


Abstract
In humans and rats, a synergistic blood pressure reduction was observed when the fibrate gemcabene (CI-1027) was coadministered with the angiotensin-converting enzyme inhibitor quinapril. In a quinapril (3 mg/kg) pharmacokinetic rat study, there was a 40% decrease in urinary excretion and a 53% increase in plasma area under the curve from 0 to 24 h of the active metabolite quinaprilat when coadministered with gemcabene (30 mg/kg). This observation revealed a possible transporter-mediated drug-drug interaction (DDI) between gemcabene and quinapril. This led to a series of studies investigating the underlying clearance mechanisms associated with these compounds intended to elucidate renal transporter interactions between quinapril and gemcabene. In vitro transporter studies using human embryonic kidney 293 cells transfected with human or rat organic anion transporter 3 (hOAT3, rOat3) revealed that quinaprilat is a substrate in both species, with a Km value of 13.4 μM for hOAT3. Subsequent studies discovered that gemcabene inhibited quinaprilat uptake by hOAT3 and rOat3 at IC50 values of 35 and 48 μM, respectively. Moreover, gemcabene acylglucuronide, the major metabolite of gemcabene glucuronidation, also inhibited hOAT3- and rOat3-mediated uptake of quinaprilat at IC50 values of 197 and 133 μM, respectively. High plasma concentrations of gemcabene (>100 μM) achieved in humans and rats upon oral dosing corroborate with gemcabene inhibition of renal OAT3-mediated secretion of quinaprilat in vitro. This investigation established that a DDI between gemcabene and quinapril involving inhibition of renal transporters and subsequent elevation in plasma concentrations of quinaprilat is responsible for the apparent synergistic blood pressure reduction observed with these compounds.
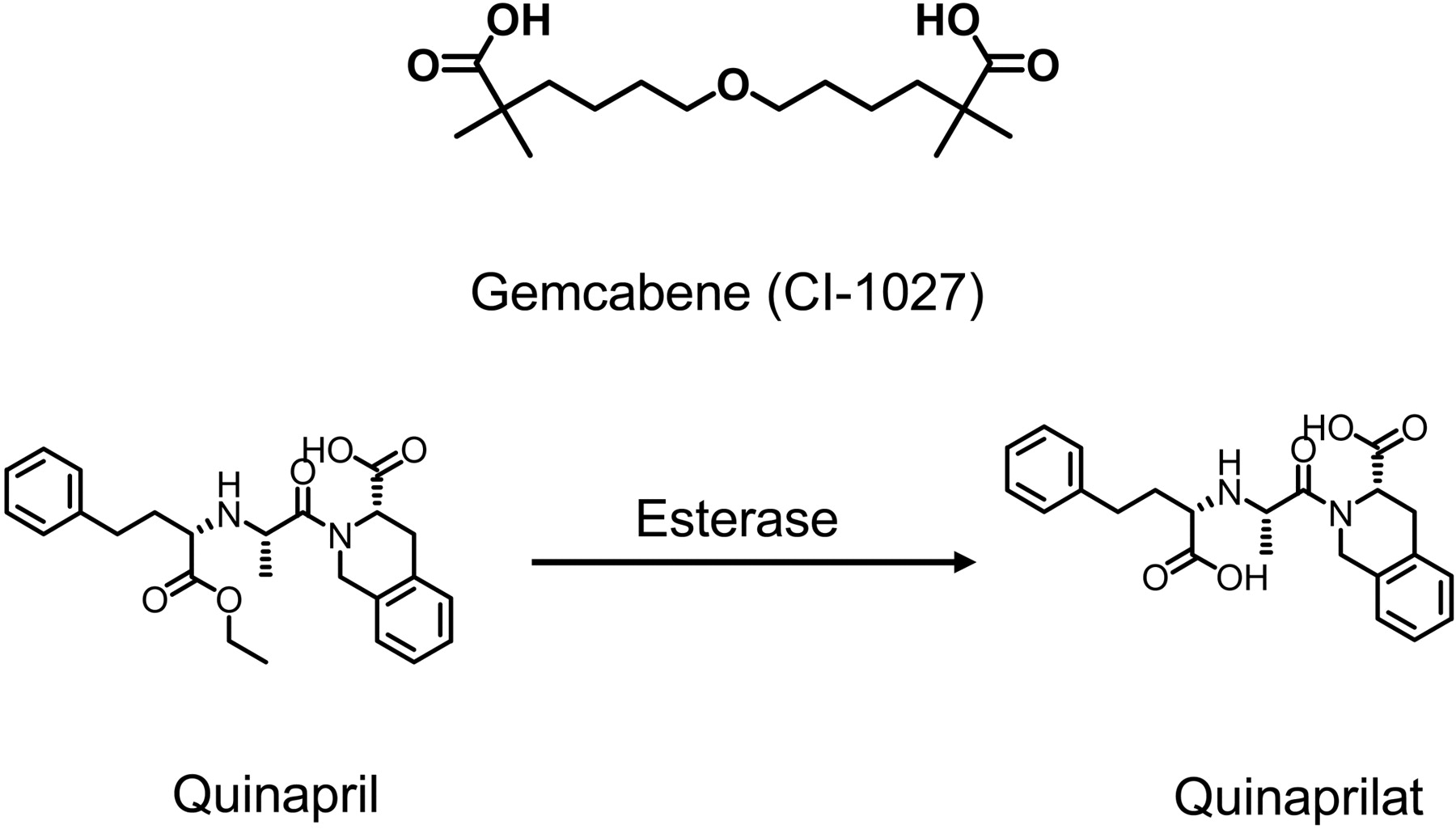
The dialkyl ether dicarboxylic acid gemcabene (CI-1027; Fig. 1) is a representative of a class of lipid-regulating compounds characterized by the presence of a gem-dimethyl carboxylate moiety. Gemcabene advanced into early clinical development as a therapeutic used to increase high-density lipoprotein while lowering serum triglyceride in dyslipidemic patients (Bays and Stein, 2003). Gemcabene exhibited near complete absorption in human and rat and achieved high plasma exposure at therapeutic doses. In rat, 30 mg/kg gemcabene reached a mean Cmax value of 376 μM and decreased low-density lipoprotein and the very-low-density lipoprotein cholesterol by 40 and 75%, respectively (Pfizer, internal data). In humans, low-density lipoprotein was reduced by 25% at a Cmax value of 884 μM with 900 mg once daily administration of gemcabene (Pfizer, internal data). It has been shown clinically that gemcabene is cleared almost exclusively by glucuronidation followed by renal elimination, with 78% of the radioactive dose (61% glucuronide and 17% unchanged parent) recovered in human urine (Pfizer, internal data). UDP-glucuronosyltransferase 2B7 is the major enzyme responsible for gemcabene glucuronidation, with the major metabolite being an acylglucuronide (Bauman et al., 2005). Therefore, the potential for a drug-drug interaction (DDI) between gemcabene with compounds metabolized by cytochromes P450 is very low.
Quinapril (Fig. 1) is an angiotensin-converting enzyme (ACE) inhibitor, used for the treatment of hypertension and congestive heart failure (DeFelice, 1987; Wadworth and Brogden, 1991). Quinapril is a prodrug that is enzymatically de-esterized in vivo to quinaprilat, the more potent and pharmacologically active diacid form (Fig. 1) (Kaplan et al., 1989; Wadworth and Brogden, 1991). Metabolism of quinaprilat is low, and it is excreted primarily unchanged in urine (Olson et al., 1989). In humans, renal clearance of quinaprilat averages 66 ml/min. Quinaprilat is 97% bound to plasma proteins in humans, which indicates that clearance driven by glomerular filtration of quinaprilat accounts for only a small fraction of the total renal clearance of quinaprilat. Active tubular secretion is the predominant mediator of its renal clearance (Olson et al., 1989). Previous studies in isolated perfused rat kidney have suggested that both quinapril and quinaprilat are actively secreted in proximal tubular cells of the kidney (Kugler et al., 1995). Still, the precise mechanistic basis for renal tubular secretion of quinapril and quinaprilat remains unknown.

Chemical structures of gemcabene (CI-1027) and the esterase-modulated conversion of quinapril to quinaprilat.
Tubular secretion of xenobiotics, a transporter-mediated pathway, is a key function of the renal proximal tubule (Wright and Dantzler, 2004). Organic anion transporters (OATs) and organic cation transporters are two major drug transporter families highly expressed in kidney (Dresser et al., 2001). Renal OAT1 and OAT3, located on the basolateral membranes of proximal tubule cells, have been shown to play a central role in the renal secretion of a wide range of anionic xenobiotics (Uwai et al., 1998; Race et al., 1999; Tojo et al., 1999; Cha et al., 2001; Hasegawa et al., 2002). Importantly, involvement of renal OATs in DDIs has been widely reported. For example, it was reported that probenecid increased the half-life and the AUC of the serum concentration of methotrexate through inhibition of OAT1-mediated renal secretion (Uwai et al., 2000). Tahara et al. (2006) also demonstrated that an inhibitory effect of probenecid on hOAT3-mediated fexofenadine transport resulted in a renal DDI between these two compounds. In addition, Nakagomi-Hagihara et al. (2007) reported that inhibition of hOAT3-mediated pravastatin transport by gemfibrozil and its metabolites led to a decrease in the renal clearance of pravastatin in a clinical setting. Consequently, renal organic anion transporter-mediated DDIs have garnered growing attention in understanding the subsequent ramifications relative to the discovery and development of new therapeutics.
Combination therapies with fibrates and ACE inhibitors are widely used in clinical practice. Therefore, during the course of clinical development, gemcabene was examined alone and in combination with quinapril for blood pressure effects in patients with essential hypertension. It was curious that when combined with quinapril, gemcabene evoked an apparent synergistic lowering of blood pressure compared with quinapril alone (Pfizer, internal data). Thus, mechanistic studies for investigating the potential for a renal transporter-mediated DDI between gemcabene along with its major metabolite, gemcabene acylglucuronide, with quinapril were undertaken using in vivo rat studies and in vitro transporter assays. The objectives of these studies were to 1) evaluate the potential for gemcabene to affect pharmacokinetics of quinaprilat in rat; 2) define the interactions of quinaprilat with rat and human major renal OATs in vitro; and 3) assess the impact of gemcabene and gemcabene acylglucuronide on the renal transport of quinaprilat in vitro.
Materials and Methods
Materials. Quinapril hydrochloride and carboxylmethylcellulose (low viscosity) were purchased from Sigma-Aldrich (St. Louis, MO). Gemcabene, gemcabene acylglucuronide, and quinaprilat were synthesized by Pfizer Global Research & Development (Ann Arbor, MI). Solvents used for analysis were of analytical or high-performance liquid chromatography grade (Thermo Fisher Scientific, Waltham, MA). The human OAT3- and rat Oat3-transfected HEK293 cell line were acquired from Dr. Kathleen Giacomini (University of California at San Francisco, San Francisco, CA), and the human OAT1-transfected HEK293 cell line was constructed internally (Pfizer Inc., Groton, CT).
In Vivo Studies in Rats. The Pfizer Institutional Animal Care and Use Committee reviewed and approved the animal use in these studies. The animal care and use program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International. During the study, the animal housing and husbandry were performed in accordance with the provisions of the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996).
Pharmacokinetic Studies in Rats. Jugular vein cannulated male spontaneous hypertensive rats (SHRs) (230–250 g) purchased from Charles River Laboratories, Inc. (Wilmington, MA) were used for these studies. All animals were fasted overnight before dosing, whereas access to water was provided. Animals were fed after collection of the 4-h blood samples. Quinapril (3 mg/kg) and gemcabene were administrated orally in 3% carboxylmethylcellulose and 0.1% Tween 80. Serial plasma samples were collected before dosing and 0.033, 0.15, 0.5, 1, 2, 4, 6, 8, and 24 h after dose. Plasma samples were generated from whole blood by centrifugation at 3000 rpm for 10 min at 4°C and stored at -80°C, pending analysis by LC-MS/MS. Urine samples were collected over intervals of 0 to 4, 4 to 8, 8 to 12, and 12 to 24 h and stored frozen before analysis.
In Vivo Pharmacological Study in SHRs. Male SHRs at 11 to 12 weeks of age underwent surgeries to implant radiotelemetry pressure transmitters (PA11-C40; Data Sciences International, Roseville, MN), followed by a 7-day recovery period before compound administration. Animals were assigned to the following four groups: vehicle, quinapril (3 mg/kg), gemcabene (loading dose 30 mg/kg, reduced to 10 mg/kg at day 3), and gemcabene + quinapril (gemcabene was dosed at the same dosing scheme with monodose; quinapril was dosed at 3 mg/kg for the whole experiment period). All dosing groups were formulated using the vehicle containing 3% carboxyl methocellulose + 0.1% Tween 80. All compounds were administered once daily via oral gavage (once a day dosing at 8:00 AM; dosing volume, 1 ml/rat; n = 8). Blood pressure and heart rate were continuously monitored throughout the experiment using a Dataquest ART computerized data acquisition and analysis system (Data Science International, St. Paul, MN). Raw data were collected every 10 min and averaged over 60-min periods. Plasma samples were collected ∼5 h after daily administration at 5 days and 3.5 weeks after the initiation of treatments for measuring drug exposure levels.
In Vitro Transporter Uptake Assays. The detailed methods have been described previously (Feng et al., 2008). In brief, HEK293 cells were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (nondialyzed), 1% penicillin/streptomycin, and 100 μg/ml phleomycin (Zeocin; Invitrogen, Carlsbad, CA). HEK293 cells transfected with rOat3, hOAT1, or hOAT3 were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and 50 μg/ml hygromycin. The assays were carried out in 24-well poly-d-lysine-coated plates (Biocoat; BD Biosciences, Franklin Lakes, NJ), with nearly confluent cells seeded 48 h before each experiment. Immediately before the experiment, the cells were washed twice with 1 ml of DPBS buffer at room temperature and then incubated with 100 μl of DPBS buffer containing the substrate at room temperature. After 5 min, the cellular uptake was terminated by washing the cells three times with 1 ml of ice-cold DPBS and then lysed directly on the plate in the presence of methanol/water (70:30) containing the analytical internal standard. The samples were processed and analyzed as described below.
Inhibition of Uptake and IC50 Determination. Incubations were performed in 24-well poly-d-lysine-coated plates using quinaprilat as a substrate. Individual serial dilutions of gemcabene and its metabolite, gemcabene acylglucuronide, at concentrations ranging from 0 to 1000 μM were prepared and individually administered to the cells containing 5 μM quinaprilat. Mean and S.D. of substrate uptake rate were calculated for each series of measurements (n = 3). These values were then converted to percentage of uptake relative to the substrate uptake without inhibitor, with the substrate uptake without inhibitor standard representing 100%. IC50 values were estimated from semilogarithmic plots of the inhibitor concentrations versus percentage of net uptake relative to the uninhibited control using Prism software (GraphPad Software Inc., San Diego, CA).
Sample Preparation for Concentration Determination in Plasma and Cell Lysate. For in vivo plasma and urine sample analysis, calibration standards were first prepared by creating a stock solution that consisted of fortifying control blank plasma or blank urine with the appropriate amount of quinapril, quinaprilat, gemcabene, and gemcabene acylglucuronide. Serial dilutions were conducted to give calibration standard concentrations ranging from 0.000305 to 5.0 μM. For quinapril and quinaprilat analysis, the standards were prepared by fortifying quinapril and quinaprilat simultaneously. For gemcabene and gemcabene acylglucuronide plasma and urine sample analysis, the calibration standards were prepared separately for each analyte. Given the high plasma concentrations of gemcabene in the pharmacokinetic studies, the samples were diluted 100-fold before analysis. For gemcabene acylglucuronide analysis in urine samples, the samples were diluted 10-fold before analysis. Aliquots of 15 μl of either the calibration standard or unknown plasma or urine samples were transferred to a 96-well plate. The internal standard working solution at a volume of 85 μl (1 μM tolbutamide in acetonitrile) was then added to all samples to precipitate the plasma proteins. The 96-well plate was sealed with the sealing mat and subjected to centrifugation at 4000 rpm for 5 min. A 90-μl aliquot of supernatant was transferred into a labeled 96-well plate using a Quadra 96-320 system (Tomtec, Hamden, CT). A 5-μl sample extract was then injected onto the LC-MS/MS system for analysis.
For in vitro samples analysis, after the cells were lysed with 300-μl volume of 70% methanol in water containing the analytical internal standard, the assay plate was centrifuged at 4000 rpm for 5 min. An aliquot of 80 μl of supernatant was transferred to a 96-well plate. The plate was sealed, and sample extracts were drawn for analysis by LC-MS/MS. The preparation of standard control samples used for establishing the calibration curve was prepared with a fortified mixture of methanol and incubation medium at ratio of 70:30.
High-Performance Liquid Chromatography-Tandem Mass Spectrometry Analysis. An AD20 quaternary micro pump (Shimadzu, Columbia, MD) and an AQUASIL C18 (50 × 2.1 mm; 3-μm particle size) high-performance liquid chromatography column (Thermo Fisher Scientific) were used for the chromatographic separation. The autosampler was an HTS-PAL from LEAP Technologies (Carrboro, NC). The mobile phase consisted of 0.1% formic acid water/acetonitrile (A:B) blend. The chromatographic gradient conditions required maintaining 90% A for 0.5 min isocratically and ramping to 90% B in 2 min. This was followed by maintaining the mobile phase at 90% B for 0.5 min before adjusting to 10% B over 0.2 min and finally maintaining at 10% B another 1.3 min before the next injection. The total run time was 4.5 min. The flow rate was kept at 0.4 ml/min for the entire gradient chromatographic separation.
An API 4000 triple quadrupole mass spectrometer (MDS Sciex, Concord, ON, Canada) with a turbo ion spray interface operated in positive ionization mode was used for the multiple reaction monitoring LC-MS/MS analyses. The mass spectrometric conditions were optimized for quinapril, quinaprilat, gemcabene, gemcabene acylglucuronide, and tolbutamide (used as analytical internal standard). The following precursor→product ion transitions were used for multiple reaction monitoring: quinapril, m/z 439→234; quinaprilat, m/z 411→206; gemcabene, 303→143, gemcabene acylglucuronide, 479→303; and tolbutamide, m/z 272→155.
Pharmacokinetic Calculations. The pharmacokinetics was characterized by peak concentration in plasma (Cmax), time to peak concentration (tmax), elimination half-life (t1/2), and AUC from 0 to 24 h [AUC(0–24 h)]. Cmax and tmax values were taken directly from the original data. The terminal log-linear phase of the plasma concentration-time curve was identified visually for each curve. The elimination rate constant (ke) was determined by a linear regression analysis with the last three to five points on the plot of the natural logarithm of the plasma concentration-time curve. The t1/2 was calculated by the equation t1/2 = ln2/ke. The AUC(0–24 h) values were calculated by the linear trapezoidal rule. The renal clearance (CLrenal) of quinaprilat was calculated as CLrenal = Ae(0–24 h)/AUC(0–24), in which Ae(0–24 h) is the amount of quinaprilat excreted into the urine within 24 h.
Km and Vmax Determination. To determine the Km value in the hOAT3 substrate assay, seven different concentrations of quinaprilat were used (0, 0.25, 1, 4, 15.6, 62.5, 250, and 1000 μM). After a 5-min incubation (optimal time point for initial rate) at room temperature, the reaction was terminated and intracellular concentration of quinaprilat was determined as described above. Substrate uptake in HEK293 control cells was subtracted from the uptake in the transfected cells. Uptake velocity versus substrate concentration was analyzed by Enzyme Kinetics in Prism software (GraphPad Software Inc.) using the Michaelis-Menten equation.
Results
Quinapril and Gemcabene on Blood Pressure Reduction in SHRs. The systolic blood pressure after administration of quinapril (3 mg/kg) and gemcabene (30 mg/kg) alone and in combination in SHRs is shown in Fig. 2. Quinapril alone lowered blood pressure 14 mm Hg (P < 0.05) relative to baseline after 6 days of dosing, whereas gemcabene alone had no effect. Interestingly, the combination drove a dramatic ∼40 mm Hg reduction in blood pressure. It is important to note that plasma concentrations of quinaprilat in the combination group were nearly 5-fold greater than that of the quinapril monotherapy group (Fig. 3).
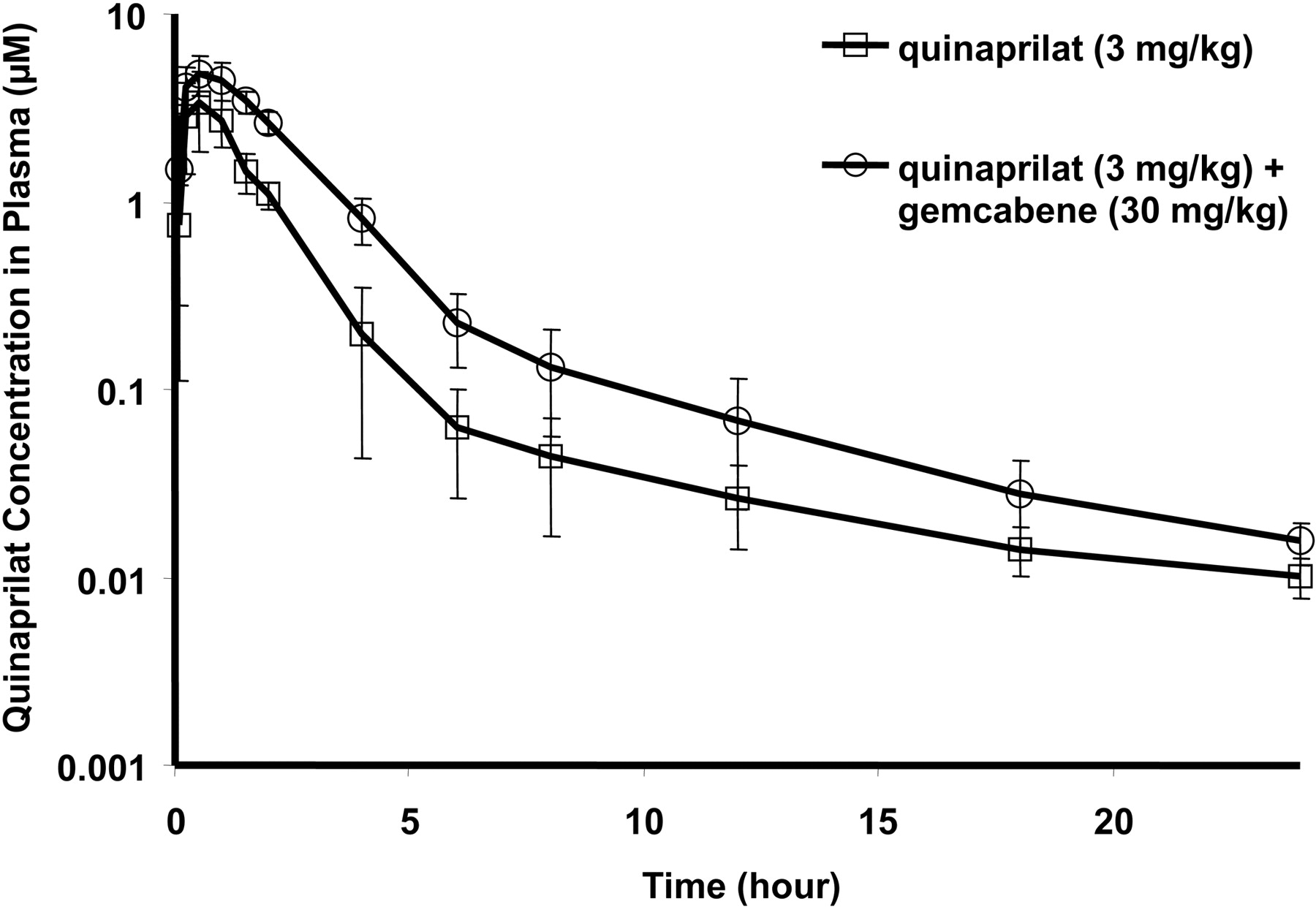
Pharmacokinetics of Quinapril with Coadministration of Gemcabene. Mean quinaprilat plasma concentrations over time with quinapril (3 mg/kg) alone and in combination with gemcabene (30 mg/kg) in Sprague-Dawley rats are shown in Fig. 4a. Total plasma quinaprilat AUC(0–24 h) was increased 53% from 3.02 to 4.62 μM · h (Fig. 4b), whereas the total amount of quinaprilat excreted in urine Ae(0–24 h) was decreased 40% from 38.4 to 23.1 μg (Fig. 4c). Consequently, renal clearance of quinaprilat in monodose and codose groups was 31.0 and 12.2 ml/h, respectively, with CL-renal equivalent to Ae(0–24 h)/AUC(0–24 h). As expected, plasma and urine concentrations of quinapril were below the limit of quantification (0.00305 μM) for all the time points sampled, due to the rapid conversion of quinapril into quinaprilat in rat. Mean plasma concentration of gemcabene and gemcabene acylglucuronide over time after coadministration of 3 mg/kg quinapril and 30 mg/kg gemcabene in SHRs is shown in Fig. 5. As expected, plasma concentrations of gemcabene parent were much higher than the acylglucuronide metabolite.

Systolic blood pressure after administration of 30 mg/kg gemcabene followed by 10 mg/kg gemcabene beginning on day 3, quinapril alone at 3 mg/kg, and concomitantly both at the same dosing scheme of each individual dose. The experiment was conducted with eight rats in each dosing group. The blood pressure reduction in gemcabene and quinapril codose group compared with quinapril monodose group was significantly different from day 1 to day 5 (*, P < 0.05, Student's t test).

Quinaprilat plasma concentration after administration of quinapril and coadministration of quinapril and gemcabene in SHRs. The data were obtained at 5 h after dose on the fifth day of administration. The experiment was conducted with eight animals per each dosing group (*, P < 0.05, Student's t test).
Quinaprilat Interacts with Renal Organic Anion Transporters. The uptake of quinaprilat in two major human renal organic anion uptake transporters, hOAT1 and hOAT3, was examined in hOAT1- and hOAT3-transfected HEK293 cells. These data indicate that the uptake of quinaprilat in hOAT1-HEK and hOAT3-HEK was significantly increased compared with that of the wild-type HEK293 cells (Fig. 6a). Moreover, the uptake of quinaprilat in hOAT3-HEK was markedly higher that that in hOAT1-HEK cells, implying that hOAT3 transports quinaprilat more effectively than hOAT1. Uptake kinetics of quinaprilat in hOAT3-HEK were further characterized (Fig. 6b), and the Km value was determined to be 13.4 ± 4.1 μM. Quinaprilat was also evaluated in a rat Oat3 transporter assay, a rat ortholog of human OAT3. As anticipated, rat Oat3 was able to effectively transport quinaprilat as well.
Gemcabene and Gemcabene Acylglucuronide Inhibition of hOAT3- and rOat3-Mediated Quinaprilat Uptake. Human OAT3 and rat Oat3 inhibition assays were used to assess the inhibition potency of gemcabene and gemcabene acylglucuronide on quinaprilat uptake. The IC50 values for gemcabene inhibition of quinaprilat uptake by hOAT3 and rOat3 were 35 and 48 μM, respectively (Fig. 7, a and b). Whereas the IC50 values for gemcabene acylglucuronide inhibition of quinaprilat uptake mediated by hOAT3 and rOat3 were 197 and 133 μM, respectively (Fig. 7, c and d).
Discussion
The compound gemcabene, a fibrate, progressed into clinical development for the treatment of dyslipidemia and associated cardiovascular risk factors. A study in essential hypertension patients discovered that although gemcabene monotherapy had no effect on blood pressure (BP), dosing in combination with the ACE inhibitor quinapril evoked a significant BP reduction relative to quinapril alone (14.1 versus 6.6 mm Hg; P < 0.05; Pfizer internal data). Unfortunately, plasma concentration of the active metabolite quinaprilat, which mediates the BP effect, was not determined in the study. Subsequent preclinical studies using the SHR replicated this apparent synergistic BP reduction (Fig. 2). In these SHR studies, plasma quinaprilat levels were evaluated and found to be significantly elevated when codosed with gemcabene. These novel clinical and preclinical observations along with the understanding that quinaprilat is predominantly cleared via the kidney led to the hypothesis that gemcabene mediates a DDI through inhibition of renal clearance of quinaprilat. To test this hypothesis, a comprehensive characterization of the effect of gemcabene on quinaprilat plasma concentration and renal clearance was undertaken in a rat pharmacokinetic (PK) study.
Previous PK studies demonstrated that gemcabene is nearly 100% bioavailable after oral administration, with a long half-life (23 h) in rat (Pfizer, internal data). Quinapril, an alkyl ester prodrug, is converted into its pharmacologically active diacid form, quinaprilat, which is present as the major metabolite in plasma and urine in both rat and human (Olson et al., 1989). Therefore, in vivo pharmacokinetic investigations as well as in vitro transporter studies were focused on characterizing the disposition of the active metabolite quinaprilat. In normal rats, excretion of quinaprilat into urine was found to be significantly decreased (∼40%) (Fig. 4c), whereas plasma concentration AUC(0–24 h) increased (53%) (Fig. 4b) with gemcabene coadministration. This reflects a 61% decrease in the renal clearance of quinaprilat when dosed with gemcabene. This finding was replicated in SHRs in which plasma concentrations of quinaprilat were nearly 5-fold higher with gemcabene codosing and aligned with a greater magnitude BP reduction (Figs. 2 and 3). These data reveal the apparent synergistic BP reduction observed in SHRs is in fact due to elevated plasma concentrations of quinaprilat caused by decreased renal clearance. We next turned our investigations toward elucidation of the mechanism underlying this DDI.

a, mean plasma concentration-time profiles of quinaprilat after administration of 3 mg/kg quinapril alone (n = 4) and after 3 mg/kg quinapril with 30 mg/kg gemcabene concomitantly in SHRs (n = 3). b, plasma AUC(0–24 h) of quinaprilat after administration of 3 mg/kg quinapril alone (n = 4) and coadministration of 3 mg/kg quinapril and 30 mg/kg gemcabene concomitantly (n = 3). c, total amount of quinaprilat excreted in urine Ae(0–24 h) was 38.4 μg with quinapril monotherapy and 23.1 μg in the codose group a. *, p < 0.05 (t test), significant difference in quinaprilat concentration obtained in the codose group compared with the monodose group.

Mean plasma concentration-time profiles for gemcabene and gemcabene acylglucuronide after coadministration of 3 mg/kg quinapril and 30 mg/kg gemcabene in spontaneously hypertensive rats.
Because quinaprilat is known to be cleared by the kidney, transporters involved in active renal secretion were considered as potential culprits of gemcabene-mediated interference with quinaprilat clearance. Based on the understanding that the organic anionic secretory system is the primary mechanism by which quinapril and quinaprilat are transported into and across the renal proximal cells (Kugler et al., 1996), quinaprilat uptake by the major human renal organic anion transporters hOAT1 and hOAT3 was determined. Quinaprilat uptake in hOAT1- and hOAT3-transfected HEK293 cells compared with wild-type cells was increased 2- and 25-fold, respectively, implying that hOAT3 plays a major role in the renal active secretion of quinaprilat (Fig. 6a). Further kinetic studies defined the apparent hOAT3 affinity (Km) for quinaprilat as 13.4 μM (Fig. 6b). These data point to hOAT3 as the major renal uptake transporter responsible for active uptake of quinaprilat into renal proximal tubule cells. Moreover, these data corroborate in vivo study data in which renal clearance of quinaprilat is much higher than the glomerular filtration rate. Quinaprilat was next evaluated in rat Oat3 assay to assess potential species differences for quinaprilat uptake. As with hOAT3, quinaprilat was found to be a substrate for rOat3, which was expected given that the rat ortholog is highly homologous to hOAT3 (Burckhardt et al., 2003). Therefore, rOat3 transport accounts for active renal secretion of quinaprilat in rat. The potential renal transporter-mediated DDI between quinaprilat with gemcabene and gemcabene acylglucuronide was investigated in hOAT3 and rOat3 inhibition assays. Gemcabene inhibited quinaprilat uptake mediated by hOAT3 and rOat3, with IC50 values of 35 and 48 μM, respectively (Fig. 7, a and b). Gemcabene acylglucuronide also inhibited quinaprilat uptake mediated by hOAT3 and rOat3, albeit in a somewhat less potent manner, with IC50 values of 197 and 133 μM, respectively (Fig. 7, c and d). Rat PK data with therapeutic doses were able to achieve plasma concentrations of gemcabene and gemcabene acylglucuronide in the range of 200 and 1 μM, respectively (Fig. 5). Although the plasma concentration of gemcabene corresponds to its in vitro potency, the gemcabene acylglucuronide plasma concentration is much lower than its in vitro IC50 value. Consequently, gemcabene is probably the predominant molecule involved in inhibiting rOat3-mediated quinaprilat transport in rat, responsible for the decreased renal clearance and the increased plasma concentrations of quinaprilat. An additional rat pharmacokinetic study was conducted in which gemcabene was dosed 1 h before quinapril to ensure the plasma exposure of gemcabene reached Cmax when quinapril was administrated. With the long t½ of gemcabene, this dosing scheme provided a more consistent gemcabene plasma exposure to the basolateral side of proximal tubule cells during the whole course quinapril was present in the systemic circulation. The plasma AUC(0–24 h) was increased 109% from 6.3 μM · h with quinapril monotherapy to 13.1 μM · h with the combination (Fig. 8). A significant elevation of plasma concentration of quinaprilat was present as early as 5 min after dose. This study further supports the contention that gemcabene modulates quinaprilat plasma concentration.

a, uptake of 5 μM quinaprilat in wild-type HEK293, hOAT1-HEK, and hOAT3-HEK cells. Data are mean values ± S.D. for three wells of cells in 24-well plates. b, kinetic studies of quinaprilat transport mediated by hOAT3. The rate of quinaprilat uptake by hOAT3 at various concentrations was measured. hOAT3-mediated transport was obtained by subtracting the transport velocity in HEK293 control cells from that in hOAT3-HEK cells. Data are mean ± S.D. (n = 3). Kinetic parameters were determined by fitting the data to the Michaelis-Menten equation using Prism software. The apparent Km value for quinaprilat was 13.4 ± 4.1 μM. *, P < 0.01 (t test), significant differences of the hOAT3-mediated and hOAT1-mediated quinaprilat uptake compared with the uptake in HEK293 cells.

Inhibition of hOAT3- (a) and rOat3 (b)-mediated quinaprilat uptake by gemcabene and inhibition of hOAT3- (c) and rOat3 (d)-mediated quinaprilat uptake by gemcabene acylglucuronide. The IC50 value was determined by measuring the inhibition of 5 μM quinaprilat uptake mediated by hOAT3-HEK and rOat3-HEK with varying concentrations of gemcabene or gemcabene acylglucuronide. Estimated IC50 values were 35 ± 4.1 and 48 ± 6.1 μM for hOAT3 (a) and rOat3 (b), respectively, for gemcabene inhibition and 197 ± 26.6 and 133 ± 23.4 μM for hOAT3 (c) and rOat3 (d), respectively, for gemcabene acylglucuronide inhibition. Data are mean values ± S.D. for three wells of cells in 24-well plates.
Because in vitro transporter studies suggest the species difference in the renal transporter interaction with quinaprilat modulated by gemcabene was minimal, and the therapeutic plasma exposure of gemcabene is well above the concentration necessary to inhibit OAT update, one could expect that a similar DDI between gemcabene and quinapril would exist in humans. In the previously described clinical study, a significantly greater reduction in BP occurred when gemcabene was coadministered with quinapril. The maximal plasma concentration of gemcabene at the 900-mg dose used in the study was approximately 900 μM, which is much higher than the IC50 value of gemcabene to inhibit hOAT3. Although quinaprilat exposure was not available in human quinaprilat and gemcabene clinical studies, based on the above-mentioned in vitro transporter studies, rat in vivo PK studies, and SHR BP studies, it is reasonable to conclude that the observed “synergistic effect” on BP was most probably due to a simple increase of quinaprilat exposure caused by gemcabene inhibition of renal transport.
In conclusion, gemcabene exhibited a significant DDI with quinapril in rat. The observed DDI was caused by decreased renal secretion of quinaprilat due to inhibition of renal organic anion transport of quinaprilat resulting in an elevation of quinaprilat plasma concentration in rat. Moreover, a profound BP reduction was observed in SHRs when treated with quinapril in combination with gemcabene. Because in vitro and in vivo data correlate well in rat and the species differences for renal transporter interactions with quinaprilat and gemcabene were not observed in rat and human, the synergistic BP reduction observed in humans after coadministration of gemcabene and quinapril can be explained by the same mechanism in rat. Renal transporter-mediated DDIs remain an emerging area. These studies reveal the importance and utility of in vitro transporter assays that can contribute to the understanding of existing clinical observations; and most importantly, they can predict transporter-mediated DDIs of experimental compounds before entering clinical development.
Mean plasma concentration-time profiles of quinaprilat after administration of 3 mg/kg quinapril alone and 3 mg/kg quinapril with 30 mg/kg gemcabene. Gemcabene was dosed 1 h ahead of quinapril.
Acknowledgments
We thank Dr. Leonid Kirkovsky for valuable scientific discussions and Steve Wene, Lesley Albin, and Kathy Mandrell for help with the in vivo pharmacokinetics studies.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.108.149476.
-
ABBREVIATIONS: CI-1027, gemcabene; DDI, drug-drug interaction; ACE, angiotensin-converting enzyme; OAT, organic anion transporter; AUC, area under the curve; h, human; HEK, human embryonic kidney; SHR, spontaneous hypertensive rat; LC-MS/MS, liquid chromatography-tandem mass spectrometry; r, rat; DPBS, Dulbecco's phosphate-buffered saline; CL, clearance; BP, blood pressure; PK, pharmacokinetic.
- Received December 6, 2008.
- Accepted April 3, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}