


Abstract
The metabolism of etoposide was investigated by using human liver microsomes and nine recombinant human cytochrome P450 (CYP) isoforms to identify the CYP isoform(s) involved in the major metabolic pathway (3′-demethylation) of etoposide as well as to evaluate the possible metabolic interactions with several antitumor or supporting agents. The 3′-demethylation of etoposide followed a Michaelis-Menten one-enzyme kinetic behavior in six human liver microsomal samples. The relationships were assessed with six different human liver microsomes between the 3′-demethylation of etoposide and metabolic activities for substrate probes of the respective CYP isoforms, showing a significant correlation (r = 0.932, P < .01) only with 6β-hydroxylation of testosterone, a marker substrate for CYP3A4. Inhibitor/substrate probes for CYP3A4, ketoconazole, troleandomycin, verapamil and cyclosporin, or supporting agents, vincristine and prednisolone, inhibited etoposide 3′-demethylation by human liver microsomes. p-Nitrophenol, a substrate for CYP2E1, also inhibited etoposide 3′-demethylation. Among the nine recombinant human CYP isoforms, CYP3A4 exhibited the highest catalytic activity with respect to etoposide 3′-demethylation, compared with the minor activities of CYP1A2 and 2E1. Collectively, these data suggest that etoposide 3′-demethylation is mediated mainly by CYP3A4 and to a minor extent by CYP1A2 and 2E1. Furthermore, some supporting agents (vincristine and prednisolone) and the substrates of CYP3A4, which may be coadministered with etoposide during the cancer chemotherapies, inhibit the etoposide 3′-demethylation activity in vitro. The results may provide clinical implications with respect to the possible metabolic interactions between etoposide and other drugs studied herein in patients with cancer undergoing etoposide concurrently with either of them.
Etoposide (4′-demethylepipodophyllotoxin-9-(4,6-O-ethylidene)-β-d-glucopyranoside) (fig. 1) is one of clinically important antitumor agents derived from 4′-demethylepipodophyllotoxin which is the extract from plants ofPodophyllum peltatum or Podophyllum emodi(Stähelin and von Wartburg, 1991; Clark and Slevin, 1987). Etoposide is used during chemotherapies for a wide range of malignancies (e.g., small cell lung cancer, acute leukemia, lymphoma, testicular cancer) as a single agent or one of the constituents of standard therapeutic regimens (Clark and Slevin, 1987).

Chemical structures of etoposide, 3′-demethyletoposide and internal standard.
The pharmacokinetic disposition and metabolism of etoposide in humans have been reviewed (Clark and Slevin, 1987; Stewart, 1994), indicating that the elimination half-live ranged between 5 and 10 hr, the urinary excretion of unchanged etoposide ranged from 30 to 40% of the intravenous dose, and several metabolites were identified in plasma and urine such as cis-(picro) lactone, hydroxy acid derivatives, 4′-O-glucuronide of etoposide or agrycon, 3′-demethyletoposide. With respect to the hepatic metabolism of etoposide, the main metabolic pathway appears to be 3′-demethylation as has been observed in in vitro metabolism study by using human liver microsomes (Relling et al., 1992). Furthermore,Relling et al. (1993) have demonstrated that human cytochrome P450 3A4 (CYP3A4) is an isoform involved in the 3′-demethylation of etoposide by using human liver microsomes or cDNA expressed recombinant CYP isoforms. Indeed, the systemic exposure to etoposide (i.e., the area under the drug concentration-time curve) and its other pharmacokinetic parameters (e.g., clearance, half-life) are significantly affected by cyclosporin, a CYP3A4 substrate (Kronbach et al., 1988), in patients with cancer (Lum et al., 1992). However, other report by Rellinget al. (1989) has shown that epipodophyllotoxins such as etoposide and teniposide inhibited mephenytoin 4′-hydroxylation, a CYP2C19-mediated reaction (Goldstein and de Morais, 1994; Wrighton and Stevens, 1992), by the respective inhibitory potencies of 25 and 83% of the control. This result has suggested that CYP2C19 may also play a certain role in the metabolism of etoposide.
In this study, we intended to perform an in vitro metabolic study of etoposide at clinically realistic concentration ranges with human liver microsomes and nine recombinant human CYP isoforms to identify the human CYP isoform(s) involved in the 3′-demethylation of etoposide. Furthermore, possible metabolic interactions were evaluated with respect to etoposide 3′-demethylation by a number of drugs which may be coadministered with etoposide in cancer chemotherapies.
Materials and Methods
Chemicals and reagents.
Etoposide, 3′-demethyletoposide and internal standard (4′-demethylepipodophyllotoxin-9-(4,6-O-propylidene-β-d-glucopyranoside) were available from Nippon Kayaku Co., Ltd. (Tokyo, Japan). Chemical structures of these compounds are shown in figure1.
Racemic mephenytoin was kindly donated by Dr. Küpfer (University of Bern, Bern, Switzerland), and S- andR-mephenytoin were separated by Chiralcel OJ column (10 μm, 4.6 × 250 mm, Daisel Chemical Co., Ltd., Tokyo, Japan) according to the method previously reported (Yasumori et al., 1990). Troleandomycin, ketoconazole and verapamil were obtained from Sigma Chemical Co. (St. Louis, MO). Furafylline was obtained from Salford Ultrafine Chemicals and Research (Manchester, UK). Quinidine, coumarin, p-nitrophenol and prednisolone were purchased from Wako Pure Chemical Industries (Osaka, Japan), and sulfaphenazole was from Meiji Yakuhin Co. (Tokyo, Japan). Bleomycin and cisplatin were obtained from Nippon Kayaku Co., Ltd. (Tokyo, Japan). Adriamycin was purchased from Mercian Co., Ltd. (Tokyo, Japan), cyclophosphamide and vincristine from Shionogi & Co., Ltd. (Osaka, Japan), cytarabine from Yamasa Corporation (Chiba, Japan), granisetron from SmithKline Beecham Seiyaku K.K. (Tokyo, Japan), methotrexate from Lederle, Ltd. (Tokyo, Japan), recombinant human G-CSF from Sankyo Co., Ltd. (Tokyo, Japan) and cyclosporin from Sandoz Pharmaceuticals, Ltd. (Tokyo, Japan). Acetonitrile, methanol and other reagents of analytical grade were purchased from Wako Pure Chemical Industries. NADP+, glucose-6-phosphate and glucose-6-phosphate dehydrogenase were obtained from Oriental Yeast (Tokyo, Japan).
Microsomal preparations of nine different recombinant human CYP isoforms (i.e., CYP1A1, 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1 and 3A4) expressed in human B lymphoblastoid cell line, AHH-1, were purchased from Gentest Corp. (Woburn, MA).
Preparation of human liver microsomes.
Human liver microsomes were prepared from livers obtained from six patients (44–64 yr old, one male and five females) who underwent partial hepatectomy for metastatic liver tumor(s) in the Division of General Surgery, Department of Surgery, International Medical Center of Japan (Tokyo, Japan), as described previously (Chiba et al., 1993b). Human liver microsomes were prepared by differential centrifugation as described by Chiba et al. (1993a). After the determination of protein concentration (Lowry et al., 1951), the suspended microsomal samples were aliquoted, frozen and stored at −80°C until used.
Incubation conditions with human liver microsomes and recombinant human CYP isoforms.
The basic incubation medium contained 0.1 mg/ml of human liver microsomes, 0.5 μM NADP+, 2 mM glucose-6-phosphate, 1 IU/ml of glucose-6-phosphate dehydrogenase, 4 mM MgCl2, 0.1 mM EDTA, 100 mM sodium-phosphate buffer (pH 7.4) and 5 to 125 μM of etoposide in a final volume of 250 μl. The mixture was incubated at 37°C for 15 min after 1 min of preincubation without the NADPH-generating system, and the reaction was stopped by adding 100 μl of ice-cold acetonitrile. After the termination of the incubation, 100 μl of 1 M sodium-phosphate buffer (pH 3.0) and 50 μl of 5 μM internal standard dissolved in acetonitrile were added to the sample. The mixtures were centrifuged at 10,000 ×g for 10 min, and the supernatant was injected onto an HPLC system as described below.
Incubation conditions used for the nine different human CYP isoforms obtained from genetically engineered B lymphoblastoid cells were the same as those used for human liver microsomes, except for the incubation time (i.e., 120 min). For incubations with those recombinant isoforms, microsomes equivalent to 5 pmol of CYP were used for assessing the metabolism of etoposide. Enzyme kinetic study with recombinant CYP3A4 isoform was performed at the same conditions as those used for human liver microsomes, except for the incubation time (i.e., 20 min) and the microsomal protein concentration (i.e., 0.3 mg/ml, 20 pmol of CYP3A4/ml). The recombinant CYP isoform activities for etoposide 3′-demethylation were expressed as picomoles per picomoles of CYP per minute.
HPLC assay.
The determination of 3′-demethyletoposide was performed by an HPLC with fluorescence detection. The HPLC system consisted of a model L-7100 pump (Hitachi Ltd., Tokyo, Japan), a model L-7480 fluorescence detector (Hitachi), a model L-7200 autosampler (Hitachi), a model D-7500 integrator (Hitachi) and a 4.6 × 75 mm Develosil ODS-HG-3 column (Nomura Chemical Co., Ltd., Aichi, Japan). The mobile phase consisted of acetonitrile-potassium dihydrogenphosphate (20 mM, pH 4.6) in a proportion of 24/76 (v/v) and was delivered at a flow rate of 0.8 ml/min. The column temperature was maintained at 25°C by a model SM-05 water circulator (Taitec, Tokyo, Japan). The eluate was monitored at the wavelength of 288 nm for excitation and 328 nm for emission by use of the fluorescence detector as mentioned above. A 60 μl of sample was injected onto the HPLC system. Calibration curve was prepared for the concentration ranges of 3′-demethyletoposide between 25 and 200 pmol/tube. The retention times of 3′-demethyletoposide, etoposide and the internal standard were 5.5, 11.0 and 22.5 min, respectively. The detection limit of 3′-demethyletoposide was 25 pmol/tube (i.e., 3 pmol on column).
The quantitation of 3′-demethyletoposide was made by comparison with the standard curve by using the peak-height ratio method. The intra- (n = 6) and interassay (n = 3) coefficients of variation were <5.0 and <3.0%, respectively, for the determination of 3′-demethyletoposide.
Kinetics of the formation of 3′-demethyletoposide.
The formation rate of 3′-demethyletoposide was linear for the incubation time of up to 15 min when 50 μM of etoposide and 0.1 mg/ml of microsomal protein coexisted at 37°C. 3′-Demethyletoposide was not formed in the absence of the NADPH-generating system. A linear relationship was observed between the production rate of 3′-demethyletoposide at protein concentrations of up to 0.2 mg/ml for 15 min. Accordingly, the kinetic studies were performed at 37°C with a 15-min incubation time and at a protein concentration of 0.1 mg/ml.
The one-compartment enzyme kinetic parameters (Km , Vmax and Vmax/Km ) for the formation of 3′-demethyletoposide were estimated by the linear regression analysis by using unweighed raw data, because they followed a simple Michaelis-Menten kinetic behavior (i.e., a one-enzyme kinetic approach) in all experiments.
Correlation study.
Correlations between the metabolic activities of substrates toward the respective distinct CYP isoforms and those of etoposide were studied by using six different human liver microsomes. Assays for phenacetin O-deethylation (CYP1A2) (Tassaneeyakul et al., 1993), coumarin 7-hydroxylation (CYP2A6) (Yun et al., 1991), diclofenac 4′-hydroxylation (CYP2C9) (Goldstein and de Morais, 1994), S-mephenytoin 4′-hydroxylation (CYP2C19) (Goldstein and de Morais, 1994; Wrighton and Stevens, 1992), desipramine 2-hydroxylation (CYP2D6) (Koyama et al., 1994), chlorzoxazone 6-hydroxylation (CYP2E1) (Peter et al., 1990) and testosterone 6β-hydroxylation (CYP3A4) (Waxmanet al., 1988) were performed in duplicate with the same sets of microsomal preparations. Metabolites of these substrate probes which were formed in the respective incubation mixtures were determined according to the respective HPLC assay methods as described elsewhere (Tassaneeyakul et al., 1993; Chiba et al., 1993c,1994; Yoshimoto et al., 1995) or developed at the Department of Clinical Pharmacology, Research Institute, International Medical Center of Japan (unpublished data, chiba, K., Koyama, E., Zhao, X-J., Kawashiro, T. and Ishizaki, T.).
Inhibition study.
The effects of coincubation of specific inhibitor/substrate probes for different human CYP isoforms on the microsomal metabolism of etoposide were studied separately. Specific inhibitor/substrate probes used were furafylline for CYP1A (Tassaneeyakul et al., 1993), coumarin for CYP2A6 (Yunet al., 1991), sulfaphenazole for CYP2C9 (Goldstein and de Morais, 1994), quinidine for CYP2D6 (Wrighton and Stevens, 1992) and ketoconazole and troleandomycin for CYP3A (Watkins et al., 1985). S-mephenytoin and p-nitrophenol were used for the inhibition studies on the CYP2C19- and 2E1-mediated metabolism of etoposide, because S-mephenytoin (Goldstein and de Morais, 1994; Wrighton and Stevens, 1992) and p-nitrophenol (Thummel et al., 1993) are the specific substrate probes for CYP2C19 and CYP2E1, respectively. We also used cyclosporin and verapamil for assessing if the metabolism of etoposide would be mediated via CYP3A4, because these two drugs are well-known substrates/inhibitors for CYP3A4 (Kronbach et al., 1988;Kroemer et al., 1993) as well as MDR modulators interacting with P-glycoprotein expressed in tumor cells (Lum et al., 1992; Lum and Gosland, 1995) which may be coadministered with etoposide in patients with cancer. A total of 50 μM of etoposide was incubated with one of inhibitor/substrate probes at the same incubation condition as described above. The effects of each compound on the formation of 3′-demethyletoposide from etoposide at the respective inhibitor concentrations were compared with the control values determined by the incubation of etoposide alone and the inhibitions were expressed as percentage of the respective control values. This part of the experiments was performed with three different microsomal preparations and the averaged values were recorded.
Metabolic interaction study.
Various drugs, which may be coadministered with etoposide in a cancer chemotherapy, were tested for their possible inhibitory effects on the 3′-demethylation of etoposide. The drugs included adriamycin, bleomycin, cisplatin, cyclophosphamide, cytarabine, granisetron, methotrexate, G-CSF, prednisolone and vincristine. This part of the experiments was performed by the same way as described in the inhibition study with three different microsomal preparations, and the averaged inhibition percentage values compared with the control were reported herein.
Statistical analysis.
Data are expressed as mean ± S.D. throughout the text. Correlations between the metabolite formation rate for the respective CYP isoform-selective substrates and 3′-demethyletoposide formation rates for etoposide were determined by the least-squares linear regression analysis. P < .05 was considered statistically significant.
Results
Metabolic profile of etoposide by human liver microsomes.
Typical HPLC-fluorescence chromatograms of standard mixture and analytes incubated with human liver microsomes are shown in figure2. Etoposide, 3′-demethyletoposide and the internal standard were well separated among each other, and there were no peaks interfering with the assay.

HPLC-fluorescence chromatograms of (A) standard mixture and (B) sample obtained after a 15-min incubation of etoposide (50 μM) with HL-34 human liver microsomes (0.1 mg protein/ml) at 37°C in the presence of NADPH-generating system. The fluorescence intensity was monitored at the wavelength of 288 nm for excitation and 328 nm for emission.
Representative Michaelis-Menten and Eadie-Hofstee plots for the formation of 3′-demethyletoposide from etoposide are shown in figure3A and B, respectively. The 3′-demethylation of etoposide gave a mono-phasic relationship in microsomes from the six human livers, suggesting that the 3′-demethylation was apparently catalyzed by one CYP isoform or isoforms which may have a similar Km value. The mean (±S.D.) apparent Michaelis-Menten kinetic parameters derived from the one-enzyme kinetic approach were: Km = 53.9 ± 6.6 μM, Vmax = 0.249 ± 0.091 nmol/mg protein/min and Vmax/Km = 4.62 ± 1.71 μl/mg protein/min.

Two representative (A) Michaelis-Menten and (B) Eadie-Hofstee plots for 3′-demethylation of etoposide obtained after a 15-min incubation of etoposide at 37°C with human liver microsomes (0.1 mg protein/ml). The solid lines indicate the computer-generated best-fit simulations.
Correlation study.
The correlation results derived from the six human liver microsomes are summarized in table1. The etoposide 3′-demethylation activity (i.e., intrinsic clearance or Vmax/Km ) was correlated significantly (r = 0.932, P < .01) with the testosterone 6β-hydroxylation activity, and there were no significant correlations between the 3′-demethylation of etoposide and the catalytic activity for each of the remaining six substrate probes for the distinct CYP isoforms (table1).
Correlation between the etoposide 3′-demethylation activities (i.e., Vmax/Km) with microsomes obtained from six human livers and metabolic reactions of seven selective substrates of distinct human CYP isoforms
Inhibition study.
Data on the effects of coincubation of distinct CYP isoform-selective inhibitor/substrate probes on the formation of 3′-demethyletoposide from etoposide at the concentration of 50 μM (i.e., around the mean Km value as described above) with human liver microsomes are shown in figure 4. Ketoconazole and troleandomycin, inhibitor probes for CYP3A4 (Watkins et al., 1985), showed a potent inhibition in a concentration-dependent manner.p-Nitrophenol, a substrate probe for CYP2E1 (Thummelet al., 1993), also showed a relatively potent inhibitory effect in a concentration-dependent manner. Other inhibitor/substrate probes for the respective CYP isoforms tested in the study (furafylline for CYP1A, coumarin for CYP2A6, sulfaphenazole for CYP2C9,S-mephenytoin for CYP2C19 and quinidine for CYP2D6) showed no inhibitory effects on the etoposide 3′-demethylation activity (fig.4).

Effects of specific CYP inhibitors/substrates on 3′-demethylation of etoposide (50 μM) after a 15-min incubation with human liver microsomes (0.1 mg protein/ml) at 37°C. The data represent the mean ± S.D. of experiments performed with microsomes obtained from three different livers.
Inhibition profiles of verapamil and cyclosporin for etoposide 3′-demethylation are also shown in figure 4. Both the drugs showed a potent inhibitory effect on the etoposide 3′-demethylation activity in a concentration-dependent manner.
Furthermore, the mechanism of inhibition was examined for etoposide 3′-demethylation by cyclosporin, which exhibited the most potent inhibition for etoposide 3′-demethylation among all of the inhibitor/substrate probes for distinct CYP isoforms (fig. 4). As a result, the inhibition mechanism by cyclosporin was a mixed-type of the competitive and non-competitive inhibition (data not shown). ItsKi was calculated as approximately 0.3 μM.
Metabolism of etoposide with recombinant human CYP isoforms.
Figure 5 shows the catalytic activities of the nine different recombinant human CYP isoforms with respect to the 3′-demethylation of etoposide at a concentration of 50 μM. Recombinant CYP3A4 gave the highest 3′-demethylation activity, and recombinant CYP1A2 and CYP2E1 revealed the much weaker activities compared with CYP3A4 (i.e., the respective activities were approximately 10% and 5% of the recombinant CYP3A4).
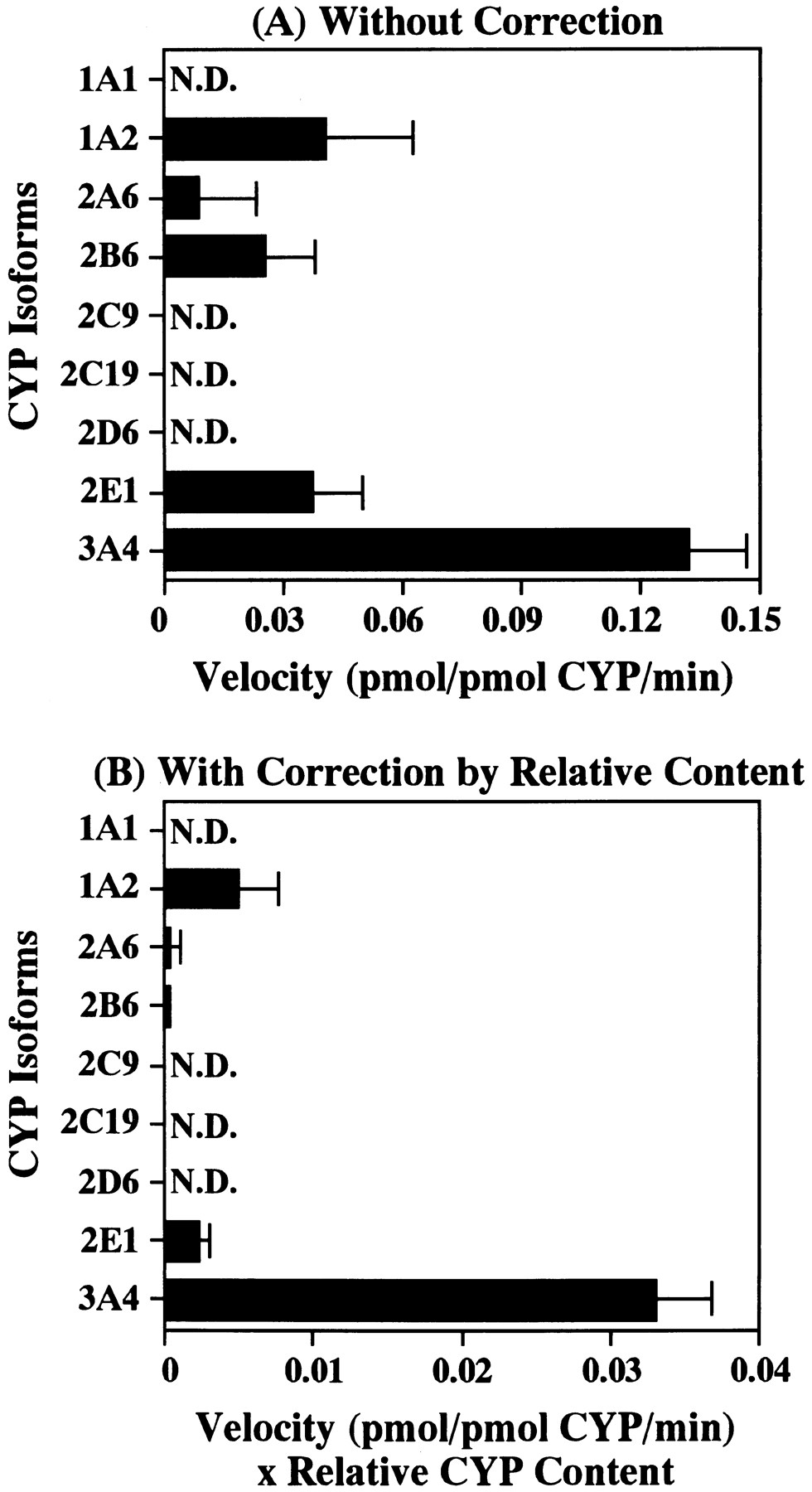
Etoposide 3′-demethylation activity after a 120-min incubation of etoposide (50 μM) at 37°C with nine recombinant human CYP isoforms expressed in human B lymphoblastoid cells (20 pmol CYP/ml). Upper graph (A) shows the activity without correction, whereas lower graph (B) shows the activity with relative content of each CYP isoform in human liver microsomes (Shimada et al., 1994). Each bar represents the mean ± S.D. from experiments performed on three different days. N.D., Not detectable.
The mean (±S.D.) apparent Michaelis-Menten kinetic parameters derived from the one-enzyme kinetic approach observed in the recombinant human CYP3A4 isoform experiments were: Km = 34.4 ± 19.7 μM, Vmax = 1.215 ± 0.262 pmol/pmol CYP/min and Vmax/Km = 40.9 ± 15.3 μl/pmol CYP/min. The mean Km value with recombinant human CYP3A4 was fairly close to that obtained from human liver microsomes (i.e., 53.9 μM).
Metabolic interaction study.
The etoposide 3′-demethylation activities in coincubation with other drugs, which may be coadministered with etoposide in cancer chemotherapies, are shown in figure 6. In cases of bleomycin, cisplatin, cytarabine, granisetron, methotrexate and G-CSF, the activity of etoposide 3′-demethylation was not appreciably changed with comparison to the respective control values, while adriamycin inhibited the etoposide 3′-demethylation to a minor extent (i.e., ≅25%) and cyclophosphamide activated it by about 30% (fig. 6). However, prednisolone and vincristine inhibited the 3′-demethylation of etoposide in a concentration-dependent manner by up to 60 and 62%, respectively (fig. 6).

Effects of various drugs on 3′-demethylation of etoposide after a 15-min incubation of etoposide (50 μM) with human liver microsomes (0.1 mg protein/ml) at 37°C. The data represent the mean ± S.D. of experiments performed with microsomes obtained from three different livers.
Discussion
The present in vitro metabolism study on etoposide 3′-demethylation with human liver microsomes and recombinant human CYP isoforms suggested that one isoform or isoforms, which may have a similar Km value, would be involved in the 3′-demethylation of etoposide, because Eadie-Hofstee plots observed with six different human liver microsomes showed a mono-phasic profile (e.g., fig. 3). In this respect, the results were quite similar to those reported by Relling et al. (1992). Furthermore, the individual Vmax/Km values of six human liver microsomes for etoposide 3′-demethylation correlated strongly only with those of testosterone 6β-hydroxylation (r = 0.932, P < .01, table 1), which is known as the metabolic reaction catalyzed by CYP3A4 (Waxman et al., 1988). All other specific substrates toward distinct human CYP isoforms did not significantly correlate with etoposide 3′-demethylation (table 1).
The contention as discussed above is also in accordance with the data derived from the inhibition study. Ketoconazole, troleandomycin, cyclosporin and verapamil, all of which are known as the inhibitor/substrate probes for CYP3A4 (Watkins et al., 1985;Kronbach et al., 1988; Kroemer et al., 1993;Wrighton and Stevens, 1992), showed a potent inhibitory effect on etoposide 3′-demethylation in a concentration-dependent manner (fig.4). Furthermore, when we have recently investigated the metabolic interaction between etoposide and quinine, a substrate drug of CYP3A4 (Zhao et al., 1996; Zhao and Ishizaki, 1997), a mutual inhibition between these two drugs was observed (Zhao et al., 1998). Thus, the 3′-demethylation of etoposide is catalyzed by human CYP3A4 isoform. p-Nitrophenol, which is the substrate probe of CYP2E1 (Thummel et al., 1993), also inhibited the etoposide 3′-demethylation activity to some extent (fig.4), suggesting that CYP2E1 plays a role in the metabolism of etoposide. All of other substrate/inhibitor probes of distinct human CYP isoforms did not inhibit the 3′-demethylation of etoposide (fig. 4).
The recombinant human CYP3A4 showed the highest activity with CYP1A2, 2A6, 2B6 and 2E1 revealing an appreciable activity for the 3′-demethylation of etoposide as shown in figure 5A. However, when the activity of each CYP isoform used in this study was adjusted for each of the respective CYP contents of human liver microsomes (fig. 5B), according to the data given by Shimada et al. (1994), CYP3A4 exhibited the greatest activity in etoposide 3′-demethylation among the nine recombinant isoforms, and CYP1A2 and 2E1 showed a certain activity (approximately 10 and 5% compared with the activity of CYP3A4, respectively). These findings provided further evidence that CYP3A4 is a principal isoform involved in the 3′-demethylation of etoposide, with the suggestion that CYP1A2 and 2E1 are the minor isoforms involved in the etoposide metabolism. However, furafylline, which is an inhibitor probe for CYP1A2 (Tassaneeyakul et al., 1993), did not inhibit etoposide 3′-demethylation in the human liver microsomal inhibition study (fig. 4). The reason for this discrepant observation remains totally obscure.
We observed that verapamil and cyclosporin, substrates/inhibitors for CYP3A4 (Kronbach et al., 1988; Kroemer et al., 1993), inhibited etoposide 3′-demethylation (fig. 4). These drugs have been investigated clinically as MDR modulators that interact with P-glycoprotein expressed in tumor cells (Lum et al., 1993;Lum and Gosland, 1995). It is well known that the pharmacokinetics and pharmacodynamics of antitumor agents (e.g., adriamycin, vincristine, etoposide) are often affected in patients treated with these MDR modulators. For instance, it has been shown that the area under the concentration-time curve of etoposide increased 1.8-fold by cyclosporin, and as a result, etoposide increased the frequency of leukopenia (Lum et al., 1992). This is because cyclosporin interacts with P-glycoprotein which is expressed normally in biliary and renal tubular epithelia (Thorgeirsson et al., 1991; Lum and Gosland, 1995). Thus, based on our findings, verapamil and cyclosporin may play a possible role as the candidate inhibitors in etoposide 3′-demethylation via CYP3A4 in addition to the MDR modulators during a cancer chemotherapy in patients undergoing etoposide and either of these drugs.
With respect to the observation that CYP3A4 is involved dominantly in the 3′-demethylation of etoposide, our results were quite similar to those demonstrated by Relling et al. (1992, 1993). However, our study using the clinically attained concentration ranges of etoposide showed that CYP1A2 and 2E1 play a minor role in the 3′-demethylation of etoposide in addition to CYP3A4, a major CYP isoform involved. Both CYP1A2 and 2E1 are well known as the inducible isoforms by smoking and alcohol drinking habits (Wrighton and Stevens, 1992), respectively. In addition, the interindividual variation of etoposide pharmacokinetics and pharmacodynamics in cancer patients has been ascribed to the variation of plasma protein binding (Stewartet al., 1990) and renal dysfunction (Hande et al., 1990). Based on our in vitro results, however, an interpatient variability in the CYP1A2 and/or 2E1 activities/activity might become one of the possible reasons for the interindividual variation of the pharmacokinetics and pharmacodynamics of etoposide.
We also performed the metabolic interaction study for the 3′-demethylation of etoposide by several antitumor agents or supporting agents. Vincristine and prednisolone inhibited the activity of etoposide 3′-demethylation in a concentration-dependent manner (fig.6). Vincristine is often used with etoposide in a chemotherapy of small cell lung cancer (Murray, 1997). The metabolism of vincaalkaloids in humans has been studied by Zhou et al. (1993)and Zhou-Pan et al. (1993) who observed that the metabolism of the vinca alkaloids was mediated by CYP 3A subfamily. Thus, on a theoretical basis, the 3′-demethylation of etoposide seems to be inhibited competitively by vinca alkaloids. However, prednisolone is usually used with etoposide in a chemotherapy of non-Hodgkin’s lymphoma (Fisher et al., 1983). Although CYP isoform(s) involved in the metabolism of prednisolone has not been reported, we assume that the major isoform that catalyzes the metabolism of prednisolone might be CYP3A4 because many steroid hormones (i.e., testosterone, androstenedione, progesterone) are metabolized to the respective 6β-hydroxy forms by CYP3A4 (Waxmanet al., 1988) and the metabolism of methylprednisolone is inhibited by ketoconazole (Kandrotas et al., 1987), a potent inhibitor of CYP3A4 (Watkins et al., 1985). Therefore, prednisolone may inhibit the metabolism of etoposide competitively. However, at the low concentration (1 μM) of vincristine or prednisolone, which is close to or higher than the realistic plasma concentration ranges attained in patients undergoing the respective therapeutic doses (Nelson et al., 1980; Henderson et al., 1979), there was no inhibitory effect by either of these drugs on etoposide 3′-demethylation (fig. 6). Although there has been, to our knowledge, no clinical study on the pharmacokinetics of etoposide in cancer patients who are administered etoposide with either of vincristine or prednisolone, we are tempted to assume that the metabolism of etoposide would not be much affected by the coadministration of vincristine or prednisolone. Otherwise, when one assumes that the hepatic tissue levels of prednisolone or vincristine might range from 1 to 10 μM as used in this study (fig. 6), the possibility exists that a significant inhibitor effect on the metabolism of etoposide in vivo would occur, leading to an interaction between etoposide and prednisolone or vincristine. Obviously, this interaction possibility must be confirmed by a pharmacokinetic analysis in cancer patients as conducted by Lumet al. (1992) for the interaction between etoposide and cyclosporin, a CYP3A4 substrate (Kronbach et al., 1988). However, because any other antitumor agents (e.g., bleomycin, cisplatin, cytarabine, methotrexate) or supporting agents (e.g., G-CSF, granisetron) did not inhibit the 3′-demethylation of etoposide in vitro (fig. 6), these drugs, which may be coadministered with etoposide during some combined cancer chemotherapies, should not affect the activity of etoposide 3′-demethylation. Although adriamycin inhibited the 3′-demethylation of etoposide by about 25% and cyclophosphamide activated it by about 30% (fig. 6), we can neither offer any reasonable explanations for nor give any clinical implications of these observations.
In conclusion, our in vitro metabolism study using human liver microsomes and recombinant human CYP isoforms suggests that CYP3A4 is a main CYP isoform involved in the 3′-demethylation of etoposide, and that CYP1A2 and 2E1 are involved as the minor enzymatic components in this metabolic pathway. Moreover, etoposide 3′-demethylation was inhibited by two well-known CYP3A4 substrates/inhibitors, verapamil and cyclosporin, in a concentration-dependent manner. The activity was also inhibited by vincristine and prednisolone only at the supratherapeutic plasma concentrations. However, the possibility of an interaction between etoposide and vincristine or prednisolone cannot totally be negated, although the hepatic tissue levels of these inhibitors attained by their therapeutic doses remain unknown. Further studies are definitely required for screening possible metabolic interactions between etoposide and other drugs that may be coadministered during combined and/or supportive cancer chemotherapies. Finally, it is emphasized that our strategy to use human liver microsomes coupled with recombinant human liver CYP isoforms seems to be a useful means to characterize and identify the CYP isoform(s) involved in the relevant metabolic pathway(s) of clinically important antitumor agents such as etoposide as well as to evaluate the possible drug-drug interactions in patients undergoing the relevant cancer chemotherapies.
Acknowledgments
The authors thank Dr. A. Küpfer, University of Bern, Bern, Switzerland, for the donation of racemic mephenytoin as the in vitro assay standard and Mr. H. Yoshikawa, Nippon Kayaku Co., Ltd., Tokyo, Japan, for the synthesis of 3′-demethyletoposide used for the HPLC assay.
Footnotes
-
Send reprint requests to: Dr. Takashi Ishizaki, Department of Clinical Pharmacology, Research Institute, International Medical Center of Japan, Toyama 1-21-2, Shinjuku-ku, Tokyo 162-8655, Japan.
-
↵1 This study was supported by a grant-in-aid from the Ministry of Human Health and Welfare and by a postdoctoral fellowship training program from the Bureau of International Cooperation, International Medical Center of Japan, Tokyo, Japan.
- Abbreviations:
- CYP
- cytochrome P450
- HPLC
- high-performance liquid chromatography
- G-CSF
- granulocyte colony-stimulating factor
- MDR
- multidrug resistance
- Received December 15, 1997.
- Accepted May 5, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}