


Abstract
In the present study, we investigated the transport of ochratoxin A (OTA) by kidney-specific organic anion transporter 1 (OAT1). When expressed in Xenopus laevis oocytes, OAT1 mediated sodium-independent uptake of OTA (Km = 2.1 μM). Piroxicam, which has been shown to prevent the nephrotoxicity of OTA, inhibited OAT1-mediated uptake of OTA. By contrast, another protective compound, aspartame, did not. Using a cell line derived from the mouse kidney terminal proximal tubule (S3) transfected with OAT1 cDNA, we investigated the transport of OTA and also its effect on cell proliferation and cell viability. S3 cells expressing OAT1 mediated the saturable transport of OTA (Km = 0.57 μM). Cell proliferation was suppressed in S3 cells expressing OAT1 when exposed to 2 and 10 μM OTA. This suppression was rescued by the coaddition of 1 mMp-aminohippurate in the media. The present study indicates that OTA is transported by OAT1 and that the accumulation of OTA via OAT1 in proximal tubular cells is the primary event in the development of OTA nephrotoxicity.
Ochratoxin A (OTA) is a secondary fungal metabolite produced by Aspergillus ohraceus and Penicillium verrucosum. Contamination of foods, especially cereals, with OTA has been noted in East and Central Europe, North Africa, North America, and Japan. In these countries, OTA was detected in the blood of human and animals. Studies being conducted for identification of the etiological factor of an endemic nephropathy (Balkan Nephropathy) have implicated OTA in the development of this disease (Kuiper-Goodman and Scott, 1989; Breitholtz et al., 1991;Maaroufi et al., 1995).
Within the body, OTA accumulates in several tissues, especially in the kidney and liver, and is excreted mainly from the kidney (Kuiper-Goodman and Scott, 1989). An in vivo study revealed that probenecid (a typical inhibitor of the renal organic anion transporter) decreased the renal clearance of OTA (Stein et al., 1985). Using rabbit renal basolateral membrane vesicles or rabbit renal tubular suspensions, it has been shown that the renal organic anion transport system mediates the uptake of OTA and may play a role in OTA toxicity (Sokol et al., 1988; Groves et al., 1998). Because more than 99% of OTA is bound to plasma proteins (Chu, 1971; Hagelberg et al., 1989), glomerular filtration of OTA is considered to be minimal. Thus, the excretion of OTA into urine is thought to be mainly by tubular secretion, presumably via the organic anion transport system (Gekle and Silbernagl, 1994).
Recently, a rat renal organic anion transporter (OAT1) was isolated (Sekine et al., 1997; Sweet et al., 1997). OAT1 is expressed predominantly in the kidney and is localized on the basolateral membrane of the middle proximal tubule (S2) (Tojo et al., 1999). As OAT1 mediates the transport of various endogenous and exogenous organic anions, including p-aminohippurate (PAH), it is considered the classical organic anion transporter responsible for the basolateral uptake of organic anions in renal epithelial cells.
In the present study, we investigated the transport of ochratoxin A by OAT1 using both Xenopus laevis oocytes for transient expression and culture cells for stable expression. Using the culture cells expressing OAT1, we also investigated the effect of ochratoxin A on cell proliferation and cell viability.
Experimental Procedures
Materials.
[3H]OTA (547.6 GBq/mmol) was purchased from Moravek Biochemicals Inc. (Brea, CA). Unlabeled OTA was obtained by purification (Jung and Endou, 1989). All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO).
cRNA Synthesis and Its Injection to X. laevisOocytes.
Capped cRNA for OAT1 and rat Na+-dicarboxylate cotransporter (rNaDC1) was synthesized in vitro using T7 RNA polymerase as described elsewhere (Sekine et al., 1997, 1998). Defolliculated oocytes were injected with 15 ng of cRNA. For coexpression experiments, both OAT1 cRNA (12 ng) and rNaDC1 cRNA (3 ng) were injected into the oocytes. After injection, the oocytes were maintained in modified Barth’s solution containing gentamicin [88 mM NaCl, 1 mM KCl, 0.33 mM Ca(NO3)2, 0.4 mM CaCl2, 0.8 mM MgSO4, 2.4 mM NaHCO3, 10 mM HEPES, and 50 μg/ml gentamicin, pH 7.4, sterilized by filtration] for 3 days at 18°C.
Uptake Experiments in Oocytes.
Three days after the injection of OAT1 and/or NaDC1 cRNA, uptake experiments were performed in ND96 solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES, pH 7.4) containing radiolabeled substrates as indicated in each experiment. The uptake was stopped by the addition of ice-cold ND96 solution, and the oocytes were washed five times with the same solution. Single oocytes were dissolved in 0.25 ml of 10% sodium dodecyl sulfate and 2.5 ml of aquasol-2 (Packard, Meriden, CT), and radioactivity was determined.
Cell Culture.
S3 cells, which were derived from the terminal portion of the mouse kidney proximal straight tubule (S3) of transgenic mice harboring the simian virus 40 large T antigen gene, have been established as described previously (Hosoyamada et al., 1996). OAT1-expressing S3 cells were obtained by transfecting S3 cells with OAT1 cDNA in the mammalian expression vector pcDNA3.1 by the electroporation method. These cells were routinely grown in RITC 80-7 medium (Kyokuto Pharmaceutical Industrial Co., Tokyo, Japan) containing 5% fetal bovine serum, 10 μg/ml transferrin, 0.08 U/ml insulin, and 10 ng/ml recombinant epidermal growth factor in a humidified incubator at 33°C and 5% CO2 (Takeda et al., 1996). S3 cells transfected with pcDNA3.1 without OAT1 cDNA were used as control cells.
Uptake Experiment in Cells.
The cells were seeded in 24-well tissue culture plates at a cell density of 0.5 × 105 cells/well. After cultivation for 3 days, the cells were washed three times with Dulbecco’s PBS, containing 5.6 mMd-glucose, and then preincubated with the same solution for 30 min in a water bath at 37°C. The cells were then incubated in Dulbecco’s PBS containing 5.6 mM d-glucose with [3H]OTA as indicated in each experiment. The uptake was stopped by the addition of ice-cold buffer, and the cells were washed three times with the same solution. The cells of each well was dissolved with 0.5 ml of 0.1 N sodium hydroxide and 2.5 ml of aquasol-2, and radioactivity was determined.
Proliferation of Cells.
OAT1-expressing S3 cells and control cells were seeded in 48-well tissue culture plates at a cell density of 0.3 × 105 cells/well and cultured for 18 h. Then, the RITC 80-7 medium was exchanged with medium with or without OTA (2 or 10 μM) and/or 1 mM PAH and then cultured for an additional 24 or 48 h. The cells were counted in a standard hemocytometer.
3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium Bromide (MTT) Assay.
OAT1-expressing S3 cells and control cells were cultured in 48-well plates with or without OTA (2 or 10 μM) and/or PAH (1 mM) as described above. After 24 h, 50 μl of 0.5% MTT was added to the media, and the cells were further incubated for 4 h. After solubilizing the cells with isopropanol/HCl solution, optical density (at 570 nm with 630 nm as a reference) was measured. Each value was expressed as the percentage of control.
Statistics.
Data are expressed as mean ± S.E.M. Statistical differences were determined using Student’s unpairedt test. Differences were considered significant at the level of p < .05.
Results
Uptake of OTA via OAT1 in X. laevis Oocytes.
Figure 1 shows the uptake of 4 μM [3H]OTA in X. laevis oocytes expressing OAT1. Compared with control oocytes (noninjected oocytes), oocytes injected with OAT1 cRNA showed significantly higher uptake of [3H]OTA. The OAT1-mediated uptake increased linearly up to 3 h.

Time dependence of [3H]OTA uptake in OAT1-expressing oocytes. Oocytes, injected with 15 ng of OAT1 cRNA, were maintained for 3 days at 18°C. After preincubation with 1 mM glutarate for 2 h, uptake of 4 μM [3H]OTA in OAT1-injected oocytes (●) and noninjected oocytes (■) was measured for 5 min to 3 h at room temperature. Values represent means ± S.E.M. of 10 oocyte-associated radioactivity determinations.
In the experiment shown in Fig. 2, we examined the properties of OTA uptake via OAT1. Replacement of extracellular sodium with choline had no effect on the rate of OAT1-mediated [3H]OTA uptake (sodium, 2.34 ± 0.37 pmol/ oocyte/h; choline, 2.29 ± 0.17 pmol/oocyte/h) (Fig. 2A). Figure 2B shows the trans-stimulation effect of dicarboxylate (glutarate) on OAT1-mediated uptake of [3H]OTA. Oocytes expressing rNaDC1 (rat sodium-dicarboxylate transporter) did not mediate the transport of OTA (control, 0.19 ± 0.01 pmol/oocyte/h versus rNaDC1, 0.18 ± 0.03 pmol/oocyte/h). When OAT1- and rNaDC1-coexpressing oocytes were preincubated with 1 mM glutarate, the oocytes showed a further significant increase in the rate of [3H]OTA uptake (3.80 ± 0.36 pmol/oocyte/h) than sole OAT1-expressing oocytes (preincubation (−), 1.82 ± 0.34 pmol/oocyte/h; preincubation (+), 2.53 ± 0.37 pmol/oocyte/h). This result means that the glutarate preloaded by rNaDC1 trans-stimulates the uptake of OTA.

Sodium and glutarate dependence of [3H]OTA uptake in OAT1-expressing oocytes. A, oocytes were injected with 15 ng of OAT1 cRNA and maintained for 3 days at 18°C. After preincubation with 1 mM glutarate for 2 h, uptake of 1 μM [3H]OTA in the oocytes was measured in a sodium or sodium-free (choline) medium for 1 h at room temperature. Values represent means ± S.E.M. of seven to nine determinations. B, oocytes were injected with 15 ng of rNaDC1 cRNA, OAT1 cRNA, or OAT1 plus rNaDC1 cRNA and maintained for 3 days at 18°C. With or without preincubation with 1 mM glutarate for 2 h, uptake of 1 μM [3H]OTA in the oocytes was measured for 1 h at room temperature. Values represent means ± S.E.M. of eight to nine determinations. **P < .01 versus OAT1-expressing oocytes without preincubation; ΨP < .05 versus OAT1expressing oocytes with preincubation.
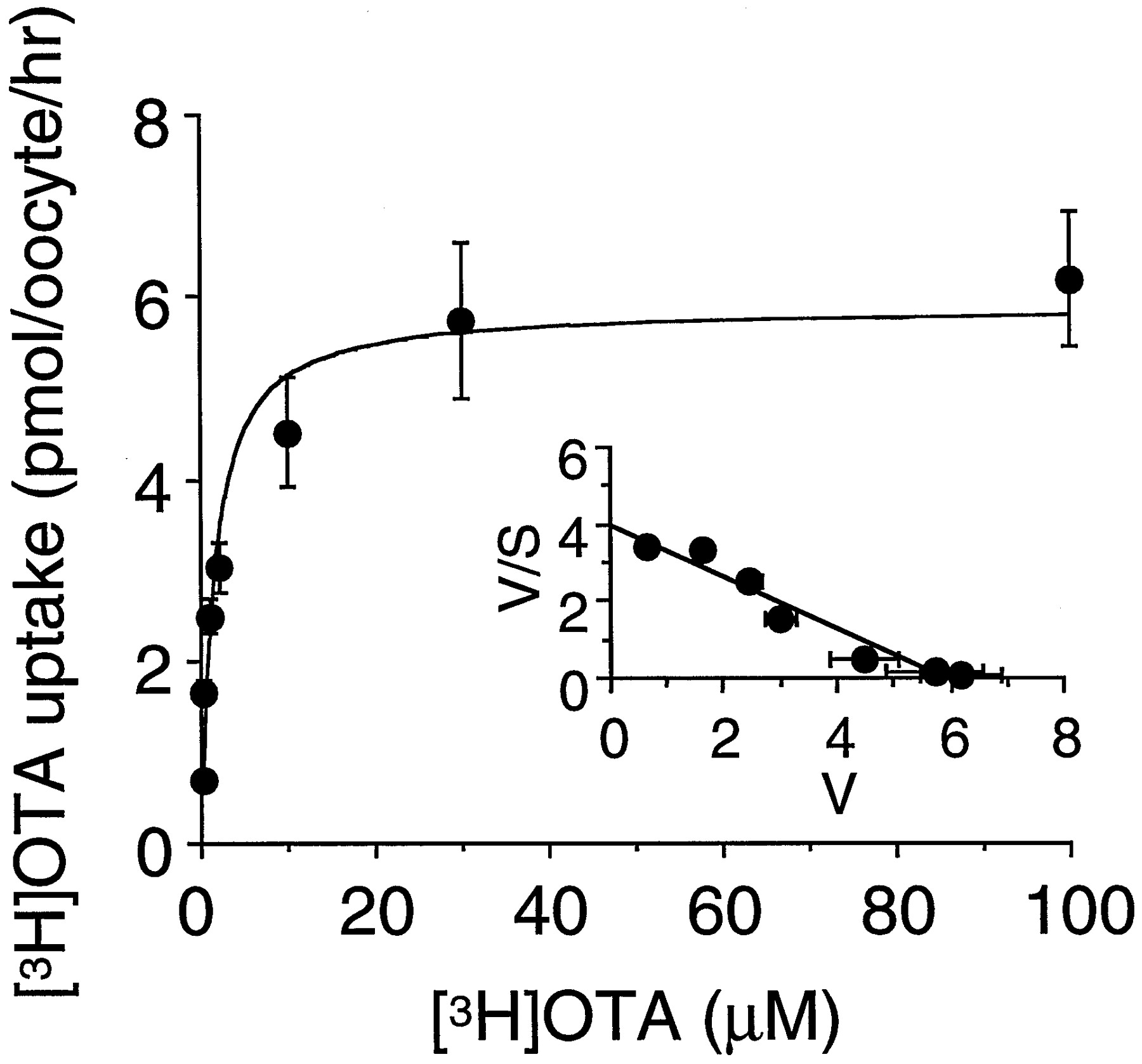
Specific uptake of [3H]OTA in OAT1-expressing oocytes revealed saturable kinetics (Fig.3), and the Eadie-Hofstee plot gave a single straight line (Fig. 3, inset). The estimatedKm andVmax values were 2.1 ± 0.3 μM and 8.0 ± 1.1 pmol/oocyte/h, respectively (n = 3). Figure 4 shows the inhibition experiments using several substrates, previously reported to prevent nephropathy or the transport of OTA, and another mycotoxin, citrinin. PAH, probenecid, piroxicam, octanoate, and citrinin at the concentration of 0.2 mM significantly inhibited [3H]OTA uptake via OAT1. In contrast, 1 mM aspartame and tetraethylammonium did not change it.
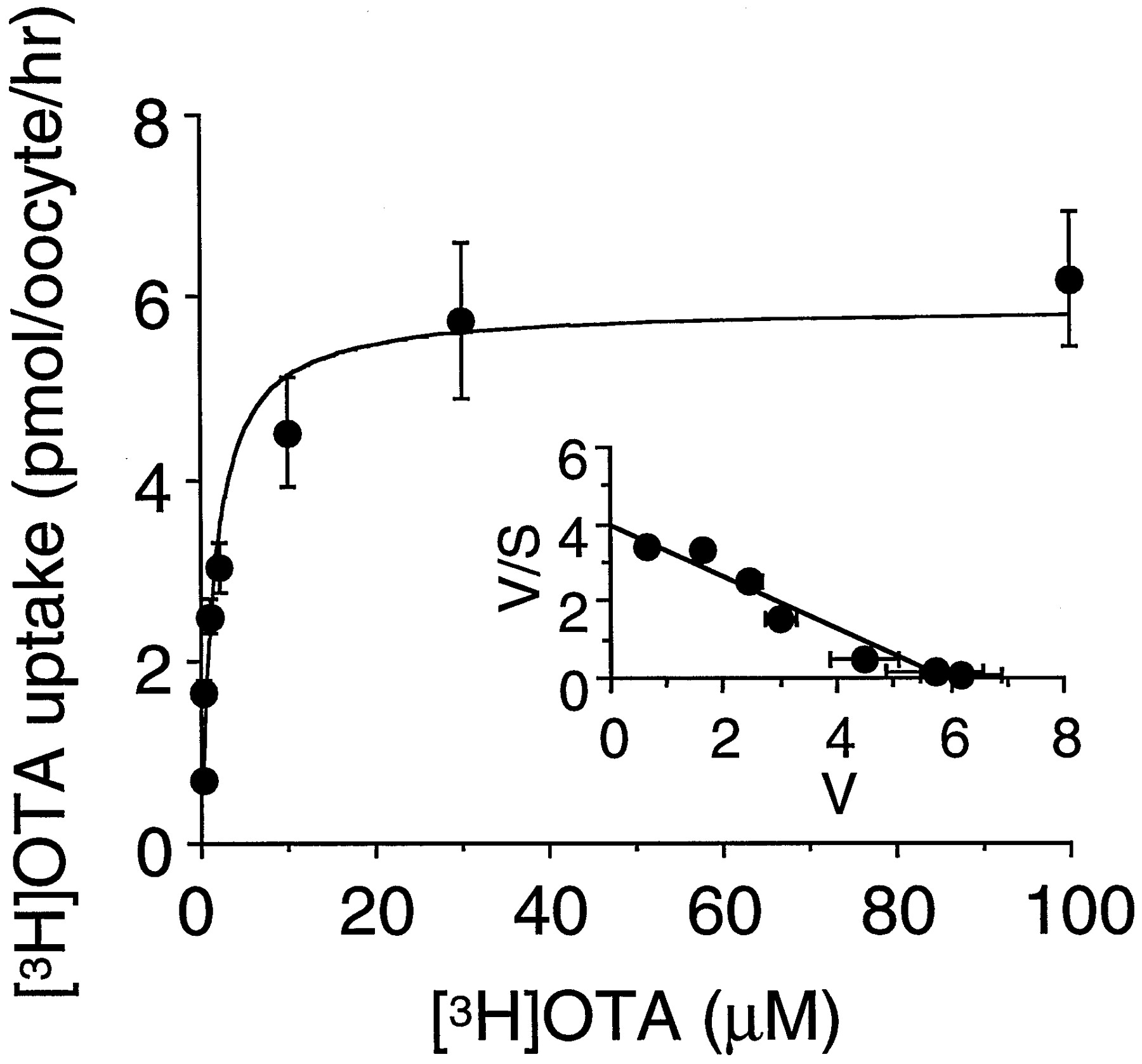
Concentration dependence of [3H]OTA uptake in OAT1-expressing oocytes. Oocytes were injected with 15 ng of OAT1 cRNA and maintained for 3 days at 18°C. After preincubation with 1 mM glutarate for 2 h, uptake of 0.2 to 100 μM [3H]OTA in the oocytes was measured for 1 h at room temperature. The results show the values after subtraction of the uptake in noninjected oocytes from that in OAT1-injected ones. Values represent means ± S.E.M. of 9 to 10 determinations. Inset, Eadie-Hofstee plot of the uptake of [3H]OTA.

Inhibition study of [3H]OTA uptake in OAT1-expressing oocytes. Oocytes were injected with 15 ng of OAT1 cRNA and maintained for 3 days at 18°C. After preincubation with 1 mM glutarate for 2 h, the oocytes were incubated with 1 μM [3H]OTA containing 0.2 mM or 1 mM of the given compounds for 1 h at room temperature. The results show the values after subtraction of the uptake in noninjected oocytes from that in OAT1-injected ones. Values represent means ± S.E.M. of 7 to 10 determinations. TEA, tetraethylammonium. ***P < .001 versus control.
Uptake of OTA in OAT1-Expressing S3 Cells.
OTA transport was examined also using culture cells stably expressing OAT1. OAT1 cDNA subcloned into pcDNA3.1 was transfected into a cell line (S3) derived from the proximal tubule of transgenic mice harboring the simian virus 40 large T antigen gene. Significant uptake of [3H]OTA in OAT1-expressing S3 cells was observed, and the specific uptake of [3H]OTA in OAT1-expressing S3 cells increased linearly up to 2 min (Fig.5A). As shown in Fig. 5B, we studied the kinetics of OAT1-mediated [3H]OTA uptake.Km andVmax values were determined to be 0.57 ± 0.06 μM and 6.4 ± 0.2 pmol/mg protein/min, respectively (n = 3).
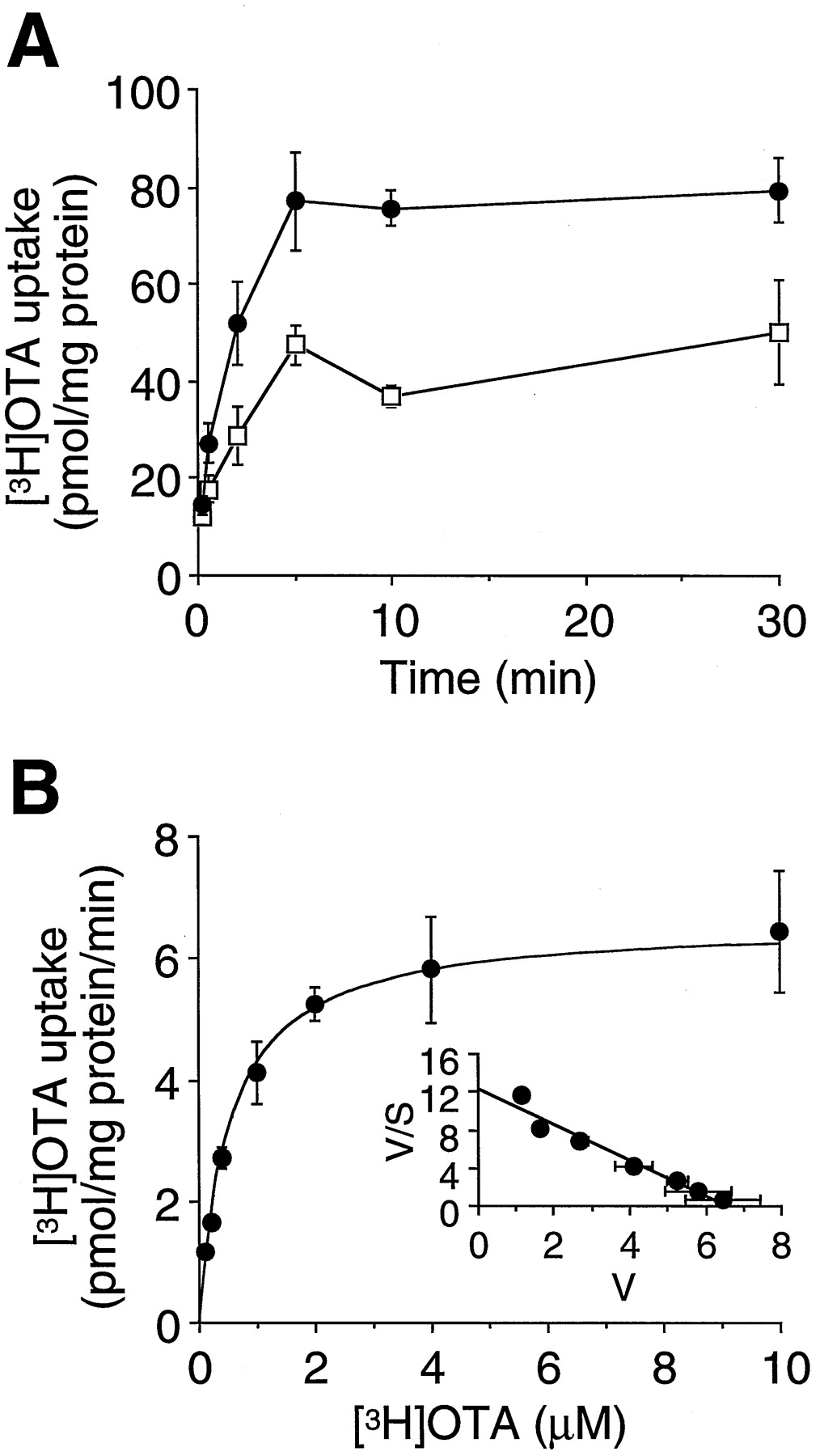
Uptake of [3H]OTA in OAT1-expressing S3 cells. A, time course of [3H]OTA uptake was measured in OAT1-expressing S3 cells (●) and S3 cells transfected with the expression vector pcDNA3.1 alone (mock cells, ■). The cells were incubated with 4 μM [3H]OTA for 10 s to 30 min at 37°C. Values represent mean ± S.E.M. of three determinations. B, concentration dependence of [3H]OTA uptake was measured in OAT1-expressing S3 cells. The cells were incubated with 0.1 to 10 μM [3H]OTA for 2 min at 37°C. The results show the values after subtraction of the uptake in mock cells from that in OAT1-transfected ones. Values represent means ± S.E.M. of three determinations. Inset, Eadie-Hofstee plot of the uptake of [3H]OTA.
Effect of OTA on Cell Proliferation and Viability of OAT1-Expressing S3 Cells.
We examined the effect of OTA on cell proliferation and cell viability. When OAT1-expressing S3 cells were cultured with 2 and 10 μM OTA in RITC 80 to 7 medium, proliferation of the cells was significantly suppressed (Fig.6B). This inhibitory effect of OTA on cell proliferation was rescued by the coaddition of 1 mM PAH. In contrast, in S3 cells, transfected with the pcDNA3.1 vector only, no suppression of cell proliferation was observed following the addition of 10 μM OTA (Fig. 6A). In the experiment shown in Fig.7, we examined the cell viability by MTT assay. After being exposed to 10 μM ochratoxin A for 24 h, the viability of OAT1-expressing S3 cells decreased significantly, and this decrease was recovered by the simultaneous addition of 1 mM PAH in the media. In this assay, the viability of the control cells was not influenced by OTA.

Effect of OTA on the proliferation of OAT1-expressing S3 cells. Mock cells (A) and OAT1-expressing S3 cells (B) were cultured in the medium not containing OTA (control, ●) and containing 2 μM OTA (▪), 10 μM OTA (▴), 10 μM OTA and 1 mM PAH (○), and 1 mM PAH (■) for 24 or 48 h at 33°C. The cell number was counted in a standard hemocytometer. Values represent means ± S.E.M. of six determinations. *P < .05 versus control; **P < .01 versus control; ***P < .001 versus control; ΨΨP < .01 versus 10 μM OTA; ΨΨΨP < .001 versus 10 μM OTA.

Effect of OTA on the cell viability of OAT1-expressing S3 cells. OAT1-expressing S3 cells (closed column) and mock cells (open column) were cultured as described in the experiment of Fig. 6. After 24 h of incubation with OTA and/or PAH, MTT assay was performed. Values represent means ± S.E.M. of six determinations. *P < .001 versus control; **P < .05 versus 10 μM OTA.
Discussion
Chronic tubulointerstitial nephropathy (Balkan Nephropathy) is developed in European countries, particularly Balkan countries (Castegnaro and Chernozemsky, 1987; Kuiper-Goodman and Scott, 1989). In this endemic nephropathy, various tubular dysfunctions, such as tubular proteinuria, and benign or malignant epidermal tumors are observed. Etiologic studies have indicated that this endemic nephropathy is caused by some environmental factor. Although the studies are not conclusive, ochratoxin A is considered to be one of the causative agents. Studies on the secretion and reabsorption of OTA in the renal proximal tubules have suggested that OTA is a substrate of the renal organic anion transport system (Sokol et al., 1988; Groves et al., 1998).
The present study demonstrated that a renal multispecific organic anion transporter (OAT1) mediates the high-affinity transport of OTA. Transport of OTA by OAT1 was sodium-independent andtrans-stimulated by dicarboxylate preloading. TheKm values of OTA transport via OAT1 were determined to be 2.1 μM and 0.57 μM using the oocyte expression system and S3 cells stably expressing OAT1, respectively. These values are identical with that reported in an experiment using renal proximal tubular cells (Groves et al., 1998), where theKm value of OTA was determined to be 1.4 μM. In S3 cells expressing OAT1, the addition of 2 and 10 μM OTA resulted in significant suppression of cell proliferation. Cell viability determined by MTT assay also decreased when the cell-expressing OAT1 was exposed to OTA. This suppression of cell growth and viability was rescued by the coaddition of PAH, a high-affinity substrate of OAT1. The present cell system is not equipped with the unidentified luminal exit transporter of organic anions; therefore, it does not mimic the physiological condition of the proximal tubule cells completely. Despite this limitation, we consider that these results suggest that the OTA accumulating in the proximal tubular cells via OAT1 causes tubular dysfunction, possibly also in Balkan Nephropathy.
It has been reported that several substrates such as piroxicam, octanoate, and aspartame prevent the nephrotoxicity of OTA or inhibit the transport of OTA (Baudrimont et al., 1995; Creppy et al., 1995;Groves et al., 1998). In this study, we demonstrated that piroxicam (a nonsteroidal anti-inflammatory drug) and octanoate (a fatty acid) inhibited OAT1-mediated uptake of OTA, like PAH and probenecid (a typical inhibitor of OAT1). The inhibitory effect of piroxicam and octanoate on OAT1-mediated OTA uptake can explain the prevention of OTA nephrotoxicity by these compounds. Because these substrates, despite their beneficial effects on the renal tubules, inhibit tubular secretion of OTA, OTA will remain in the body for a longer period. In contrast, aspartame (an artificial sweetener) showed no inhibition of OAT1-mediated OTA uptake. Zingerle et al. (1997) and Schwerdt et al. (1997) suggested that the reabsorption of OTA by renal proximal tubules was, in part, mediated by a H+-coupled peptide transporter. Aspartame (aspartyl-phenylalanine methyl ester) is a dipeptide derivative and a candidate for substrate of a peptide transporter of the proximal tubules. Thus, aspartame is considered to prevent nephrotoxicity by reducing the rate of OTA reabsorption via peptide transporter(s). Aspartame may be preferable to piroxicam or octanoate, as it facilitates the urinary excretion of OTA without the increased accumulation of OTA in the proximal tubule cells.
Citrinin is also a nephrotoxic mycotoxin, whose structure is related to OTA. Citrinin has been shown to cause renal dysfunction, such as glucosuria and proteinuria (Phillips et al., 1980), and is considered to be a substrate of the renal organic anion transporter (Berndt and Hayes, 1982; Berndt, 1983). Because radiolabeled citrinin is commercially unavailable, whether or not citrinin is a transportable substrate of OAT1 could not be determined. In this study, citrinin inhibited OAT1-mediated uptake of OTA. This result, along with previous reports, demonstrates that OAT1 may transport citrinin and may also play a crucial role in the development of citrinin nephrotoxicity.
Recently, it was reported that an organic anion-transporting polypeptide-1 (oatp-1) mediated the transport of OTA (Kontaxi et al., 1996) and that oatp-1 is a member of a distinct organic anion transporter family (oatp family). oatp-1 is localized at the sinusoidal membrane of hepatocytes and the luminal membrane (S3) of renal proximal tubular cells (Jacquemin et al., 1994; Bergwerk et al., 1996). Therefore, it is believed that in the liver, oatp-1 mediates the uptake of OTA from blood; in the kidney, oatp-1 reabsorbs OTA across the luminal membrane. Thus, both OAT1 and oatp-1 transport OTA in the proximal tubules. However, the Kmvalue of OTA uptake via oatp-1 in oocytes was 16.6 μM (Kontaxi et al., 1996), which is eight times higher than that via OAT1 (2.1 ± 0.3 μM in Fig. 3). In addition, because the main route of renal OTA excretion is tubular secretion, the primary step in the accumulation of OTA in renal proximal tubular cells is considered to be the basolateral uptake of OTA from the blood. Considering these findings and the inhibitory effects of piroxicam and octanoate, the OAT1 contribution seems to be more important to the accumulation of OTA in proximal tubules than that of oatp-1.
The toxicokinetics of OTA have been investigated in several animal species (Hagelberg et al., 1989; Galtier, 1991). Although more than 99% of OTA was bound to plasma proteins in all species including human, the plasma half-life of OTA is variable among species. It is about six times longer in monkey (510 h) than in rat (58–120 h), and the half-life of OTA in human is unknown but is believed to be similar to that in monkey (Kuiper-Goodman and Scott, 1989). It is possible that transport rate of OTA via OAT1 is different among species, which causes the different toxicokinetics. Excretion rate of OTA, however, is also related to the reabsorption rate of OTA via H+-peptide cotransporter and the efflux rate via unidentified transporter at the luminal membrane. Further investigations will help to elucidate the mechanisms underlying the toxicokinetic differences of OTA. From the pathophysiological point of view, it is very important to investigate the transport properties of human OAT1 and other transporters responsible for the renal handling of OTA.
In conclusion, we reported that a nephrotoxic mycotoxin, OTA, was transported by OAT1. Proximal tubular cells stably expressing OAT1 showed suppressed cell proliferation when cultured in media containing OTA. Another mycotoxin, citrinin, was capable of inhibition of OAT1-mediated uptake of OTA. The present study suggests that OAT1 plays a pivotal role in the development of nephrotoxicity of mycotoxins.
Footnotes
-
Send reprint requests to: Hitoshi Endou, M.D., Kyorin University School of Medicine, 6-20-2 Shinkawa, Mitaka, Tokyo 181-8611, Japan. E-mail: endouh{at}kyorin-u.ac.jp
- Abbreviations:
- OAT
- organic anion transporter
- OTA
- ochratoxin A
- PAH
- p-aminohippurate
- rNaDC1
- rat Na+-dicarboxylate cotransporter
- oatp
- organic anion transporting polypeptide
- MTT
- 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- Received November 3, 1998.
- Accepted February 4, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}