerebral blood flow (Ohno et al., 1978). The 20-ml/min infusion rate yielded a cerebral F approximately 4 times greater (0.092 ± 0.024 ml/s/g) and in the same range as the initial Takasato method (Takasato et al., 1984). Blood-brain barrier PSu values at the two infusion rates are summarized in Fig. 2. The PSu values matched between the two infusion rates (Fig. 2) (p > 0.05); thus, the lower infusion rate with the more physiologic F was used in all subsequent serum and protein perfusion experiments. Brain Vv did not vary significantly with F (p > 0.05).
No statistically significant differences were observed in brain regional Kin, PSu, F, or Vv, with the exception of the cerebellum, which had significantly lower F. The cerebellum receives most of its blood flow from the basilar artery and thus was poorly perfused with the carotid artery brain perfusion technique (Takasato et al., 1984). Because brain regions with adequate F showed little or no regional differences in Kin or PSu, regional Kin and PSu values (including cerebral cortex, hippocampus, striatum, midbrain/colluculi, and thalamus/hypothalamus) were pooled to obtain combined cerebral hemisphere values to simplify presentation. Based on the linear uptake study, a tracer perfusion time of 30 s was used in all subsequent experiments.
Effect of Plasma Albumin on Brain Uptake of Sudlow Site I Ligand: Warfarin.Figure 3 illustrates the influence of albumin from three different species on brain up-take of the Sudlow site I-specific drug, warfarin from saline perfusion fluid. For each albumin, warfarin uptake Kin decreased with increasing albumin concentration. The magnitude of the reduction matched that in perfusion fluid fu as measured in vitro. Blood-brain barrier uptake PSu for warfarin in the presence of protein did not differ significantly from that in the absence of protein (p > 0.05). Similar trends were observed for brain uptake of warfarin in the presence of human, bovine, and rat serum albumin. Vv was affected by the presence of albumin in the perfusion fluid (p > 0.05). For all three albumins, measured Kin matched that predicted by eq. 4 from separately measured fu,PSu, and F. Although data in Fig. 3 are presented as pooled cerebral hemisphere values, the same pattern was observed when data were analyzed as individual brain regions (e.g., cerebral cortex, hippocampus, striatum, midbrain/colluculi, and thalamus/hypothalamus). In all instances, the results obtained with the individual regions matched that of the pooled hemisphere.
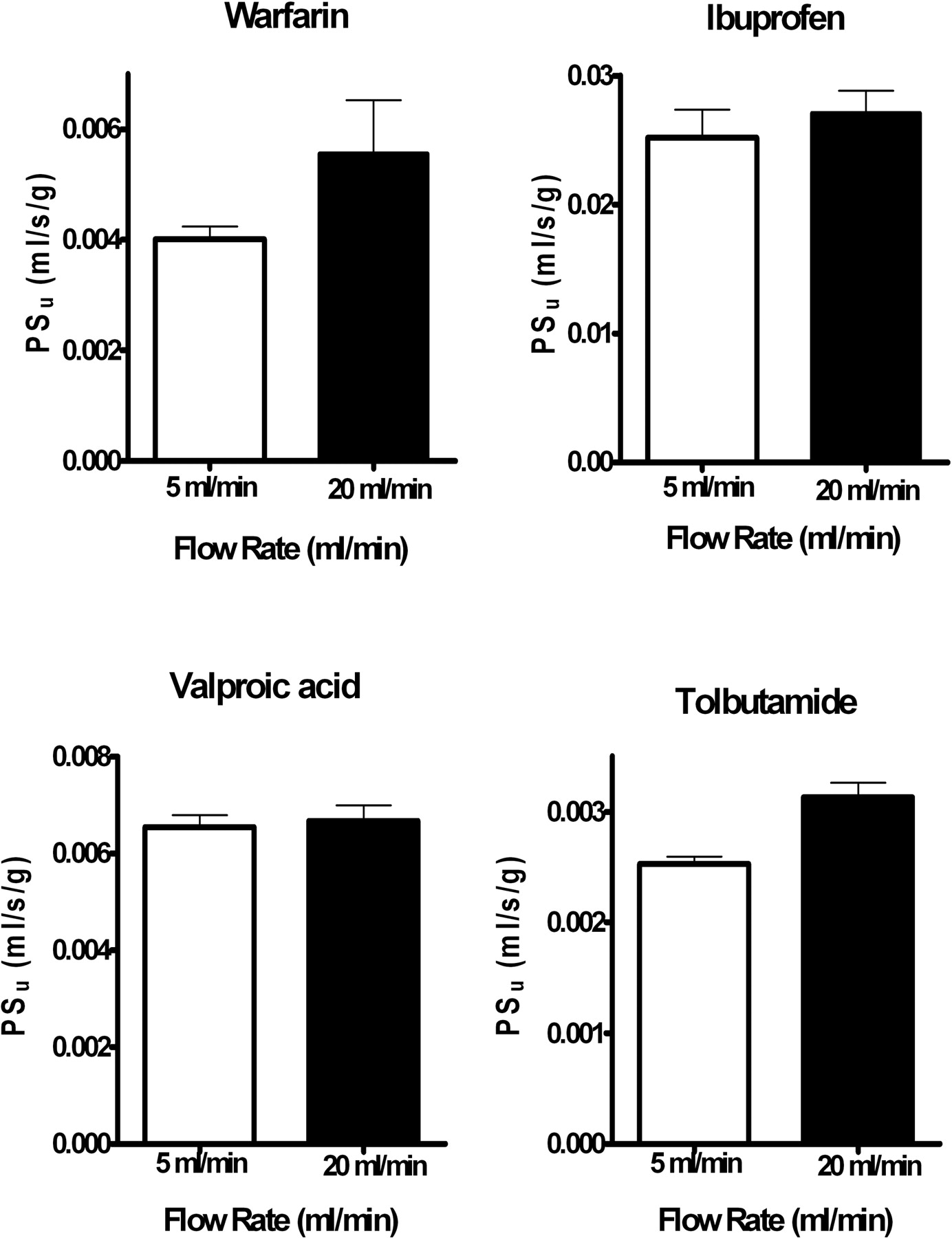
Blood-brain barrier PSu measured at 5 and 20 ml/min infusion rates. Values represent mean ± S.E.M. (n = 3–9) for the pooled left cerebral hemisphere (regions as noted in legend for Fig. 1). Values did not differ significantly between the two flow rates by ANOVA (p > 0.05).
Effect of Plasma Albumin on Brain Uptake of Sudlow Site II Ligand: Ibuprofen.Figure 4 illustrates the influence of albumin from different species on brain uptake of the Sudlow site II-specific drug, ibuprofen from saline perfusion fluid. As was found for warfarin, brain uptake Kin for ibuprofen decreased with increasing perfusion fluid albumin concentration. Measured Kin values in the presence of protein matched that predicted by the modified Kety-Crone-Renkin equation (eq. 4) using fu in vitro. Species differences did exist in the affinity of albumin to ibuprofen, but in general, the model fit the brain uptake data. Blood-brain barrier PSu calculated in the presence of protein did not differ significantly from that in the absence of protein (p > 0.05). Data for the cerebral hemisphere matched that of the individual brain regions.
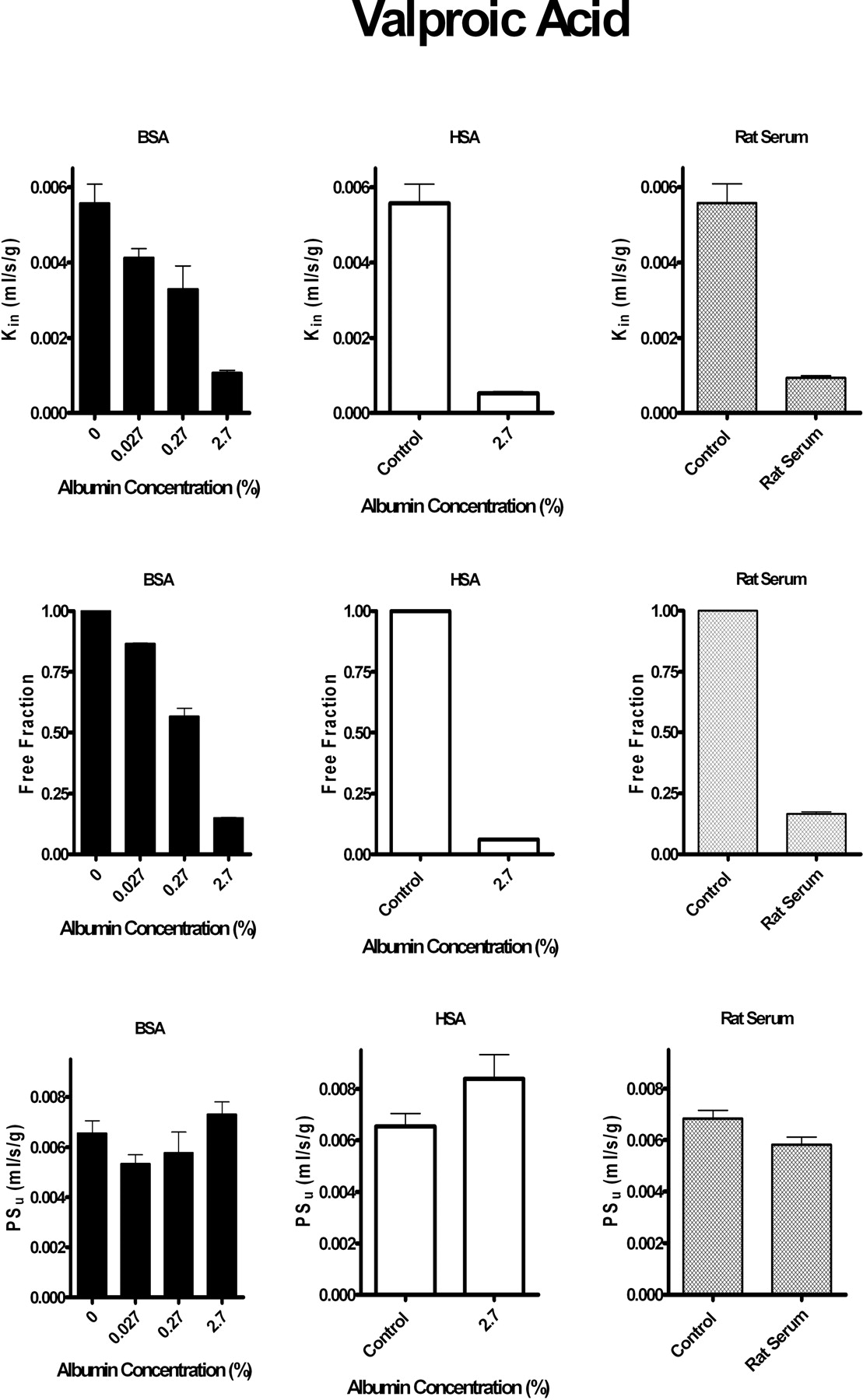
Effect of Plasma Albumin on Brain Uptake of Sudlow Site I and II-Mixed Ligands: Tolbutamide and Valproic Acid. Figures 5 and 6 illustrate the influence of albumin from different species on brain uptake of the mixed site I and II ligands, tolbutamide and valproic acid, respectively. For each compound, the addition of albumin reduced blood-brain barrier Kin in a manner that matched in vitro perfusion fluid fu. No statistically significant differences were obtained in blood-brain barrier PSu from protein-free controls (p > 0.05).
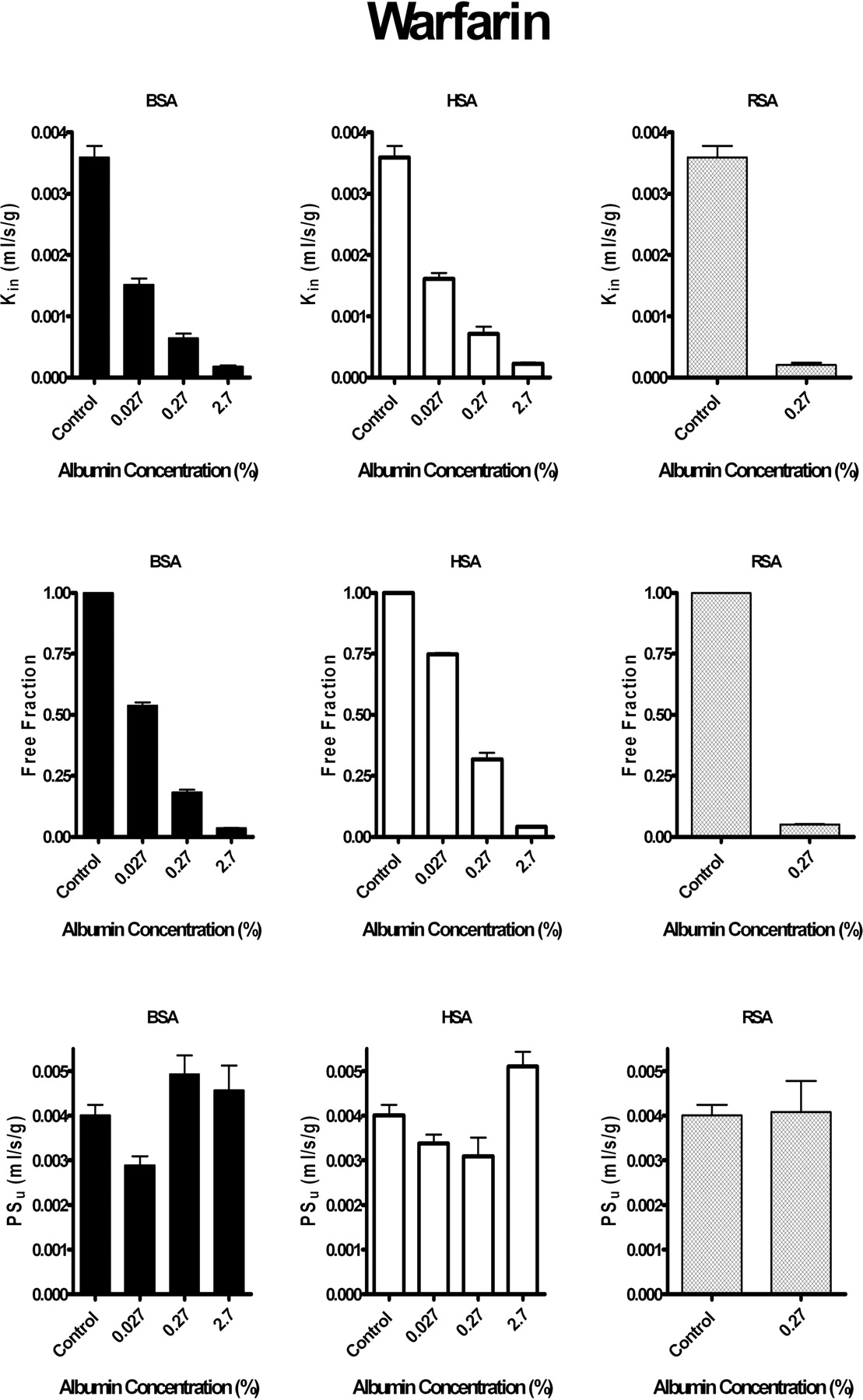
Brain uptake Kin and PSu and perfusion fluid fu of [3H]warfarin (≤0.07 μM) in the presence and absence of albumin from different species. Left, bovine serum albumin (BSA). Center, human serum albumin (HSA). Right, rat serum albumin (RSA). Top, Kin at increasing protein concentrations. Middle, fu obtained in vitro at different protein concentrations. Bottom, blood-brain barrier free drug PSu calculated at different protein concentrations from the measured Kin corrected for flow and in vitro fu using the modified Kety-Crone-Renkin equation (eq. 5). Values represent mean ± S.E.M. (n = 3–5). Blood-brain barrier Kin and PSu values are for the pooled left cerebral hemisphere (regions as noted in legend for Fig. 1). All Kin and fu values measured in the presence of albumin (0.027, 0.27, or 2.7%) differed significantly (p < 0.05) from matching controls (no albumin).
Because purified proteins may not fully model natural proteins in blood, brain uptake of tolbutamide and valproate was also measured from freshly collected rat serum (Figs. 5 and 6). With whole-rat serum, perfusion fluid in vitro fu for tolbutamide equaled 0.049 ± 0.006 with a measured brain uptake Kin of 1.3 ± 0.2 × 10–4 ml/s/g (n = 3). Calculated PSu from rat serum using the modified Kety-Crone-Renkin equation (eq. 5) (2.7 ± 0.4 × 10–3 ml/s/g, n = 3) did not differ significantly from that obtained in the absence of plasma protein (2.4 ± 0.2 × 10–3 ml/s/g) at the same infusion rate. Likewise, in vitro fu for valproic acid in rat serum equaled 0.16 ± 0.01 with a measured Kin of 9.4 ± 0.5 × 10–4 ml/s/g (n = 3). The valproic acid PSu with serum (5.9 ± 0.3 × 10–3 ml/s/g, n = 3) compared well with that determined at the same infusion rate in the absence of protein (6.6 ± 0.7 × 10–3 ml/s/g, n = 6). The same pattern was noted between pooled hemisphere and individual brain regions. The results suggest that the modified Kety-Crone-Renkin model provides accurate predictions of Kin, even in the presence of whole-rat serum.
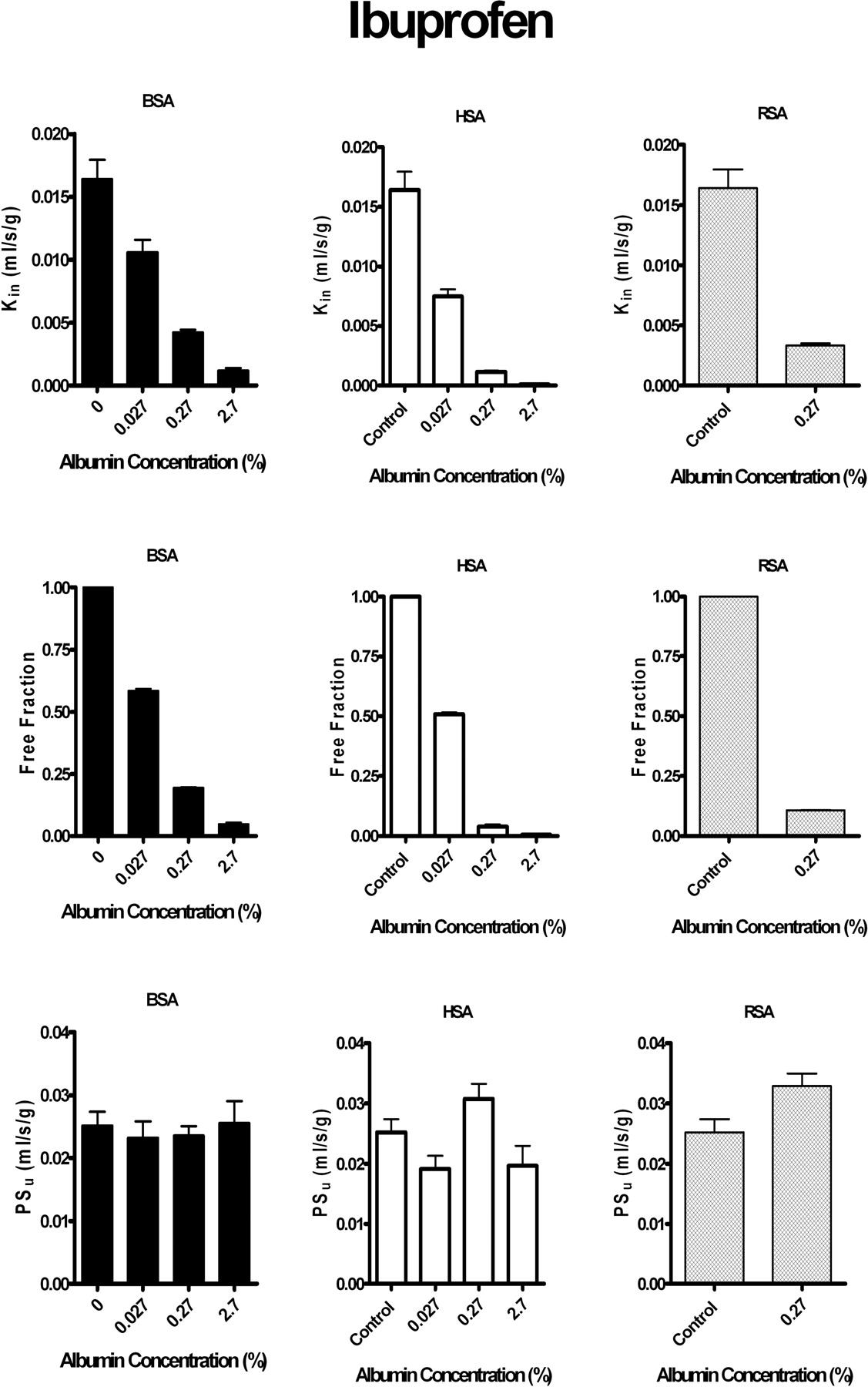
Brain uptake Kin and PSu and perfusion fluid fu of [14C]ibuprofen (3–11 μM) in the presence and absence of albumin from different species. Left, bovine serum albumin (BSA). Center, human serum albumin (HSA). Right, rat serum albumin (RSA). Top, Kin at increasing protein concentrations. Middle, fu obtained in vitro at different protein concentrations. Bottom, blood-brain barrier free drug PSu calculated at different protein concentrations from the measured Kin corrected for flow and in vitro fu using the modified Kety-Crone-Renkin equation (eq. 5). Values represent mean ± S.E.M. (n = 3–5). Blood-brain barrier Kin and PSu values are for the pooled left cerebral hemisphere (regions as noted in legend for Fig. 1). All Kin and fu values measured in the presence of albumin (0.027, 0.27, or 2.7%) differed significantly (p < 0.05) from matching controls (no albumin).
Relationship between Blood-Brain Barrier Log PS and logDoct 7.4.Figure 7A illustrates the relationship between blood-brain barrier log PSu and log Doct 7.4 for the four studied drugs in the absence of plasma protein binding. For all of the tested drugs, log PSu, based upon free drug, fell close to or within the 95% prediction limits of the line of permeability reference compounds (Smith, 2003). However, when uptake data in the presence of plasma protein (2.7% albumin) were expressed as a PStot, neglecting protein binding (i.e., assuming fu = 1.0), log PStot calculated (Fig. 7B) for the four values fell sharply below the line of the permeability reference compounds by >2 to 4 log units, illustrating the large effect of plasma protein binding on brain uptake. The magnitude of the dropoff in log PStot for these compounds was proportional to the decrease in fu. Drugs that are actively effluxed across the blood-brain barrier (e.g., AZT, vincristine, and vinblastine) also fall markedly below the line of permeability reference compounds. The results illustrate the importance of accurately incorporating the contribution of plasma protein binding in brain uptake PS calculations. If fu is ignored, the derived PStot may give the impression that the solute is actively effluxed out of brain at the blood-brain barrier.

Brain uptake Kin and PSu and perfusion fluid fu of [14C]tolbutamide (3–7 μM) in the presence and absence of albumin from different species Left, bovine serum albumin (BSA). Center, human serum albumin (HSA). Right, rat serum. Top, Kin at increasing protein concentrations. Middle, fu obtained in vitro at different protein concentrations. Bottom, blood-brain barrier free drug PSu calculated at different protein concentrations from the measured Kin corrected for flow and in vitro fu using the modified Kety-Crone-Renkin equation (eq. 5). Values represent mean ± S.E.M. (n = 3–5). Blood-brain barrier Kin and PSu values are for the pooled left cerebral hemisphere (regions as noted in legend for Fig. 1). Kin and fu values measured in the presence of 0.27 or 2.7% albumin or rat serum differed significantly (p < 0.05) from matching controls (no albumin).

Brain uptake Kin and PSu and perfusion fluid fu of [14C]valproic acid (1.5–3 μM) in the presence and absence of albumin from different species. Left, bovine serum albumin (BSA). Center, human serum albumin (HSA). Right, rat serum. Top, Kin at increasing protein concentrations. Middle, fu obtained in vitro at different protein concentrations. Bottom, blood-brain barrier free drug PSu calculated at different protein concentrations from the measured Kin corrected for flow and in vitro fu using the modified Kety-Crone-Renkin equation (eq. 5). Values represent mean ± S.E.M. (n = 3–5). Blood-brain barrier Kin and PSu values are for the pooled left cerebral hemisphere (regions as noted in legend for Fig. 1). Kin and fu values measured in the presence of 0.27 or 2.7% albumin or rat serum differed significantly (p < 0.05) from matching controls (no albumin).
Relationship betweenfu andKin Ratio (Presence versus Absence of Plasma Protein).Figure 8 illustrates the relationship between the magnitude of Kin reduction in the presence of protein and the magnitude of perfusion fluid drug fu. For slow- to moderate-penetrating solutes, such that PSu < F, the modified Kety-Crone-Renkin equation reduces from Kin = F (1 – e–fu×PSu/F) to Kin ≈ fu × PSu. Therefore, for slow- to moderate-penetrating compounds at normal flow, Kin is predicted to be proportional to perfusion fluid fu. This was indeed the case for the four compounds studied. The line of identity indicates perfect agreement between fu and brain uptake Kin. Most of the tested drugs fell on or close to the line of identity, indicating a restrictive effect of plasma protein binding on brain uptake for these compounds.

Relationship between lipophilicity and brain uptake. A, log PSu versus log octanol/water distribution coefficient (pH 7.4). B, log PStot versus log octanol/water distribution coefficient (pH 7.4) where blood-brain barrier PStot indicates uptake in the presence of plasma proteins (2.7% albumin) without correction for fu. Values represent means for n = 3 to 9 for the pooled cerebral hemisphere. ▵, permeability reference compounds, including sucrose, mannitol, fructose, glycerol, urea, methylurea, ethylene glycol, trimethylene glycol, thiourea, acetamide, butanediol, nicotinamide, water, methanol, iodoacetamide, ethanol, antipyrine, aminopyrine, butanol, and iodoantipyrine (Smith, 2003). Filled line indicates the linear regression line for the permeability reference compounds with ± 95% confidence intervals (dotted lines). •, tested compounds of interest as ligands for site-specific binding. ⋄, brain uptake of carrier-mediated influx substrates. □, actively effluxed substrates (Smith, 2003).
Discussion
In this study, we have reinvestigated the role of plasma protein binding on brain uptake of four drugs that bind selectively to Sudlow site I, site II, or both Sudlow sites of albumin. The results find no significant deviation from the modified Kety-Crone-Renkin equation. Measured brain drug uptake Kin, with the brain perfusion technique, matched that predicted by the modified Kety-Crone-Renkin model at different protein levels and also with protein from different species. No evidence was found for enhanced in vivo dissociation. The results suggest that accurate predictions can be made with the modified Kety-Crone-Renkin model for drugs that bind reversibly to albumin at Sudlow sites I and II.
The in situ rat brain perfusion technique allows clear determination of drug uptake rates into brain in the presence and absence of protein. For the four agents used in this study, brain uptake of free drug was moderately rapid reaching brain-to-perfusion ratios of 0.08 to 0.68 ml/g in 30 s. These brain uptake ratios (i.e., spaces) exceeded vascular volume by 10–75-fold, thus providing a strong signal against which to determine the effect of plasma protein binding. However, brain uptake was not so rapid that it depended critically on F. Single-pass free drug extractions (Eu) were less than 25% for all compounds studied, with the exception of one. With the Kety-Crone-Renkin model, with Eu < 25%, brain Kin would be expected to change <13% with F changes of 2-fold or less. Thus, brain uptake for the tested compounds is essentially independent of small changes in F. Consistent with this, blood-brain barrier PSu values matched at two different perfusion rates (normal and 4× elevated), with an average PSu difference of ≤16%, which was not statistically significant (p > 0.05). Brain capillary diameter has been reported to change only ∼20% over a broad range of blood flow as produced by hypercapnia and hypocapnia (Duelli and Kuschinsky, 1993).
The role of plasma protein binding at Sudlow site I of albumin has not been closely investigated previously. With the brain uptake index technique, Urien et al. (1989) noted greater brain uptake of warfarin in the presence of human serum albumin than could be accounted for using the Kety-Crone-Renkin equation. Warfarin is a defining substrate for Sudlow site I, at which it binds rapidly and reversibly (Loun and Hage, 1994). With the brain perfusion technique and albumin from different species (human, bovine, and rat), we found no statistically significant deviation from predicted Kin using three different albumin concentrations (∼1, 10, and 100% of normal albumin levels for the rat). Warfarin binds highly to plasma proteins, with a free fraction of only ∼1% at physiological protein concentrations. Brain warfarin uptake Kin was consistent with perfusion fluid in vitro fu at all three albumin concentrations. Warfarin fu calculated from in vivo brain uptake experiments did not differ from matching values determined in vitro. The addition of albumin to perfusion fluid did not significantly modify brain Vv. Vv was determined simultaneously with Kin in the presence of albumin to maximize accuracy of the vascular correction. Mean Vv in these experiments matched that reported in the literature (Takasato et al., 1984), suggesting an intact blood-brain barrier.

Measured Kin ratio (presence/absence of plasma protein) plotted versus measured fu of drug in the presence of albumin. A, Sudlow site I ligand warfarin in the presence of albumin from three different species. B, Sudlow site II ligand ibuprofen in the presence of albumin from three different species. C, mixed binding site ligands—valproic acid and tolbutamide—in the presence of albumin from different species and rat serum. Values equal means ± S.E.M. for n = 3 to 9. Kin ratios are for the pooled cerebral hemisphere (regions as noted in legend for Fig. 1). Dotted lines are the lines of identity (slope = 1.0; intercept = 0).
Serum albumins from mammalian species differ in amino acid sequence and protein structure. Consistent with this, binding-site affinity and selectivity vary from species to species. Site I on human serum albumin consists of two overlapping subregions, one for warfarin and one for azapropazone (Fehske et al., 1982). These subregions overlap in rat and are separate in bovine and absent in dog (Kosa et al., 1997). We observed differences in binding affinity of warfarin with rat, bovine, and human albumin that correlated with reported KD constants (Seller et al., 1977). Because of these differences, albumins from separate species should not be considered interchangeable. Nonetheless, no statistically significant differences were observed between predicted and measured up-take Kin with any of the three albumins.
A number of studies have been published suggesting enhanced dissociation of ligands that bind fairly selectively to Sudlow site II of albumin (e.g., benzodiazepines and tryptophan) (Jones et al., 1988; Fenerty and Lindup, 1989; Lin and Lin, 1990; Pardridge, 1998; Tanaka and Mizojiri, 1999). In all instances, greater uptake was reported than could be accounted for by the Kety-Crone-Renkin equation. In our study, the site II-selective agent ibuprofen showed no significant evidence for enhanced dissociation. Measured and predicted Kin values agreed over a 100-fold range in perfusion fluid albumin concentrations using albumin from three species. Furthermore, no deviation was detected using two additional ligands (tolbutamide and valproate) that have been reported to bind at both sites (Sudlow I and II) (Sjoholm et al., 1979). The latter statement holds both for perfusion with purified albumin, as well as whole-rat serum collected less than 1 h before perfusion.
At least one other study reports findings in agreement with ours. Dubey et al. (1989) used microdialysis to study the distribution of diazepam in brain interstitial fluid and plasma. They found no evidence for an elevated brain interstitial fluid concentration of diazepam resulting from enhanced dissociation. Diazepam is a high-extraction drug with a transfer rate that depends critically on F (Takasato et al., 1984; Smith and Nagura, 2001). Therefore, in this study, we used ibuprofen, which has a lower E and has been shown to bind to Sudlow site II of albumin for all three species, unlike diazepam (Kosa et al., 1997). Furthermore, we used in situ brain perfusion and preperfused the brain for 30 s to wash out endogenous blood elements and obtain steady-state conditions prior to uptake determination. We agree with Dubey et al. (1989) that the discrepancies with the prior reports may relate to mixing of the injectate bolus with circulating blood as the bolus passes through the brain vascular system. If blood-brain barrier PSu is underestimated from protein-free saline injectate due to mixing and binding to endogenous blood constituents, subsequent measurements in the presence of protein would give the appearance of exceeding that expected based upon the Kety-Crone-Renkin equation. Adkison and Shen (1996) reported on brain uptake of valproic acid in the presence of albumin as determined by brain perfusion. Their values, when plugged into the Kety-Crone-Renkin model, are consistent with ours and suggest that uptake can be explained by perfusion fluid fu without the need to hypothesize enhanced dissociation.
Because in situ brain perfusion with saline may wash out critical vascular or endothelial elements that are essential for enhanced dissociation, brain uptake for two drugs, valproate and tolbutamide, was determined in the presence of whole-rat serum. Whole-rat serum offers the advantage of including all of the binding proteins present in serum and also avoids conformational changes due to protein purification. The obtained Kin values agree with that predicted by the Kety-Crone-Renkin equation based upon input fu,PSu, and F. The findings suggest that the prior saline results are not artifacts of the perfusion model system. The in situ rat brain perfusion technique offers the advantage that it can accurately determine blood-brain barrier Kin and PSu over a broad range at defined F.
The results highlight the importance of plasma protein binding in determinations of brain uptake and blood-brain barrier permeability. If correction is not made for in vivo drug fu, estimated blood-brain barrier PS values can greatly underestimate true values by 1 to 3 orders of magnitude (Fig. 7). Good agreement was obtained between blood-brain barrier PSu determined from in situ rat brain perfusion and that predicted based upon the log Doct 7.4. Most log PSu values for the tested drugs fell near (±1 log unit) that predicted from the permeability reference markers based upon the measured drug log Doct 7.4 (Smith, 2003). However, in the absence of protein binding correction, the derived LogPS values in the presence of plasma protein fell 1 to 3 log units lower, indicating the importance of the plasma protein binding correction in brain uptake measurements.
Finally, the linear relation between Kin ratio and fu shows that protein binding for the studied compounds is restrictive. For such compounds, blood-brain barrier PSu < F, and thus blood-brain barrier Kin ≈ fu × PSu. We have previously determined that nonrestrictive plasma protein binding requires PSu/F > 1 and becomes more important as the ratio is enhanced. For ibuprofen, PSu/F ≈ 1 at normal F; thus, the compound is right on the edge between restrictive and nonrestrictive. The modified Kety-Crone-Renkin equation used in this study assumes that binding and dissociation rates of drug from albumin (t1/2 <150 ms) are much greater than the transit time of brain capillaries (400–1000 ms). Some evidence exists to support this. For example, the t1/2 for tryptophan dissociation from albumin is 50 to 100 ms (Yang and Hage, 1997; Talbert et al., 2002). However, for compounds with PSu < F, this assumption is not important. It would only become critical for drugs with PSu ≫ F, such that significant redistribution occurs as blood passes through the cerebral capillary.
Footnotes
-
This work was supported in part by Grant NS052484 from the National Institutes of Health (to Q.R.S.).
-
doi:10.1124/jpet.105.097402.
-
ABBREVIATIONS: PSu, permeability-surface area product; LSC, liquid scintillation counting.
- Received October 18, 2005.
- Accepted January 9, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}