


Abstract
Antibody-drug conjugates (ADCs) made with auristatin antimitotic agents have shown significant preclinical and clinical oncology activity. SGN-75 is composed of the anti-CD70 antibody h1F6 conjugated to monomethylauristatin F through a noncleavable maleimidocaproyl linkage. To understand the pharmacologic basis of the activity of this ADC, its pharmacokinetics and biodistribution were evaluated in a mouse xenograft model with use of a dual-radiolabeled ADC. The concentrations of antibody, total auristatin (conjugated plus unconjugated), and unconjugated auristatin were measured simultaneously in serum, tumor, and 16 normal tissues. Serum pharmacokinetic parameters for antibody and total auristatin were similar with very little unconjugated auristatin observed, demonstrating a high degree of stability. The kinetic values in normal tissues generally tracked with serum: the first time point (1 h) had the highest antibody and total auristatin concentrations with low unconjugated auristatin concentrations, with the exception of organs expected to be involved in hepatobiliary clearance of the ADC, where total and unconjugated auristatin concentrations peaked at 4 h and then rapidly decreased. In tumors, antibody concentrations were maximal at 1 day, with total auristatin increasing until 2 days. Intratumoral unconjugated auristatin was a substantial fraction of the total auristatin and reached concentrations much higher than in normal tissues. The exposure of the tumor to total and unconjugated auristatin was tens to hundreds times higher than normal tissue exposure. The data establish the pharmacologic basis of activity of the ADC through specific tumor targeting, intratumoral auristatin retention, and ADC stability in the systemic circulation.
A great deal of interest in the use of monoclonal antibodies (mAbs) for anticancer drug delivery has been generated from the recent positive clinical data emerging from SGN-35 (Younes et al., 2008) and trastuzumab-DM1 (Lewis Phillips et al., 2008; Vukelja et al., 2008) in patients for whom conventional chemotherapeutic regimens failed. These antibody-drug conjugates (ADCs) consist of potent anticancer drugs covalently linked to mAbs that bind to tumor-associated antigens. This binding leads to cellular uptake and intracellular drug release. Aside from SGN-35 and trastuzumab-DM1, several other ADCs are in various stages of clinical testing for a variety of different types of cancers (Ma et al., 2006; Tse et al., 2006; Carter and Senter, 2008; Ishitsuka et al., 2008).
Although ADCs offer the opportunity to widen the therapeutic window of cancer therapies by increasing the amount of drug that gets to the tumor and sparing normal tissues from systemic drug exposure, only a small percentage of administered mAb will localize within tumors (Wu and Senter, 2005; Carter, 2006; Kovtun and Goldmacher, 2007; Carter and Senter, 2008). The rest is catabolized in the liver and several other organs, such as intestine, muscle, and skin (Henderson et al., 1982; Moldoveanu et al., 1988; Ferl et al., 2006). To design maximally effective and well tolerated ADCs, it is critical to understand how the conjugate is distributed throughout the body, and how much various tissues are exposed to the unconjugated drug.
Previous biodistribution studies with ADCs have focused exclusively on the mAb or drug components, and have not provided separate measurements of conjugated and free drug concentrations at tumor and nontumor sites (Spearman et al., 1987a,b; Mosure et al., 1997; Xie et al., 2004). These studies investigated the impact of drug conjugation on mAb localization or the distribution of conjugated drug, but they did not evaluate the release and retention of the free drug and the species responsible for pharmacologic activity in tumors and normal tissues. To more fully understand the basis for antitumor activity and dose-limiting toxicities, it is essential to know how much ADC and antibody and drug components individually accumulate in both target and nontarget tissues.
We have shown that h1F6-mcMMAF, known as SGN-75, has pronounced in vivo activity against CD70 antigen-positive renal cell carcinoma xenografts at well tolerated doses (Law et al., 2006; McDonagh et al., 2008; Oflazoglu et al., 2008b). The underlying assumption was that the ADC localized to tumor tissues and that normal tissues had minimal unconjugated auristatin exposure. To assess this hypothesis, we prepared a dual-radiolabeled version of SGN-75 consisting of 3H-labeled h1F6 (McEarchern et al., 2008) C4v2 variant (McDonagh et al., 2006) and 14C-labeled monomethylauristatin F (MMAF), which was attached to h1F6 C4v2 through a maleimidocaproyl (mc) linker (Doronina et al., 2006). This drug linker yields cys-mcMMAF after complete degradation of the ADC inside cells (Fig. 1) (Doronina et al., 2006). This unconjugated auristatin contains the mc linker and an amino acid along with the MMAF drug, in contrast to systems that release the drug without the linker (Doronina et al., 2003). The C4v2 variant allows a maximum of four drugs to be conjugated to the reduced interchain disulfides of the mAb. This strategy prevents formation of highly loaded drug conjugates that could clear more rapidly than lower loaded drug conjugates and complicate analysis (Hamblett et al., 2004). Nude mice with subcutaneous CD70-positive 786-O renal cell carcinoma xenografts were injected with the dual-labeled ADC, and tumor, serum, and 16 normal tissues were collected and analyzed for mAb, total auristatin (conjugated plus unconjugated), and cys-mcMMAF. In this way, the unique kinetics of antibody localization, drug release from the ADC, and cys-mcMMAF retention were monitored. We demonstrate that the ADC achieves both effective tumor targeting and preferential tumor exposure of cys-mcMMAF compared with all normal tissues investigated. This provides a pharmacologic rationale for the large preclinical therapeutic window observed for SGN-75.
Materials and Methods
Radiolabeling and Conjugation. h1F6 variant C4v2 (McDonagh et al., 2006) (40 ml, 1 mg/ml) in 50 mM sodium borate, pH 8, 50 mM NaCl was treated with 1 mCi of 3H-succinimidyl propionate (GE Healthcare, Little Chalfont, Buckinghamshire, UK) for 1 h at 25°C. The radiolabeling reaction was treated with 1 mM hydroxylamine to remove weakly conjugated radiolabels, and then purified and buffer changed to PBS by use of an Amicon Ultra 30,000 NMWL centrifugal concentration device (Millipore, Billerica, MA).

Structure of h1F6 C4v2-mc-MMAF ADC and cys-mcMMAF. The location of the 14C radiolabels is indicated by asterisks on the ADC. The antibody backbone is 3H-radiolabeled. The mc linker and MMAF drug are indicated by arrows. Degradation of the ADC yields cys-mcMMAF as the unconjugated auristatin.
[3H]h1F6 C4v2 was conjugated to 14C-mcMMAF (Doronina et al., 2006) (custom synthesized by Moravek Biochemicals, Brea, CA) via the interchain disulfide cysteine residues as described previously (Sun et al., 2005). In brief, 2.7 Eq of TCEP were added to 5.8 mg/ml antibody in 50 mM sodium borate, pH 8, 50 mM NaCl, 1 mM Diethylenetriaminepentaacetic acid for 100 min at 37°C. The resultant reduced antibody was conjugated by the addition of 14C-mc-MMAF in 10% molar excess for 90 min at 0°C. N-Acetyl cysteine was added to quench the unreacted drug linker (15 min), and the ADC was purified and the buffer changed to PBS by use of an Amicon Ultra 30,000 NMWL centrifugal concentration device (Millipore). The specific activity of the ADC was determined by UV/visible spectroscopy and liquid scintillation counting on a Beckman Coulter (Fullerton, CA) LS6000IC with automatic deconvolution into 3H and 14C dpm signals. The ADC drug loading was determined from the specific activity of the drug linker.
Xenograft Model, Sample Collection, and Sample Processing. All animal experiments were conducted under Institutional Animal Care and Use Committee guidelines. Female nude mice (Harlan, Indianapolis, IN) were implanted subcutaneously with 786-O renal cell carcinoma tumor fragments as described previously (McDonagh et al., 2008) and grown until the tumors were 100 to 150 mm3. Dual-radiolabeled h1F6 C4v2-mcMMAF was given at 1.5 mg/kg i.v. and three animals were sacrificed at each time point. Serum, tumor, and normal tissues were collected at the following time points for all animals: 1 h, 4 h, 8 h, 1 day, 2 days, 4 days, 7 days, and 10 days. For bone marrow, the marrow core was obtained from one femur (cleaned of connective tissue) by flushing 500 μl of PBS through the bone with a 30-gauge needle. The resulting marrow-free femur was used as the bone sample.
Serum (9.5 μl) was mixed with 4 ml of Ecoscint A (National Diagnostics, Atlanta, GA) scintillation fluid and analyzed by dual-channel liquid scintillation counting. Tumor and tissue samples (except bone and skin) were mixed with 4 volumes of methanol and homogenized on ice with a Tissue Terror tissue homogenizer (Bio-Spec Products, Bartlesville, OK). Equal volumes of tissue homogenate before (total counts) and after (soluble counts) centrifugation were mixed with 4 ml of Ecoscint A and analyzed as above. Total counts include conjugated plus unconjugated auristatin, whereas soluble counts include just the unconjugated auristatin because the ADC is insoluble in methanol. Bone samples were pulverized into small pieces and extracted with methanol. The supernatant was then mixed with 4 ml of Ecoscint A and analyzed as above. Skin samples (∼100 mg) were solubilized with 1 ml of quaternary ammonium hydroxide in toluene (Beckman Coulter) at 50°C until dissolved, quenched with 600 μl of 30% hydrogen peroxide and 70 μl of glacial acetic acid, mixed with 10 ml of Ready Organic (Beckman Coulter) scintillation fluid, and analyzed as above.
Pharmacokinetics and Biodistribution. Background corrected dpm data were converted into percentage of injected dose per gram or nanomolar concentrations of antibody or conjugated drug by use of the dose and specific activity of the parent ADC. Individual serum concentrations were fitted to a two-exponential equation (two-compartment open model) with 1/y2 weighting with use of GraphPad Prism (GraphPad Software Inc., San Diego, CA), and the rates and amplitudes of the two exponentials were used to mathematically determine pharmacokinetic parameters (Shargel et al., 2005). The number of drugs per antibody was calculated by dividing the concentration of conjugated auristatin by the concentration of antibody for individual animals. The exposure to total and unconjugated auristatin for individual animals was calculated by use of the area-undercurve analysis function of GraphPad Prism.
Results
Conjugation of Dual-Radiolabeled ADC. To enable simultaneous detection of both antibody and drug, an ADC with two distinct radiolabels was prepared. The h1F6 C4v2 variant, where two of the interchain disulfides between the heavy chains were mutated to serines (McDonagh et al., 2006), demonstrated activity against CD70-positive 786-O renal cell carcinoma cells equal to h1F6 when conjugated to mcMMAF (Oflazoglu et al., 2008b) (data not shown). h1F6 C4v2 was first trace labeled with [3H]succinimidyl propionate (0.013 3H atoms/mAb). The [3H]h1F6 C4v2 was then reduced and conjugated to [14C]mcMMAF (Doronina et al., 2006). The resulting conjugate has closely matched 3H- and 14C-specific activities (8.0 and 7.2 μCi/mg mAb) and 2.3 mcMMAF molecules/mAb.
Pharmacokinetics and Drug-Linker Stability. Mice with subcutaneous 786-O renal cell carcinoma xenografts were injected with a single 1.5 mg/kg dose of the dual-radiolabeled ADC. The serum concentrations of h1F6 C4v2-mc-MMAF are shown in Fig. 2A, with data plotted as nanomolar concentrations to allow comparison of the mAb and drug components. The initial drug concentration is higher than the mAb, reflecting the conjugation level, and remains higher than the mAb throughout the collection period. The overall shapes of the concentration curves are similar for the drug and mAb components. Both sets of data were analyzed for their pharmacokinetic parameters by fitting to a biexponential equation (two-compartment open model) describing the more rapid distribution and slower terminal clearance phases. Table 1 shows that the terminal half-lives for the mAb and drug components are similar (2.54 ± 0.87 and 2.27 ± 0.44 days, respectively), suggesting that the linkage between antibody and drug is quite stable over this time period. A plot of the number of drugs per antibody (Fig. 2B) shows that this is indeed the case, because the measured half-life of the drug linker is approximately 10 days, consistent with previous reports for this drug linker (Alley et al., 2008). Additional pharmacokinetic parameters are also similar for the antibody and drug components, again consistent with a highly stable linkage between the antibody and the drug.
Pharmacokinetic parameters derived from serum concentrations Average of three animals per time point ± S.D.

Pharmacokinetics and drug-linker stability. A, concentration of mAb and auristatin components in serum after a 1.5 mg/kg injection of dual-radiolabeled h1F6 C4v2-mcMMAF to mice with subcutaneous 786-O tumors. Solid lines are biexponential fits to the data (two-compartment open model) with 1/y2 weighting. B, drugs per antibody in serum. Error bars are standard deviations for three animals per time point. mAb = monoclonal antibody.
Biodistribution. Tumor and normal tissues were mechanically homogenized in methanol and analyzed by dual-channel liquid scintillation counting to obtain the total 3H-antibody signal, the total [14C]auristatin signal (conjugated plus unconjugated), and the 14C-unconjugated auristatin (cys-mcMMAF) signal. The molar tumor mAb and auristatin concentrations are shown in Fig. 3. The mAb concentration rose quickly and reached a plateau from 4 h to 2 days after dose, followed by a slow decrease. The total auristatin concentration instead rose over the first 2 days, and then decreased in a similar manner. Unconjugated auristatin rose more slowly than the total auristatin, with the data clearly showing a time-dependent increase in ratio of unconjugated to total auristatin. This is consistent with a slow conversion of ADC to cys-mcMMAF. After the 2-day peak, the tumor unconjugated auristatin concentration decreased with kinetics similar to the terminal phase of the mAb in serum.
The concentrations of mAb and auristatin in several normal tissues are shown in Fig. 4. mAb concentrations at all time points were lower in all the normal tissues than in the tumor (Fig. 3), and, unlike in the tumor, all declined from the earliest first or second time points with kinetics similar to that of the serum (Fig. 2A). For almost all normal tissues, the amount of unconjugated auristatin present at all time points was substantially lower than the total auristatin. The exception for this was the liver and intestine contents, where the concentrations of unconjugated and total auristatin were much closer. After the 4-h peak, the liver unconjugated auristatin concentrations decreased with kinetics similar to the distribution phase of the mAb in serum. Although this study did not attempt to quantitate the amount of mAb and auristatin in the entire animal (only 14% of the weight of the animals was removed for analysis), at the first time point the tissues collected accounted for 48 and 46% of the injected dose for mAb and total auristatin, respectively.
The maximal percentage of injected dose per gram of both mAb and auristatin components for tumor, serum, or tissue is shown in Table 2. For the 3H antibody signal, the peak serum concentration was at the first time point (1 h) at 42 ± 5.4% ID/g. In the normal tissues, the peak was generally at 1 or 4 h, with the exception of skin and intestine (8 h). Lung showed the highest peak at 8.6 ± 1.2% ID/g. In contrast to normal tissues, the tumor reached a peak mAb concentration of 40.9 ± 9.7% ID/g at 1 day, and was substantially higher than all normal tissues. The total [14C]auristatin signal (conjugated plus unconjugated) peaked at 1 h at 38.2 ± 6.6% ID/g in the serum, closely matching the mAb value. Normal tissues again peaked at either 1 or 4 h, with the liver and intestine contents achieving the highest values (10.3 ± 0.8 and 11.3 ± 4.4% ID/g). The tumor peak was twice as high as the serum peak (82.3 ± 27.2% ID/g), and occurred at 2 days after dose. For the soluble 14C-auristatin signal (cys-mcM-MAF), very little was observed for serum (peak of 0.3 ± 0.1% ID/g), indicating that the vast majority of circulating auristatin is bound to the ADC. Substantial amounts were observed in the tumor, peaking at 2 days at 51.9 ± 13.1% ID/g. In most normal tissues, almost no soluble 14C signal was observed, with the exception of liver and intestine contents, which peaked at 4 h (3.6 ± 0.7 and 9.0 ± 3.5% ID/g, respectively). The intestine contents contained soluble [14C]auristatin signal almost exclusively and almost no 3H antibody signal, consistent with hepatobiliary catabolism of the ADC and excretion of cys-mcMMAF.
Peak percentage of injected dose per gram Three animals per group ± S.D.
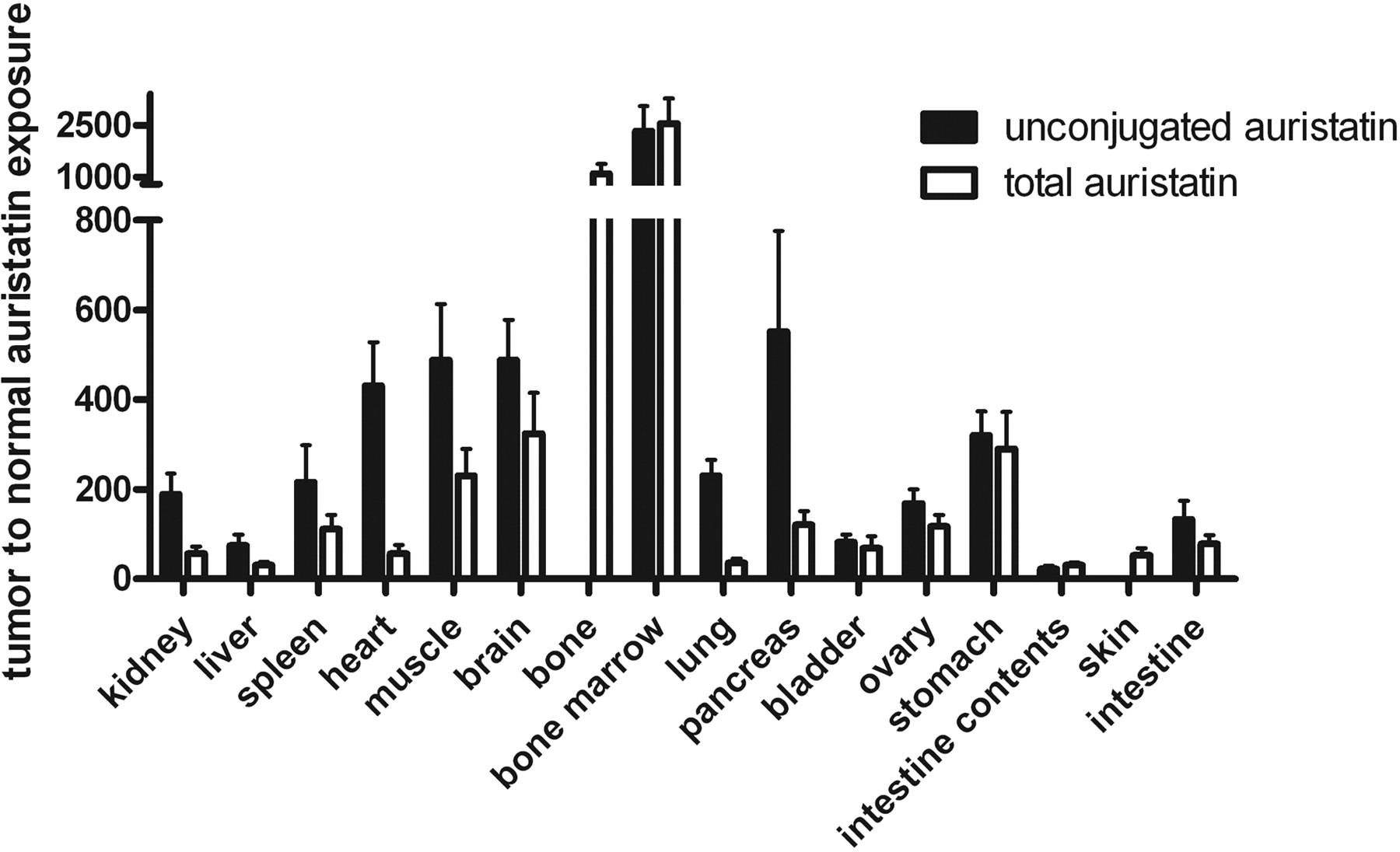
Tumor and Tissue Exposure to Free Auristatin. The exposure of the tumor and normal tissues to total and unconjugated auristatin from 0 to 10 days are shown in Table 3. Exposure to total auristatin ranged from 1.0 ± 0.1 nM * day for bone marrow to 2540 ± 590 nM * day for tumor. All normal tissues were less than 100 nM * day, whereas serum was 319 ± 28 nM * day. Exposure to unconjugated auristatin ranged from 0.7 ± 0.1 nM * day for bone marrow to 1510 ± 360 nM * day for tumor, with all normal tissues and serum less than 70 nM * day. In the tumor, the fraction of auristatin exposure contributed by unconjugated auristatin was 0.593 ± 0.010, demonstrating that the tumor is effective in converting the ADC to cys-mcMMAF. In contrast in the serum, this fraction was 0.0415 ± 0.0060, consistent with the earlier measurement of drug-linker stability. The intestine contents had the highest fractional exposure to unconjugated auristatin (0.810 ± 0.054). Normal tissue values for fractional exposure to unconjugated auristatin were all between the values for serum and tumor, except for bone marrow (0.651 ± 0.009), which had the lowest exposure to both total and unconjugated auristatin. Expressing auristatin exposure as tumor-to-normal ratios (Fig. 5), total auristatin exposure ratios ranged from 31.0 ± 6.4 for intestine contents to 2550 ± 740 for bone marrow, with most values falling between 30 and 300. For unconjugated auristatin, the exposure ratios ranged from 22.9 ± 6.7 for intestine contents to 2330 ± 730 for bone marrow, with most values falling between 70 and 550. Overall, the average unconjugated auristatin exposure ratio is 2.7-fold higher than the average total auristatin exposure ratio, because auristatin in normal tissues is found more often in the form of ADC, whereas in the tumor it is found more often in the form of cys-mcMMAF. The lowest unconjugated auristatin ratios included the intestine contents, intestine (132 ± 42), and liver (75.5 ± 23.9), again consistent with hepatobiliary excretion of cys-mcMMAF. Overall, these exposure ratios are extremely high, demonstrating that delivery of ADC and retention of cys-mcMMAF are substantially greater in the tumor than in all of the normal tissues.
Exposure to total and unconjugated auristatin from 0 to 10 days

Tumor concentrations of antibody, total auristatin (conjugated plus unconjugated), and unconjugated auristatin. Error bars are standard deviations for three animals per time point.
Discussion
ADCs provide a platform where mAb activity can be further empowered by the addition of a cytotoxic agent that is delivered with appreciable selectivity to cancer cells. Early attempts at making ADCs were unsuccessful for a variety of reasons, including immunogenicity of murine mAbs, weakly potent drugs, and unstable drug linkers. Auristatins, including MMAE (Doronina et al., 2003) and the related molecule MMAF (Doronina et al., 2006), represent a class of highly potent antimitotic agents that have shown substantial preclinical activity at well tolerated doses (Law et al., 2006; Ma et al., 2006; Tse et al., 2006; Oflazoglu et al., 2008a,b). Four auristatin ADCs are currently being evaluated in early and pivotal clinical trials.
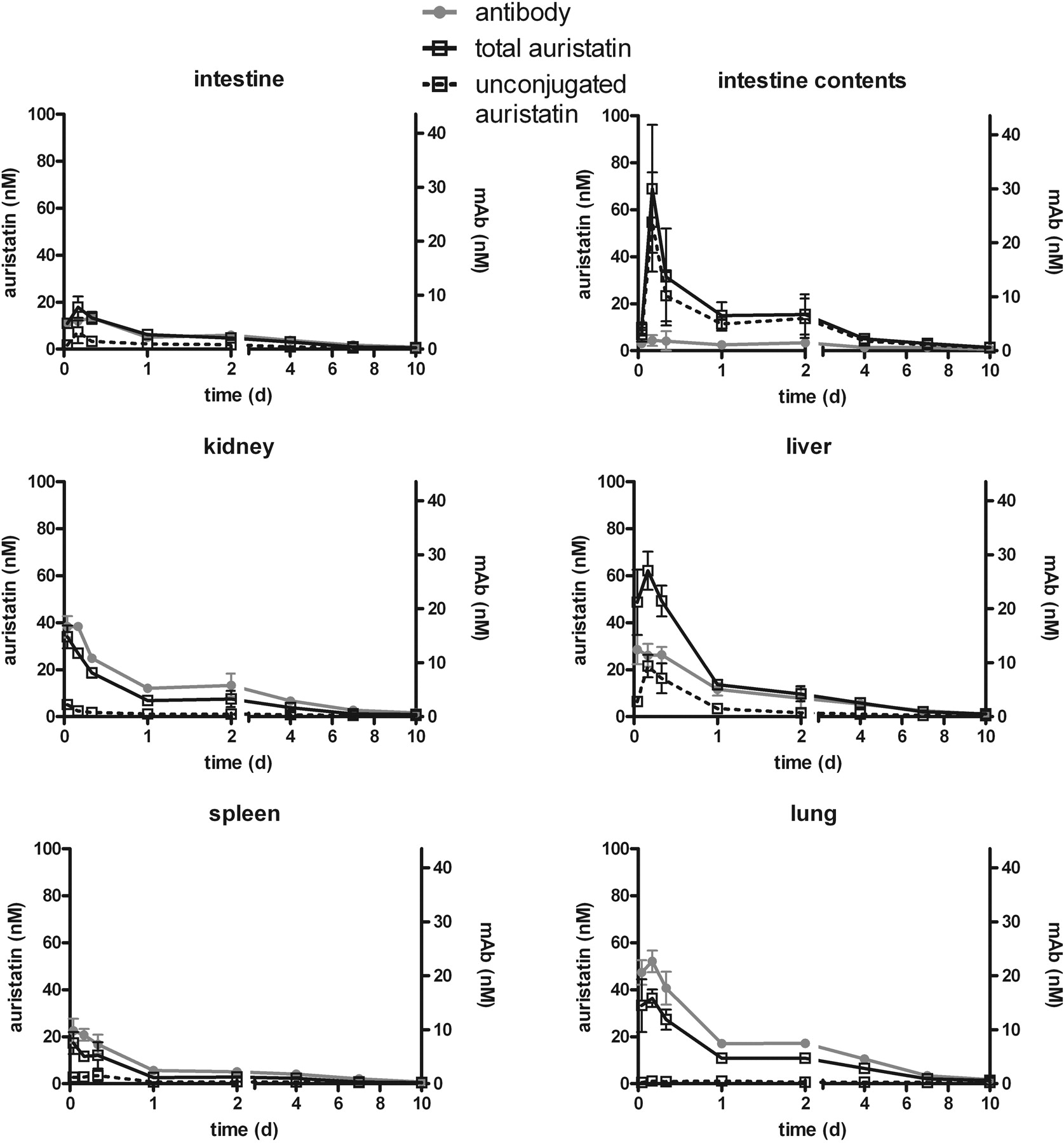
Selected normal tissue concentrations of antibody, total auristatin (conjugated plus unconjugated), and unconjugated auristatin. Error bars are standard deviations for three animals per time point.
Biodistribution experiments allowed evaluation of the tumor-targeting properties of the anti-CD70 mAb h1F6 C4v2 and the ability of the mcMMAF drug linker to selectively accumulate in the tumor rather than in normal tissues. The amount of mAb taken up by tumors depends on several factors that are specific to the mAb, antigen, and tumor model, including mAb pharmacokinetics, antigen density, internalization and re-expression rates, and vascularization (Carter, 2006; Ferl et al., 2006; Thurber et al., 2008). Tumor targeting by the anti-CD70 mAb h1F6 C4v2 showed substantial accumulation in CD70-positive tumor xenografts. The peak mAb concentration of 40.9 ± 9.7% ID/g was achieved 1 day after dose, a very high value for tumor targeting by mAbs (Ferl et al., 2006; Cai et al., 2007; Kim et al., 2008; Smith-Jones et al., 2008). Serum drug-linker stability measurements indicate that about half the drug is lost from the ADC during the 10-day period, which is considerably longer than the circulating half-life of the conjugate. The use of stable drug linkers allows tumor delivery of conjugated drug over an extended time frame, unlike the relatively unstable disulfide and hydrazone linker systems with half-lives typically of 1 to 2 days (Sanderson et al., 2005; Boghaert et al., 2008; Rodon et al., 2008).
The biodistribution data also demonstrate that cys-mc-MMAF, the degradation product of mcMMAF ADCs, is extremely effective in achieving sustained high concentrations in tumors but not in normal tissues. The drug is strongly retained in the tumor, reaching peak concentrations of 82.3 ± 27.2% ID/g and 51.9 ± 13.1% ID/g (total and unconjugated auristatin, respectively) at 2 days after dose. In contrast, peak concentrations for normal tissues are reached in 1 to 4 h, and are at concentrations much lower than in the tumor. The earlier peak in normal tissues suggests that ADC internalization, cys-mcMMAF release, and cys-mcMMAF clearance are faster than in the tumor, resulting in differences in cys-mcMMAF accumulation. This may be indicative of targeted delivery, where the tumor is using a CD70-dependent internalization mechanism, whereas murine CD70 does not bind h1F6, and CD70 expression in normal tissues is rare (McEarchern et al., 2008) and would therefore require nonspecific and presumably less efficient ADC internalization mechanisms. In addition, unconjugated dolastatin 10, a molecule that is related to MMAE and MMAF, has been shown to be effectively retained in cancer cells (Verdier-Pinard et al., 2000), a property that may make this drug class ideal for targeted delivery.
The exposure of the tumor to drug during the 10-day evaluation time was much higher than the exposure of normal tissues to drug. The tumor was also very effective in converting exposure to total auristatin into exposure to cys-mc-MMAF (fractional unconjugated auristatin exposure of 0.593 ± 0.010). This indicates that the tumor is not only effective in converting ADC to cys-mcMMAF, but also is effective in retaining cys-mcMMAF where it could have an extended cytotoxic effect. In contrast, normal tissues had less fractional exposure to unconjugated auristatin. Exceptions were those tissues previously shown to be involved in hepatobiliary clearance of antibodies (Henderson et al., 1982; Moldoveanu et al., 1988; Ferl et al., 2006). The intestine contents were almost exclusively unconjugated auristatin, but the fraction of liver exposure to unconjugated auristatin was only 0.252 ± 0.036, suggesting that once cys-mcMMAF is generated in the liver, it is quickly eliminated into the intestine contents rather than retained as in the tumor.
The tumor-to-normal exposure ratios were extremely high, with the lowest values found in the liver, intestine, and intestine contents. Differences in exposure are a reflection of both absolute concentrations and retention, and the superiority of the tumor relative to normal tissue in both aspects results in extremely high exposure ratios. These tumor-to-normal ratios, in general, are lower for total auristatin than unconjugated auristatin. The mice in this study were not perfused, and will have some residual blood pool after terminal bleeds. ADC was found in high concentrations in the serum and may result in increase of ADC concentrations in highly vascularized organs without concomitant increase in cys-mcMMAF concentrations. Auristatin ADCs with stable linkers require intracellular processing to release active drug (Sutherland et al., 2006), and normal tissue exposure to ADC will not lead to off-target toxicity unless there is nonspecific intracellular ADC uptake coupled with sustained exposure.
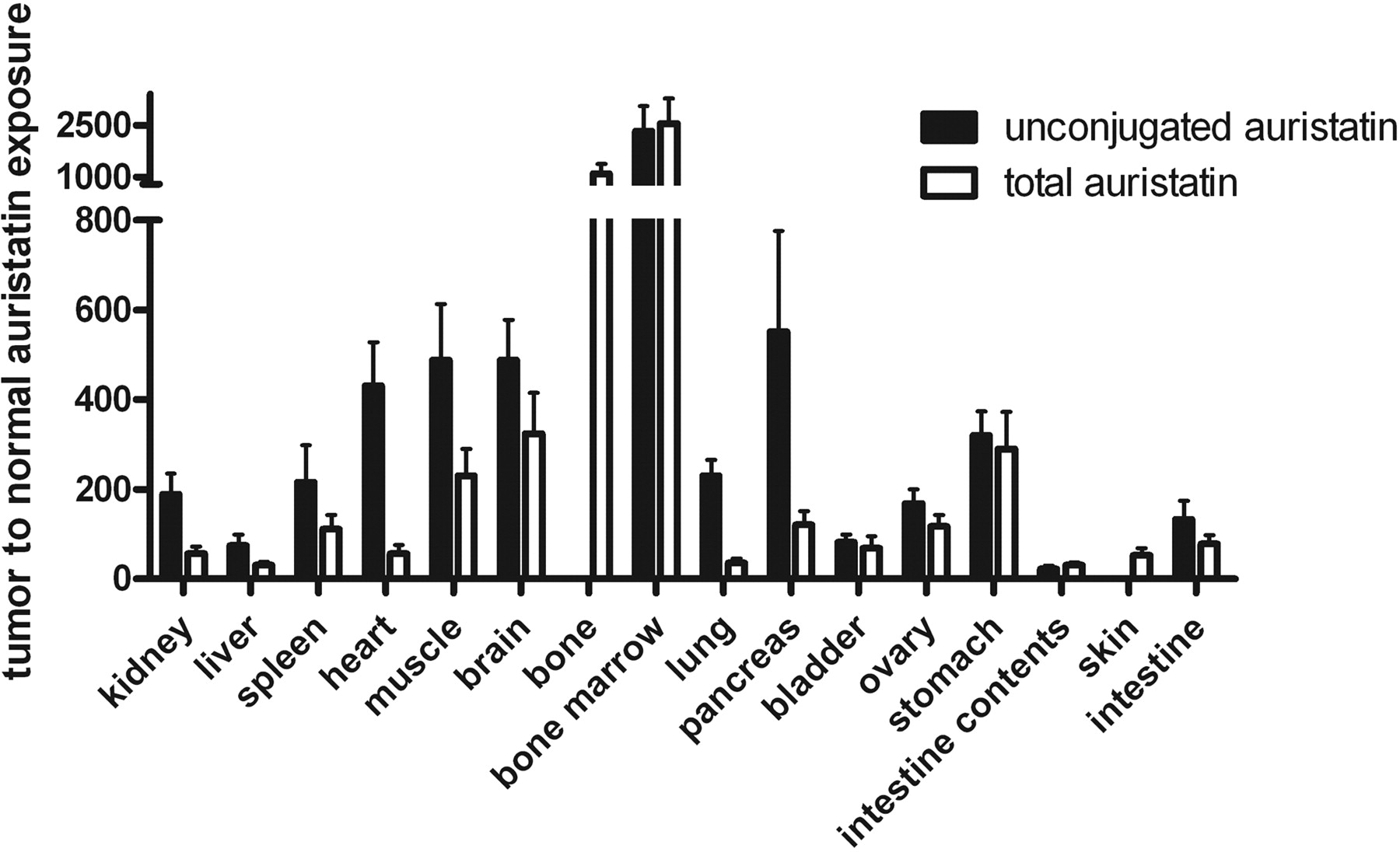
Exposure ratios of tumor to normal tissue for total and unconjugated auristatin. Error bars are standard deviations for three animals per time point. For bone and skin, the processing method allowed only total auristatin to be collected.
The use of two orthogonal radiolabels enabled the demonstration that mAb and cys-mcMMAF have unique uptake and retention kinetics. Previous biodistribution experiments with ADCs, in general, have only radiolabeled the antibody backbone (Xie et al., 2004; Sapra et al., 2005), and did not allow characterization of unconjugated drug. Because efficacy and tolerability are due to release of unconjugated drug, its evaluation is critical in the understanding of the pharmacology of ADCs. Dual-radiolabeled h1F6 C4v2-mcMMAF demonstrated that the ADC benefits from a combination of specific mAb uptake, systemic drug-linker stability, intratumoral accumulation and retention of cys-mcMMAF, and rapid clearance of cys-mcMMAF from nontarget tissues. This combination of properties provides a detailed basis for understanding the pronounced activities of mAb-auristatin conjugates.
Footnotes
-
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
-
doi:10.1124/jpet.109.155549.
-
ABBREVIATIONS: ADC, antibody-drug conjugate; mAb, monoclonal antibody; mc, maleimidocaproyl; MMAF, monomethyl auristatin F; ID, injected dose; PBS, phosphate-buffered saline.
- Received April 24, 2009.
- Accepted June 3, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}