


Abstract
5-Fluorouracil (5-FU) is a widely used antineoplastic agent. 5-FU therapy often causes gastrointestinal toxicity, which is suppressed by concomitant administration of potassium oxonate (Oxo). Here, we investigated the effect of 5-FU on the small-intestinal drug-metabolizing enzymes, which play important roles in the first-pass metabolism of drugs, in rats, by enzyme measurements and immunoblot analyses. During repeated administration of a combination of 1-(2-tetrahydrofuryl)-5-fluorouracil, an oral 5-FU-derivative drug, and 5-chloro-2,4-dihydroxypyridine (FCD), an inhibitor of 5-FU degradation, the activities of 7-ethoxyresorufin-O-deethylase, testosterone 6β-hydroxylase, 4-methylumbelliferone UDP-glucuronyltransferase, and 1-chloro-2,4-dinitrobenzene glutathioneS-transferase decreased significantly on day 4, and the activity of NADPH-cytochrome P450 (CYP) reductase decreased significantly on day 7. These effects were found to be attributable to a reduction in the enzyme protein contents in the small-intestinal mucosa. The enzymatic alterations significantly increased the plasma concentrations of orally administered nifedipine, which was prevented by concomitant administration of Oxo with FCD. However, consecutive administration of FCD for 4 days did not cause any alterations in the activity of the hepatic CYP isozyme-supported testosterone hydroxylase. These results suggest that continuous exposure to 5-FU leads to a decrease in the activities of drug-metabolizing enzymes in the intestinal mucosa by decreasing their enzyme protein contents, and increases the plasma concentrations of orally administered nifedipine, and that the sensitivity of these enzymes to the drug is greater than that of the enzymes of the liver. These effects were prevented by concomitant administration of Oxo.
The pyrimidine analog 5-fluorouracil (5-FU) is an essential component of chemotherapeutic regimens used for the treatment of gastrointestinal (GI), head and neck, and breast cancer. 5-FU is by itself not cytotoxic, but it requires bioactivation, including phosphorylation, by multistep pathways, and inhibits the enzyme thymidylate synthase (TS) (Danenberg and Lockshin, 1982), which provides the only de novo source of thymidylate for DNA synthesis, or incorporates into RNA (Ullman and Kirsch, 1979). However, intravenous bolus administration of 5-FU is associated with low response rates and a short duration of remission because of the rapid degradation of the drug by the enzyme dihydropyrimidine dehydrogenase (DPD) (EC 1.3.1.2) in the liver. On the other hand, continuous intravenous infusion of 5-FU has been found to be associated with improved response rates in patients with gastric, colorectal, and breast cancer (Moynihan et al., 1988; Barbounis et al., 1989; Huan et al., 1989), and long-term infusion of 5-FU has been reported to be associated with higher response rates than intravenous bolus administration as adjuvant chemotherapy in patients with metastatic colorectal cancers (Lokich et al., 1989).
Combination of 1-(2-tetrahydrofuryl)-5-fluorouracil (tegafur; FT), which is an oral prodrug of 5-FU, and 5-chloro-2,4-dihydroxypyridine (CDHP), which is a competitive inhibitor of DPD (Tatsumi et al., 1987) that does not have any intrinsic antitumor activity by itself, at a molecular ratio of 1:0.4 (FCD), results in prolonged retention of an effective concentration of 5-FU in the blood, mimicking continuous intravenous infusion of the drug (Shirasaka et al., 1996; Fukushima et al., 1998); however, these methods of administration of 5-FU is limited by the high incidence of GI toxicity, which is related to the phosphorylation of the drug in the GI mucosa (Houghton et al., 1979) and the consequent potent cytotoxic action against all rapidly growing cells, including those of the GI mucosa. Monopotassium 1,2,3,4-tetrahydro-2,4-dioxo-1,3,5-triazine-6-carboxylate (potassium oxonate; Oxo) is mainly distributed to the cells of the small intestine after oral administration, and it competitively inhibits the activity of pyrimidine phosphoribosyltransferase (EC 2.4.2.10) (Shirasaka et al., 1993; Yoshisue et al., 2000b), which converts 5-FU to fluorouridine monophosphate in the small intestine. The combination of FCD + Oxo resulted in a reduction of the incidence of GI toxicity without loss of antitumor activity (Shirasaka et al., 1993; Yoshisue et al., 2000a), and better therapeutic effects were obtained on various rat tumors and human xenografts than that obtained with other p.o. fluoropyrimidines (Shirasaka et al., 1996; Fukushima et al., 1998).
The site of first-pass metabolism is generally the liver because of its size, the relatively high level of drug-metabolizing enzyme activities, and its anatomic location relative to the site of absorption. However, studies on cyclosporin (Kolars et al., 1991) indicate that, in general, cytochrome P450 (CYP) 3A activity in the intestinal mucosa also substantially contributes to first-pass metabolism. It has been reported that when dihydropyridine calcium antagonists such as nifedipine are taken along with grapefruit juice, their concentration level in the blood increases significantly, associated with potential serious adverse reactions such as hypotension. This interaction has recently drawn much attention, because it is believed to be caused by inhibition of the activities of the CYP3A subfamily of enzymes (Bailey et al., 1998) and P-glycoprotein-mediated drug transport (Edwards et al., 1999) in the intestinal mucosa. Thus, alterations in the activities of the drug-metabolizing enzymes in the small-intestinal mucosa could be expected to have a significant impact on the pharmacokinetics of drugs subject to significant small-intestinal first-pass metabolism. There are several reports on the effects of 5-FU on the hepatic CYP isozymes (Stupans et al., 1995; Afsar et al., 1996;McLeod et al., 1998), but so far there is little knowledge regarding the activities of the drug-metabolizing enzymes in the more sensitive target tissue of the drug, namely the small intestine.
In this study, we characterized the alterations in activities of the drug-metabolizing enzymes in the small intestine during continuous exposure to 5-FU by repeated administration of FCD alone or concomitantly with Oxo, and furthermore, investigated the interaction of this drug with nifedipine.
Experimental Procedures
Chemicals.
FT, CDHP, and Oxo were synthesized by Taiho Pharmaceutical Co. (Tokushima, Japan). The structures of FT, CDHP, and Oxo are shown in Fig. 1. Trypsin inhibitor, NADPH, cytochrome c, glucose 6-phosphate, glucose-6-phosphate dehydrogenase, Brij 58, UDP-glucuronic acid, phosphatidylcholine, peroxidase-conjugated rabbit anti-goat immunoglobulin, and peroxidase-conjugated rabbit anti-mouse immunoglobulin were purchased from Sigma Chemical Co. (St. Louis, MO). Resorufin, 1-chloro-2,4-dinitrobenzene (CDNB), testosterone, glutathione, saccharic acid 1,4-lactone, 4-methylumberiferone (4-MU), 4-MU-β-d-glucuronide, and polyethylene glycol 400 (PEG) were obtained from Nacalai Tesque, Inc. (Kyoto, Japan), (p-amidinophenyl)methylsulfonyl fluoride (APMSF), nifedipine, methyltestosterone, and phenol red from Wako Pure Chemicals Industries Ltd. (Osaka, Japan), 2α-, 6α-, 6β-, 7α-, 16α-, and 16β-hydroxy-testosterone from Ultrafine (Manchester, England), and 7-ethoxyresorufin from FLUKA AG (Buchs, Switzerland). The enhanced chemiluminescence kit was purchased from Amersham Pharmacia Biotech (Arlington Heights, IL). Polyvinylidene difluoride (PVDF) membrane was obtained from Bio-Rad Laboratories (Hercules, CA). Mouse monoclonal anti-CYP3A1 was purchased from Xenotech (Kansas City, KS), and goat polyclonal anti-CYP1A and anti-NADPH-CYP reductase from Daiichi Pure Chemical (Tokyo, Japan). All the reagents and solvents used were commercially available and guaranteed to be reagent-grade or high-performance liquid chromatography (HPLC)-grade.
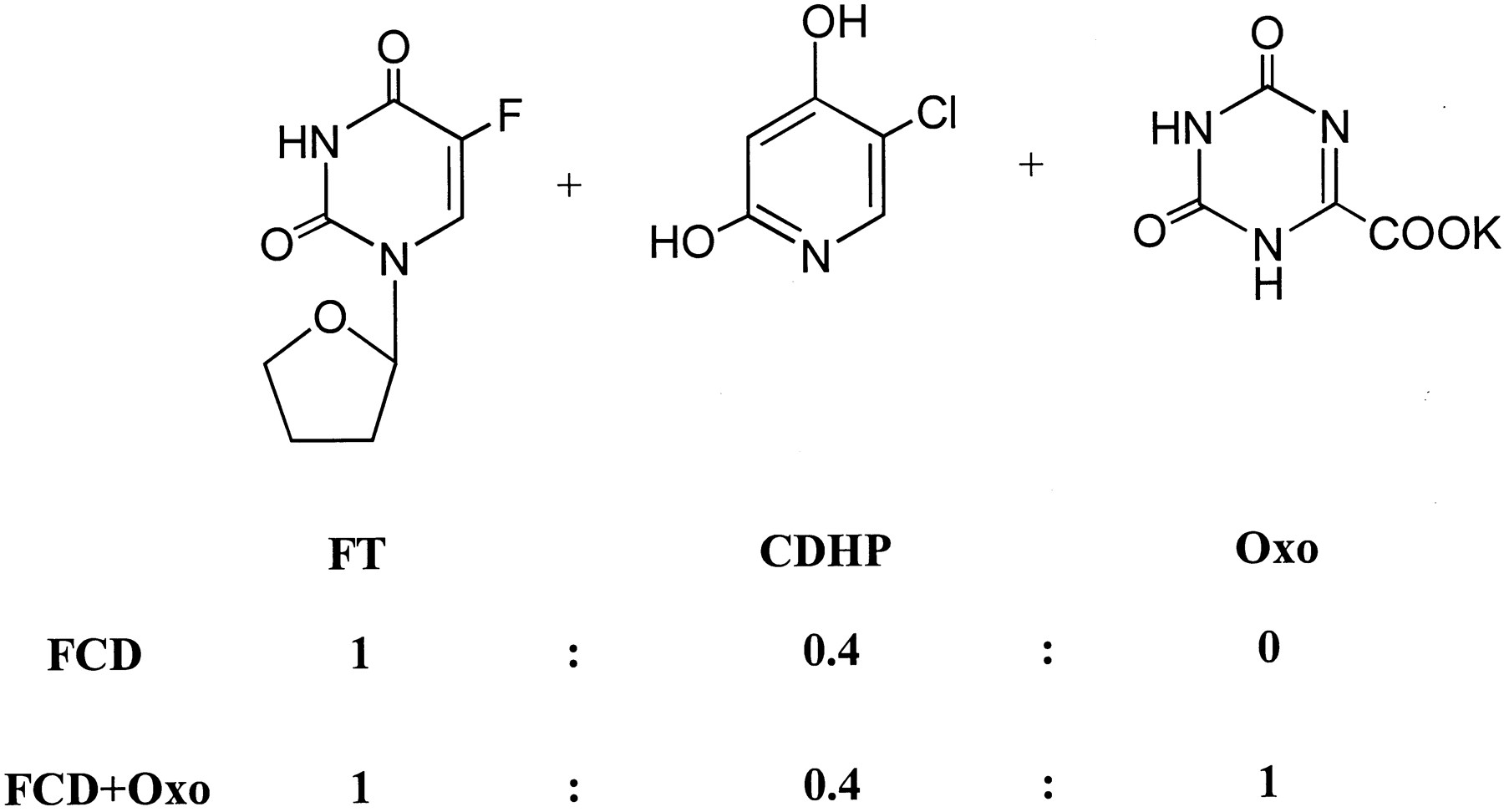
Chemical structure of the components of FCD and FCD + Oxo.
Preparation of Test Solutions.
FCD was prepared by mixing FT and CDHP at a molar ratio of 1:0.4, and FCD + Oxo was prepared by adding Oxo to FCD at a molar amount equal to that of FT. Since the active component was FT, the dose is indicated as FT dose. FCD + Oxo and FCD were dissolved at 20 mg/5 ml in 0.5% (w/v) hydroxypropylmethylcellulose solution. Nifedipine was dissolved in PEG to a final concentration of 3 mg/5 ml for oral administration and of 1 mg/1 ml for intravenous administration.
Treatment of Animals.
Six-week-old Donryu-strain male specific pathogen-free rats purchased from Charles River Japan Inc. (Shiga, Japan) were used for the experiments. The animals had free access to tap water and commercially available chow (CE-2, Clea Japan Inc., Tokyo, Japan). Each rat received FCD + Oxo or FCD at a dose of 20 mg/kg/day once daily for a maximum of 7 days. Hydroxypropylmethylcellulose solution (0.5%, w/v) was administered in the same manner as a control vehicle.
Preparation of Microsomes.
Five rats from each treatment group were sacrificed on days 1, 4, and 7 under ether anesthesia, within 3 to 6 h of the last administration. The small intestines (distal to the pylorus) were immediately excised and perfused with ice-cold solution A (physiological saline containing 0.5 mM dithiothreitol, 0.1 mM EDTA, 2 mM APMSF, and 0.5 mg/ml trypsin inhibitor). Thereafter, these small intestines were slit open and the upper villous mucosal layers were gently scraped off with the edge of a glass slide and pulverized using a microdismembrator (Braun, Melsungen, Germany) in 15 ml of solution A. The pulverized samples were centrifuged at 9,000g for 20 min at 4°C, and the supernatants were centrifuged at 105,000g for 60 min at 4°C to separate the cytosolic (supernatant) and microsomal (pellet) components. The microsomal pellets were resuspended in buffer A (0.1 M Tris-HCl buffer, pH 7.4, containing 0.5 mM dithiothreitol, 0.1 mM EDTA, 2 mM APMSF, 0.5 mg/ml trypsin inhibitor, and 20% glycerol). Hepatic microsomes were prepared from rats after 4 consecutive days of treatment with vehicle, FCD + Oxo, or FCD by differential centrifugation (Murray et al., 1983) followed by perfusion with ice-cold physiological saline and homogenization in a glass-Teflon Potter Elvehjem homogenizer (Asahi Techno Glass Co., Tokyo, Japan). The microsomal pellets were resuspended in 0.1 M Tris-HCl buffer (pH 7.4) containing 0.5 mM dithiothreitol, 0.1 mM EDTA, and 20% glycerol. The protein content of the microsomes and the cytosol were determined according to the method of Bradford (1976) with a Bio-Rad protein assay kit using bovine serum albumin as the standard. The samples were stored frozen at −80°C until further analysis.
Enzyme Assay.
Testosterone hydroxylase activity was determined by HPLC analysis, using an LC-6 dual pump system with an SPD-6A variable wavelength UV detector (Shimadzu, Kyoto, Japan) operated at 254 nm. An incubation mixture containing 0.1 or 1 mg of protein/ml, respectively, of the hepatic and small-intestinal microsomes, along with 1 mM testosterone, was preincubated for 5 min at 37°C. Metabolism was initiated by the addition of a NADPH-generating system (1 mM NADPH, 10 mM glucose 6-phosphate, 1 IU/ml glucose-6-phosphate dehydrogenase). The reaction was conducted at 37°C for 10 and 30 min, respectively, for hepatic and small-intestinal microsomes, and stopped by the addition of 1 N hydrochloride (100 μl). An internal standard (methyltestosterone) was added to each hepatic and small-intestinal microsome sample at a final concentration of 20 and 2 μM, respectively, followed by the addition of 2 ml of ethyl acetate to extract the metabolites. The resultant mixture was vortex-mixed and the ethyl acetate layer was separated by centrifugation, followed by evaporation to dryness under nitrogen. The residue was reconstituted in 150 μl of 50% methanol in water. HPLC separation was conducted using an Inertsil ODS-2 column (15 cm × 4.6-mm i.d., GL Sciences; Tokyo, Japan). The mobile phase was composed of 1% acetic acid and acetonitrile (80:20–70:30 over 0–25 min, and further 70:30–40:60 over 25–35 min) at the flow rate of 0.9 ml/min. Under these conditions, the retention times were 11.4, 13.1, 13.9, 17.2, 22.1, 24.7, and 37.3 min for 6α-, 7 α-, 6β-, 16 α-, 16 β-, 2α-, and internal standard (methyltestosterone), respectively.
UDP-glucuronyltransferase (UGT) activity directed at 4-MU was measured fluorometrically. The incubation medium consisted of 50 mM Tris-HCl buffer (pH 7.4), 0.5 mM 4-MU, 10 mM MgCl2, 150 μg of phosphatidylcholine, 2 mM UDP-glucuronic acid, 1.5 mg of bovine serum albumin, 1 mM saccharic acid 1,4-lactone, and microsomes treated with Brij 58 (0.15 μg/mg of protein) in 0.3 ml. The incubation was performed for 5 min, and the reaction was stopped by the addition of 0.5 ml of 0.5 M trichloroacetic acid, followed by centrifugation of the protein precipitate. A 10-μl aliquot of the obtained supernatant was mixed into 3 ml of 1.6 M glycine buffer (pH 10.3), and the fluorescence intensity was measured at 370 nm with the excitation wavelength of 310 nm. Under the fluorometric assay conditions employed, the relative fluorescence intensities of 4-MU and 4-MU-β-d-glucuronide were 0.99 and 100, respectively.
Microsomal 7-ethoxyresorufin-O-deethylase (EROD) activity was measured by the fluorometric assay method described by Matsubara et al. (1983). The NADPH-CYP reductase activity of the small-intestinal microsomes was assayed by measuring the reduction of cytochromec (Williams and Kamin, 1962). GlutathioneS-transferase (GST) activity in the cytosol was estimated by the method described by Habig et al. (1974), using CDNB as the substrate.
Immunoblot Analysis.
Microsomal proteins were separated by SDS-polyacrylamide gel electrophoresis, as described by Laemmli (1970)in 10% polyacylamide gels. Liver microsomes were loaded at 10 to 50 μg of protein/well. The proteins were electrophoretically transferred to a PVDF membrane (Towbin et al., 1979) blocked with 4% nonfat dry milk in phosphate-buffered saline (pH 7.5) containing 0.1% (v/v) Tween 20 (PBS-T) for 1 h at room temperature, incubated with an anti-P450 antibody in PBS-T containing 0.4% milk for an additional hour, washed with PBS-T containing 0.4% milk, and then incubated with a secondary antibody at 1/5,000 to 1/10,0000 dilution in PBS-T containing 0.4% milk. The primary antibodies were localized with peroxidase-conjugated rabbit anti-mouse IgG or rabbit anti-goat IgG. Staining of the antigen-antibody complexes was carried out using the enhanced chemiluminescence kit according to the manufacturer's instructions. The optical density of each stained band was determined with a Pharmacia-LKB densitometer using the Image Master Software.
Absorption Experiments.
The phenol red absorption experiment was performed by an in situ closed loop intestine technique in rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. A small-intestinal loop was prepared by cannulation of the proximal and distal ends of the small intestine with a silicone tubing. Two ml of phenol red at the concentration of 1 mg/ml at 37°C was injected into the small-intestinal loop. The jugular artery was also cannulated with a polyethylene tube. After the phenol red injection, blood samples were periodically drawn from the jugular artery. The plasma was separated by centrifugation, and the phenol red concentration in the plasma was determined.
Urinary Excretion of Phenol Red.
The urinary excretion of phenol red was measured in rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. Phenol red at the concentration of 1 mg/ml of physiological saline was administered by gavage using steel-ball-tipped feeding needles. The animals were placed in a metabolic cage and urine was collected until 8 h after the administration. Blank determinations were made in the same manner, except that physiological saline instead of phenol red was administered orally. The urinary excretion of phenol red was expressed as a percentage of the dose administered.
Analytical Methods for Phenol Red Determinations.
A spectrometric method was used for the determination of phenol red concentrations. A 200-μl plasma sample was alkalinized with 2 ml of 1 N sodium hydroxide and the concentration of phenol red determined spectrophotometrically at 560 nm. Urine samples were mixed with 10 ml of distilled water and centrifuged. One milliliter of each urine sample was then alkalinized with 5 ml of 1 N sodium hydroxide and the phenol red concentration determined spectrophotometrically at 560 nm.
Pharmacokinetic Studies.
All the procedures of drug preparation, dosing, and blood collection were performed under sodium lamps to prevent photodegradation of nifedipine (Grundy et al., 1994a). Nifedipine was dissolved in PEG and administered by gavage at the dose of 3 ml/kg to rats (n = 5) treated with vehicle, FCD + Oxo, or FCD for 4 consecutive days. A different group composed of similarly treated animals (n = 5) received nifedipine dissolved in PEG by bolus injection at a dose of 1 mg/kg through the jugular vein. Blood samples (0.5 ml) were collected from animals administered the drug intravenously at 5, 10, 15, and 40 min and 1, 1.5, 2, 3, and 4 h after the dose; and from animals administered the drug orally at 5, 15, and 30 min and 1, 1.5, 2, 3, 4, and 6 h after the dose. The blood sample volume withdrawn was immediately replaced with an equal volume of physiological saline. Plasma was separated from each blood sample by centrifugation and stored in light-resistant bags at −80°C until further analysis. The nifedipine concentrations in the rat plasma samples were determined according to a previously reported HPLC method (Grundy et al., 1994b) with some modifications. Samples of 0.05 ml were diluted with water to 1.0 ml and vortex-mixed after the addition of 100 μl of 1.0 M sodium hydroxide and 5 ml of t-butylmethyl ether-isooctane (75:25, v/v); the organic layer was then separated by centrifugation. After removal of the upper organic layer, the extraction was repeated three more times, and the pooled extract was evaporated to dryness. To the resulting residue was added 200 μl of the mobile phase and aliquots of 150 μl were injected onto the HPLC column. Analytical separation was accomplished using a Develosil ODS-5 column (25 cm × 4.6-mm i.d.; Nomura Chemical Co., Ltd., Aichi, Japan), and the mobile phase consisted of acetonitrile/water/acetic acid/triethylamine (50:49:1:0.03). The HPLC system consisted of an LC-6 dual pump system equipped with an SPD-6A variable wavelength UV detector (Shimadzu) operated at 350 nm. A standard calibration curve (50–1000 ng/ml) was drawn by adding a known amount of the drug to 0.05 ml of blank rat plasma and diluting it with water to 1.0 ml as described above. The calibration curve best fit the regression using a 1/x2 weighting factor (where xcorresponds to the amount of nifedipine added;r2 values greater than 0.99 were always obtained).
Pharmacokinetic Analysis.
Standard pharmacokinetic parameters obtained from each of the individual rat plasma concentration-time profiles of nifedipine were calculated by noncompartmental methods using the computer program WinNonlin, Standard Edition, version 3.1 (Pharsight Inc., Cary, NC). The area under the plasma concentration-time curve from time 0 to infinity (AUC) was calculated using the linear trapezoidal rule from time 0 to the time of the last quantifiable concentration, followed by extrapolation to infinity. The half-life (t1/2) was determined by linear regression of the log-linear portion of the plasma concentration-time profile. The apparent plasma clearance (CL) was calculated by dividing the dose by the AUC. The oral bioavailability was determined according to the equation
Statistical Analysis
Data storage and statistical analyses were performed with the Statistical Analysis System, version 6.12 software (SAS Institute Inc., Cary, NC). Comparison between groups was performed by Dunnett's multiple range test.
Results
Effects of Administration of 5-FU and Oxo on the Amounts of Cytosolic and Microsomal Protein in the Small-Intestinal Mucosa.
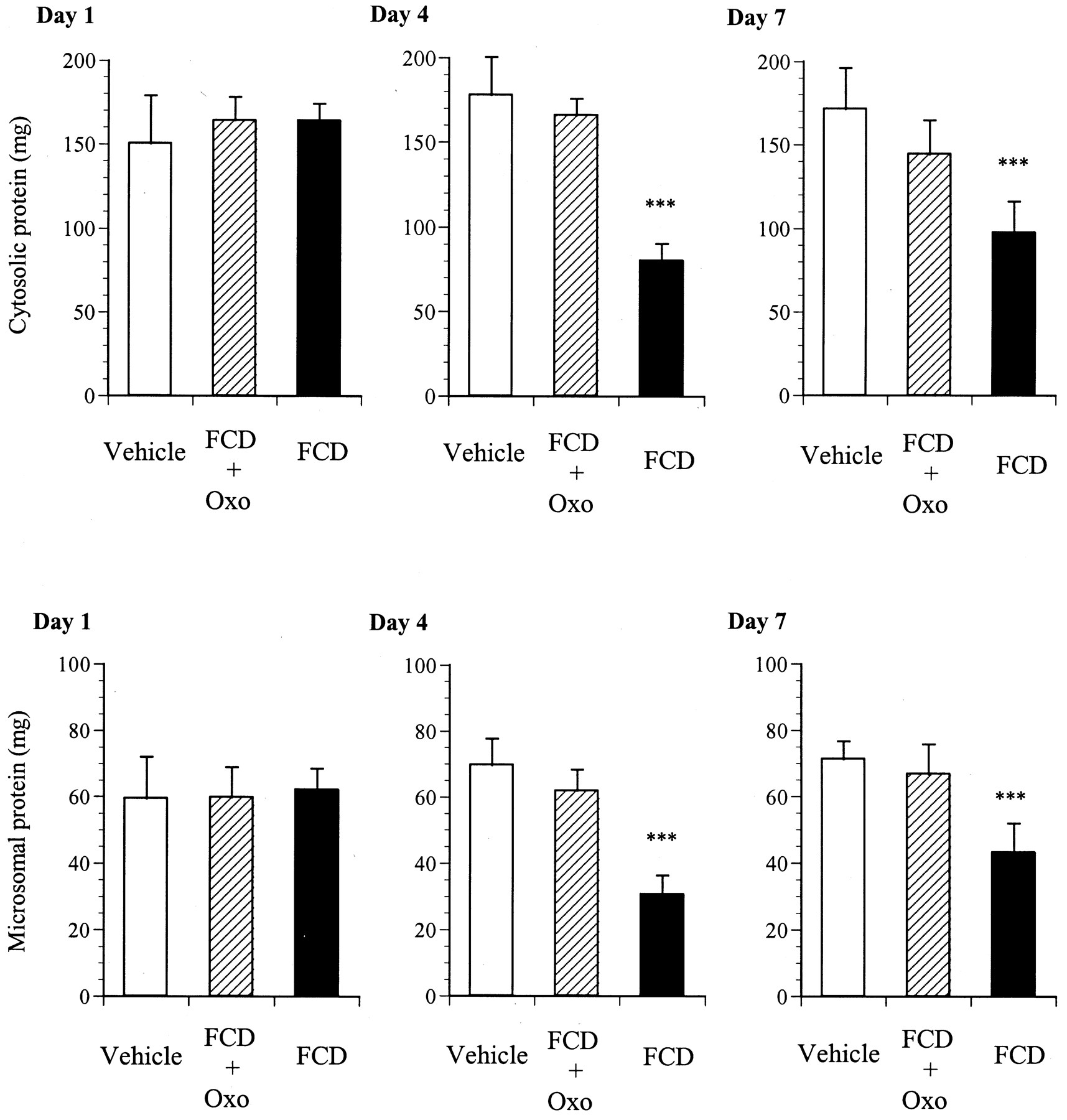
Figure 2 shows the changes in the amounts of protein in the cytosolic and microsomal fractions prepared from the small-intestinal mucosa during repeated administration of vehicle, FCD + Oxo, or FCD to rats for a maximum period of 7 days. The amounts of protein in the cytosolic and microsomal fractions prepared from FCD + Oxo-treated rats were not significantly different from those in the vehicle-treated control group throughout the experimental period. In FCD-treated rats, however, the amounts of both cytosolic and microsomal protein decreased simultaneously and significantly after day 4; no significant alterations in the amounts of protein in either fraction were observed on day 1.

Effect of administration of FCD + Oxo or FCD on the amounts of protein contained in the cytosolic and microsomal fractions prepared from the small-intestinal mucosa of rats, on days 1, 4, and 7. The rats received oral administration once daily of FCD + Oxo or FCD at a dose of 20 mg/kg. Small-intestinal cytosolic and microsomal fractions were prepared on days 1, 4, and 7 as described underExperimental Procedures. The results are expressed as mean ± S.D. for five rats in each treatment group on the respective days. ***Significantly different (P < 0.001) from vehicle-treated group on the respective day.
Effects of Administration of 5-FU and Oxo on the Activities of Drug-Metabolizing Enzymes in the Small-Intestinal Mucosa.
Figure3 shows the changes in the activities of EROD, testosterone 6β-hydroxylase, and NADPH-CYP reductase in the small-intestinal mucosa during repeated administration of vehicle, FCD + Oxo, or FCD to rats for a maximum period of 7 days. As compared with those in the vehicle-treated rats, the activities of EROD and testosterone 6β-hydroxylase in the small-intestinal mucosa in FCD-treated animals were significantly decreased on day 4 (a 56 and 26% decrease, respectively), and that of NADPH-CYP reductase was also significantly decreased on day 7 (48% decrease). On the other hand, the activities of these enzymes in FCD + Oxo-treated rats did not significantly differ from those in the vehicle-treated animals throughout the experimental period. Figure4 shows the changes in the activities of GST and UGT during repeated administration of vehicle, FCD + Oxo, or FCD to rats for a maximum period of 7 days. There were no significant differences in the activities of these enzymes between vehicle-treated rats and FCD + Oxo-treated rats throughout the experimental period; however, the activities of both GST and UGT in the small-intestinal mucosa of rats treated with FCD alone for 4 days were significantly decreased to 48 and 54%, respectively, as compared with those in the vehicle-treated rats.

Effect of administration of FCD + Oxo or FCD on the activities of the small-intestinal microsomal enzymes, EROD, testosterone 6β-hydroxylase, and NADPH-CYP reductase in rats on days 1, 4, and 7. The rats received oral administration once daily of FCD + Oxo or FCD at a dose of 20 mg/kg. Small-intestinal microsomal fractions were prepared on days 1, 4, and 7 as described underExperimental Procedures. Results are expressed as mean ± S.D. for five rats per treatment group on the respective days. ***Significantly different (P < 0.001) from vehicle-treated group on the respective day.

Effect of treatment with FCD + Oxo or FCD on the activity of the small-intestinal cytosolic enzyme, GST, and the small-intestinal microsomal enzyme, UGT, in rats on days 1, 4, and 7. The rats received oral administration once daily of FCD + Oxo or FCD at the dose of 20 mg/kg. Small-intestinal cytosolic and microsomal fractions were prepared on days 1, 4, and 7, and the activities of GST or UGT were measured as described under Experimental Procedures using CDNB and 4-MU as the substrate, respectively. Results are expressed as mean ± S.D. for five rats per treatment group on the respective days. ***Significantly different (P < 0.001) from vehicle-treated group on the respective day.
Effects of Administration of 5-FU and Oxo on the Contents of CYP1A, CYP3A, and NADPH-CYP Reductase in the Small-Intestinal Mucosa.
Figure 5 shows the results of immunoblot analysis for CYP1A, -3A, and NADPH-CYP reductase in the microsomal fractions prepared from rats administered vehicle, FCD + Oxo, or FCD for a consecutive period of 4 or 7 days, and Fig.6 shows the levels of these proteins as determined by densitometric analysis of the immunoblots. In regard to the CYP1A and -3A isoforms, treatment with FCD for 4 days resulted in a decrease in the amount of immunodetectable -1A and -3A isoforms to almost 66% and 40%, respectively, as compared with the levels in the vehicle-treated group; however, no significant changes were observed in the group treated with FCD + Oxo for 4 days. In regard to the NADPH-CYP reductase, the administration of FCD alone for 4 days did not cause any alterations of its content in the small-intestinal mucosa, however, that for 7 days resulted in a decrease in the amount of immunodetectable reductase to almost 48% of the level in the vehicle-treated group. On the other hand, treatment with FCD + Oxo for 7 days had no effect on the levels of this enzyme in the intestinal mucosa.

Immunoblots of CYP1A, -3A, and NADPH-CYP reductase proteins in the small-intestinal microsomal fractions prepared from rats administered vehicle, FCD + Oxo or FCD for 4 or 7 consecutive days. Microsomes, loaded at 10 to 50 μg of protein/well, were subjected to SDS-polyacrylamide gel electrophoresis, on 10% acrylamide gels, and transferred electrophoretically onto the PVDF membrane. The immunoblots were probed with anti-CYP1A1/2, -3A1, and NADPH-CYP reductase immunoglobulin G, followed by peroxidase-conjugated anti-goat secondary antibody (for CYP1A1/2 and NADPH-CYP reductase), or anti-mouse secondary antibody (for CYP3A1). Staining of the antigen-antibody complexes was carried out using an enhanced chemiluminescence detection kit. The microsomal samples were loaded as follows: lanes 1 and 2, vehicle-treated rats; lanes 3 and 4, FCD + Oxo-treated rats; lanes 5 and 6, FCD-treated rats. The immunoblot analysis was repeated for the other individuals with similar results.
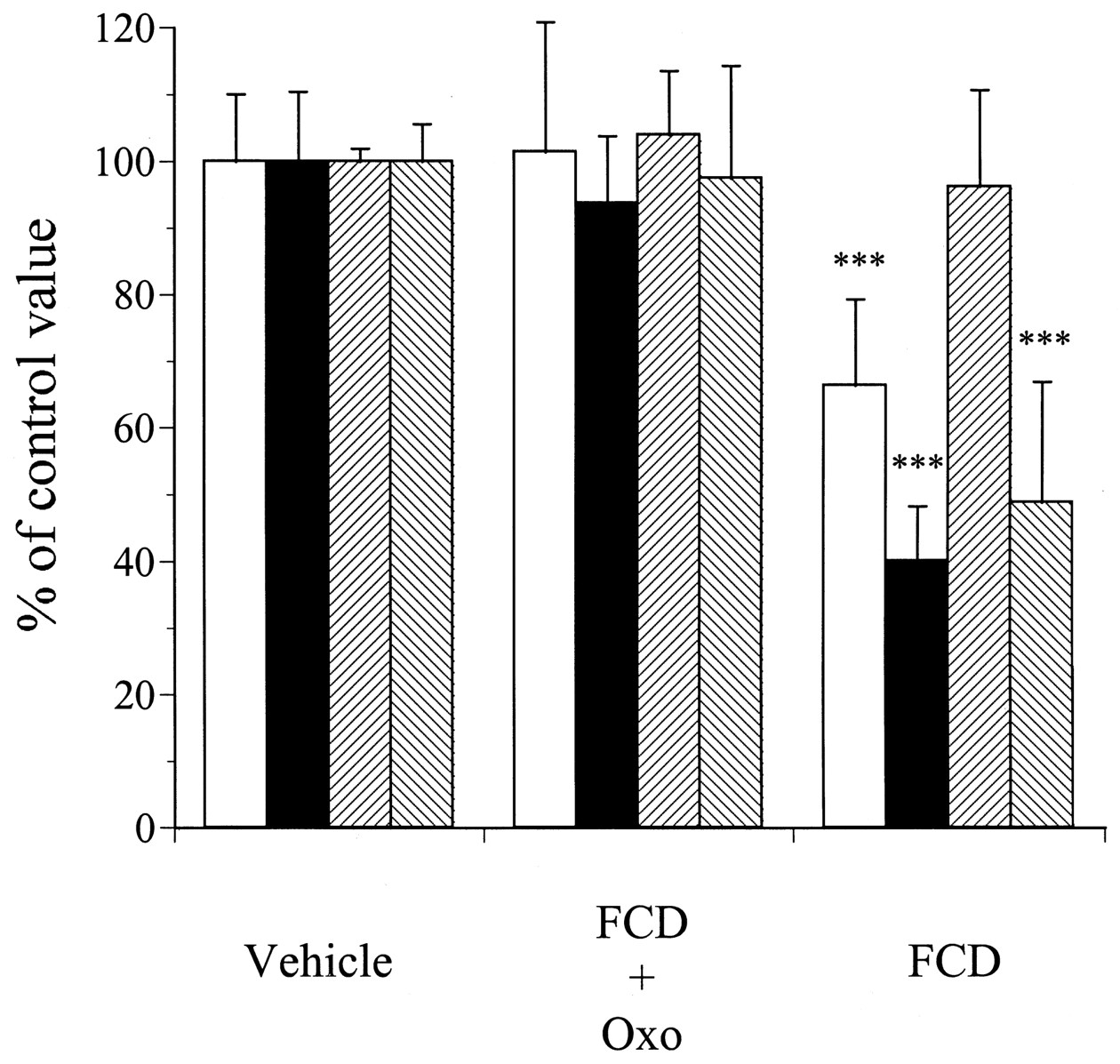
Quantitative immunoblot analysis to determine the levels of CYP1A, -3A, and NADPH-CYP reductase in small-intestinal microsomal fractions prepared from rats treated with vehicle, FCD + Oxo, or FCD for 4 or 7 consecutive days. The data represent the mean ± S.D. of arbitrary densitometric units from five individual rats per treatment group and the results for the FCD + Oxo- and FCD-treated rats are expressed as a percentage of the control. ***Significantly different (P < 0.001) from vehicle-treated rats. □, CYP1A, day 4; ▪, CYP3A, day 4; ▨, NADPH-CYP reductase, day 4; ▧, NADPH-CYP reductase, day 7.
Effects of Administration of 5-FU and Oxo on the Absorption of Phenol Red throughout the Small-Intestinal Mucosa.
The absorption of phenol red was evaluated by the in situ closed-loop-intestine technique in the small-intestine preparations of rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. The plasma phenol red concentrations and their AUC0–2 h of rats administered vehicle, FCD + Oxo, or FCD are shown in Fig.7. During administration of FCD + Oxo or FCD for 4 days, the concentration of phenol red, absorbed via the small intestine in the plasma, was similar to that in the vehicle-treated rats, and the AUC0–2 h of phenol red was also not significantly different among the three groups of animals. Figure8 shows the effects of administration of vehicle, FCD + Oxo, or FCD for 4 consecutive days on the urinary recovery of phenol red in rats. During administration of FCD + Oxo or FCD for 4 days, the urinary recovery of phenol red was 4.11 ± 0.54 and 4.06 ± 0.77% of the dose, respectively, which were not significantly different from the percentage in the vehicle-treated group (3.72 ± 0.93% of dose).

Phenol red absorption throughout the small intestine of rats after administration of vehicle, FCD + Oxo, or FCD for 4 consecutive days. FCD + Oxo or FCD was administered orally at the dose of 20 mg/kg to rats once daily for 4 consecutive days. The absorption experiment was performed by the in situ closed-loop intestine technique as described under Experimental Procedures. The values are expressed as mean ± S.D. for three rats. A and B, plasma concentration-time profiles and AUC0–2 h, respectively. No significant differences were observed as compared with the values in vehicle-treated rats.
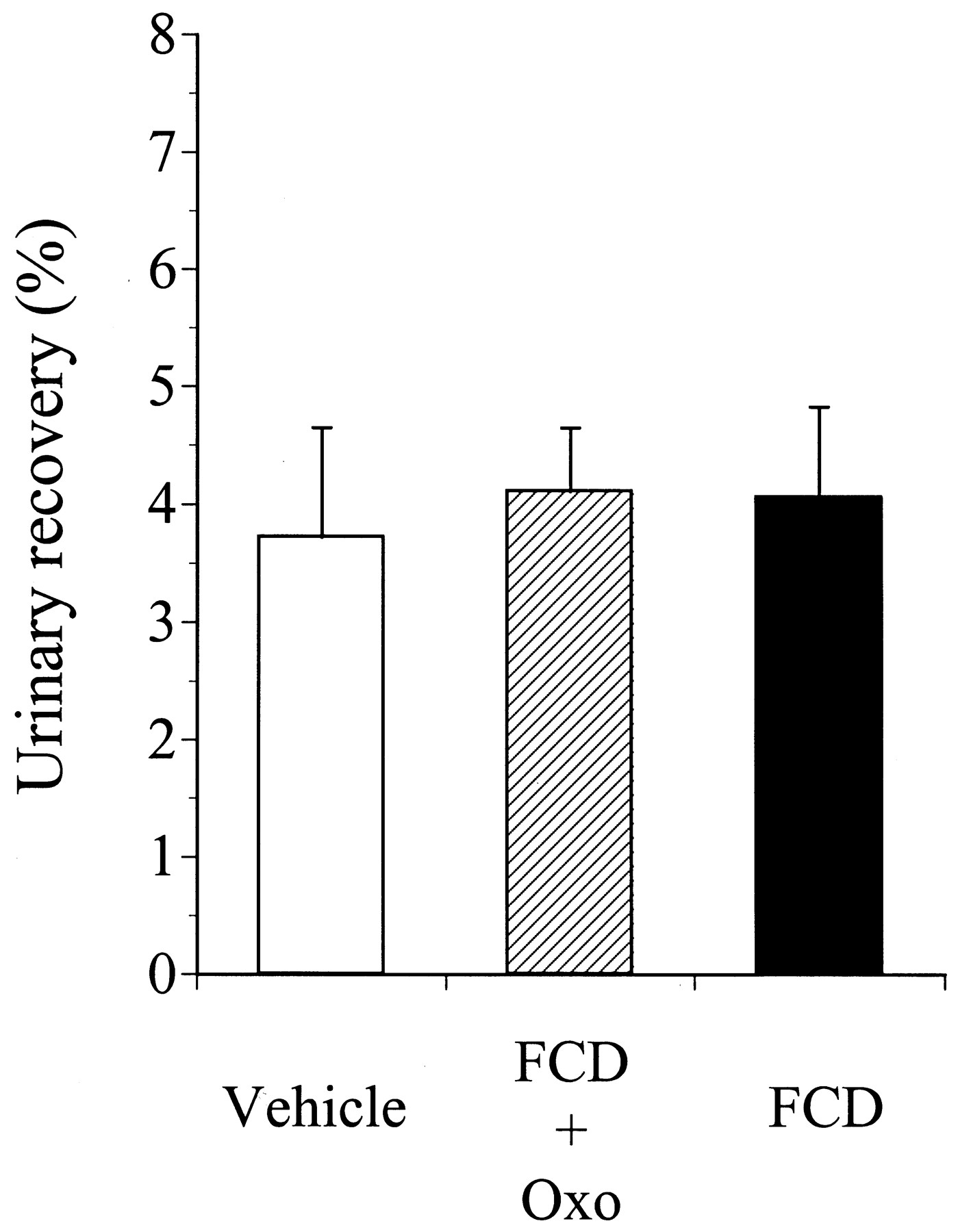
Urinary recovery of phenol red for 8 h after oral administration in rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. FCD + Oxo or FCD was administered orally at the dose of 20 mg/kg to rats once daily for 4 consecutive days. The values are expressed as the mean ± S.D. for three rats. There were no significant differences as compared with the values in the vehicle-treated group.
Effects of Administration of 5-FU and Oxo on the Activities of Testosterone Hydroxylase at the CYP-Specific Position in the Liver.
Table 1 shows the activities of testosterone hydroxylase at selected positions, indicating the selective activities of CYP isozymes, in the livers of rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. It was noted that the administration of FCD + Oxo or FCD at the dose of 20 mg/kg/day for 4 days did not cause any statistically significant alterations in the activities of testosterone 6α-, 7α-, 6β-, 16α-, 16β-, or 2α-hydroxylase in the liver.
CYP-dependent regio- and stereo-selective testosterone hydroxylation by liver microsomal enzymes in rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days
Effects of Administration of 5-FU and Oxo on the Bioavailability of Nifedipine.
Figure 9 shows the mean plasma concentration-time profile of nifedipine after intravenous and oral administration to rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. After intravenous administration, the plasma concentration-time profiles of nifedipine were almost the same among the three groups. On the other hand, the plasma concentrations of nifedipine after oral administration were higher in FCD-treated animals than in vehicle- or FCD + Oxo-treated animals. Table2 shows the AUC,t1/2, CL, andCmax of nifedipine after intravenous and oral administration to rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. The AUC, t1/2, and CL of nifedipine after intravenous administration were not significantly different among the three groups of rats. However, while the t1/2 remained almost unchanged, the AUC and Cmax after oral administration of the drug were significantly increased in the FCD-treated animals. The AUC, Cmax, and t1/2 after oral administration in FCD + Oxo-treated rats did not differ significantly from the corresponding values in the vehicle-treated animals. The oral bioavailability of nifedipine in vehicle-, FCD + Oxo-, and FCD-treated rats was 46.2, 41.9, and 74.7%, respectively.

Plasma nifedipine concentration-time curve following intravenous (A) and oral (B) administration of the drug at the dose of 1 and 3 mg/kg, respectively, to rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. Each point represents the mean ± S.D. for five rats.
Pharmacokinetics parameters obtained following i.v. administration (1 mg/kg) or p.o. administration (3 mg/kg) of nifedipine to rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days
Discussion
Previous animal studies have shown that exposure to 5-FU can alter the expression of the hepatic CYP isozymes CYP3A and -2C11, depending on the dose schedule and route of administration of the drug (Stupans et al., 1995; Afsar et al., 1996; McLeod et al., 1998). In the present study, we characterized the changes that occur in the amounts of protein in the cytosolic and microsomal fractions and the activities of the drug-metabolizing enzymes, including CYP, in the small-intestinal mucosa during repeated administration of FCD + Oxo or FCD for a maximum period of 7 days.
Single administration of FCD + Oxo or FCD (day 1) did not cause any alterations in the amounts of cytosolic or microsomal protein or the activities of the drug-metabolizing enzymes in the small-intestinal mucosa. Repeated administration of FCD led to a significant decrease in the amounts of cytosolic and microsomal protein in the small-intestinal mucosa on day 4; however, concomitant administration of FCD and Oxo did not cause any alterations in the protein amounts throughout the experimental period. The important cytostatic effect of 5-FU is mediated through stable ternary complex formation of its phosphorylated metabolite 5-fluorodeoxyuridine 5′-monophosphate, which together with methylenetetrahydrofolic acid binds to TS, causing inhibition of DNA synthesis (Danenberg and Lockshin, 1982). Furthermore, the formation of 5-fluoro-2-uridinetriphosphate results in the incorporation of the drug into RNA, leading to a disruption in RNA processing and function (Ullman and Kirsch, 1979). We reasoned that these cytotoxic actions, which occurred preferentially in rapidly growing cells, including those of the small-intestinal mucosa (Houghton et al., 1979), led to a reduction in the protein content in the small-intestinal mucosa as a result of the inhibition of protein synthesis by repeated administration of FCD. The therapeutic efficacy of fluoropyrimidines depends on the duration of TS inhibition (van Laar et al., 1996); depression of the hepatic CYP enzyme is also observed after multiple doses of 5-FU, but not after a single-dose administration of the agent (Stupans et al., 1995). The similar effect of 5-FU was also observed in the small intestine. We previously reported (Yoshisue et al., 2000a) that whereas the TS activity in the small intestine decreased during repeated administration of FCD, when the drug was administered concomitantly with Oxo, the small-intestinal TS activity remained unchanged, perhaps because Oxo, distributed only to the small-intestinal mucosa (Yoshisue et al., 2000b), inhibited the phosphorylation of 5-FU in this tissue (Shirasaka et al., 1993). We thus concluded that this action of Oxo was the reason that the amounts of the cytosolic and microsomal protein in the small-intestinal mucosa were not affected by repeated administration of FCD + Oxo throughout the experimental period.
We have shown that CYP1A-mediated EROD activity, CYP3A-dependent testosterone 6β-hydroxylase activity, 4-MU UGT activity, and CDNB GST activity in the small-intestinal mucosa decreased significantly on day 4 during FCD administration. The activities of all of these enzymes remained unchanged throughout the experimental period, as compared with those in the vehicle-treated animals, when Oxo was also administered concomitantly with FCD. We concluded from these results that continuous exposure to 5-FU resulted in a decrease in the activities of the drug-metabolizng enzymes in the small-intestinal mucosa, regardless of whether the enzyme was contained in the cytosolic or microsomal fractions or whether it mediated oxidation or conjugation, and that this decrease in the enzymatic activities was prevented by concomitant administration of Oxo. The activity of NADPH-CYP reductase in the small-intestinal microsomes was significantly decreased during repeated administration of FCD on day 7, but remained unaltered on day 4. Furthermore, the immunoblot analysis indicated that the decrease in the activity of EROD and testosterone 6β-hydroxylase on day 4, and that of NADPH-CYP reductase on day 7 during the administration of FCD, were closely related to the reduction in the CYP1A, -3A, or NADPH-CYP reductase protein content in the small-intestinal mucosa on the respective days. Our findings provide the first evidence that the depression in the small-intestinal enzyme activities during continuous exposure to 5-FU results from a reduction in their enzyme protein content. The different sensitivity of these drug-metabolizing enzymes and NADPH-CYP reductase to 5-FU might be related to the different turnover cycle time of each of these enzymes in the small intestine.
In regard to the small-intestinal damage resulting from the administration of FCD + Oxo or FCD, we previously reported that histopathological examination of the GI tissues during repeated administration of FCD + Oxo or FCD indicated that the small-intestinal damage caused by FCD + Oxo or FCD was still slight on day 4 (Yoshisue et al., 2000a). We also estimated the small-intestinal damage from the change in the urinary recovery of orally administered phenol red in vivo as described by Nakamura et al. (1982) and furthermore examined the absorption of the poorly absorbed marker, phenol red, throughout the small intestine using the in situ-loop-intestinal method in rats administered vehicle, FCD + Oxo, or FCD for 4 days. The results indicated that the small-intestinal damage occurring during the administration of FCD + Oxo or FCD, such as dysfunction of the epithelial barrier, was not significantly different from that occurring during the administration of vehicle. In addition, examination of testosterone hydroxylation at selected, CYP-specific positions in the liver indicated that CYP3A-dependent 6β-hydroxylase activities (Arlotto et al., 1991) were not affected by the administration of FCD + Oxo or FCD for 4 days; nor was any impact noted on the activities of CYP2A1/2-linked 6α- or 7α-hydroxylase, CYP2B1/2-catalyzed 16α- or 16β-hydroxylase, or CYP2C11-supported 16α- and 2α-hydroxylase in the liver (Waxman, 1991; Shimada et al., 1995; You et al., 1999). We concluded from these results that continuous exposure to 5-FU affected the small-intestinal drug-metabolizing enzymes even prior to causing epithelial barrier dysfunction, and that the CYP of the intestinal mucosa were even more sensitive to the drug than hepatic CYP enzymes.
Furthermore, to investigate the drug interactions resulting from the alteration of CYP3A activity in the small intestine induced by 5-FU, we intravenously and orally administered nifedipine, which undergoes significant first-pass metabolism in the intestine mediated by CYP3A after oral administration, in rats administered vehicle, FCD + Oxo, or FCD for 4 consecutive days. Pretreatment with FCD + Oxo or FCD did not cause any changes of the plasma concentration-time profile or pharmacokinetic parameters of nifedipine after intravenous administration of the drug. After oral administration of nifedipine, pretreatment with FCD significantly increased theCmax and AUC of nifedipine, but did not change the t1/2 of the drug, and pretreatment with FCD + Oxo did not affect any of these parameters. We concluded that the reduction in the small-intestinal first-pass metabolism of nifedipine, resulting from the decrease in the small-intestinal CYP3A activity induced by continuous exposure to 5-FU, was responsible for the increase in the plasma concentration of nifedipine after oral administration. This interaction between 5-FU-derivative drugs and nifedipine was prevented by concomitant administration of Oxo. The small-intestinal CYP content is approximately 0.055 to 0.14 nmol/mg of protein (Watkins et al., 1987;Iatsimirskaia et al., 1997) and 0.046 to 0.116 nmol/mg of protein (Rich et al., 1989; Sesardic et al., 1990) in humans and rats, respectively, and the predominant CYP isoenzyme in the human small intestine is CYP3A4, which accounts for almost 50% of the small-intestinal CYP content (Paine et al., 1997). On the other hand, the predominant isoenzyme in rats is CYP1A, and the CYP3A content is small (Zhang et al., 1996). These estimates of the small-intestinal contents of the isoenzymes of the nifedipine-metabolizing enzyme, CYP3A, in rats and humans, indicate that the interaction between 5-FU-derivative drugs and nifedipine might more seriously increase the plasma concentration of nifedipine in humans.
In conclusion, the present study indicates that continuous exposure to 5-FU results in a reduction of activity of almost all the drug-metabolizing enzymes in the small-intestinal mucosa, more sensitively than that in the liver, attributable to a decrease of their enzyme protein contents. These alterations consequently caused increase in the plasma concentration of drugs subject to substantial small-intestinal first-pass metabolism, such as nifedipine. However, these alterations in the activities of the small-intestinal drug-metabolizing enzymes and the consequent drug interactions could be prevented by concomitant administration of Oxo with the 5-FU-derivative drugs. Thus, knowledge of the alterations of activities of drug-metabolizing enzymes by antineoplastic agents could be expected to aid in the prediction and prevention of drug interactions, which in turn will lead to a more rational and efficacious application of combination chemotherapy for cancer.
Footnotes
-
Send reprint requests to: Dr. Kunihiro Yoshisue, Pharmacokinetics Research Laboratory, Taiho Pharmaceutical Co., Ltd., 224-2, Ebisuno, Hiraishi, Kawauchi-cho, Tokushima 771-0194, Japan. E-mail: kuni-yosisue{at}taiho.co.jp
- Abbreviation:
- 5-FU
- 5-fluorouracil
- GI
- gastrointestinal
- TS
- thymidylate synthase
- DPD
- dihydropyrimidine dehydrogenase
- FT
- 1-(2-tetrahydrofuryl)-5-fluorouracil
- CDHP
- 5-chloro-2,4-dihydroxypyridine
- FCD
- a mixture of FT and CDHP at a molar ratio of 1:0.4
- Oxo
- 1,2,3,4-tetrahydro-2,4-dioxo-1,3,5-triazine-6-carboxylate
- FCD + Oxo
- a mixture of FT, CDHP, and Oxo at a molar ratio of 1:0.4:1
- CYP
- cytochrome P450
- CDNB
- 1-chloro-2,4-dinitrobenzene
- 4-MU
- 4-methylumberiferone
- PEG
- polyethylene glycol 4000
- APMSF
- (p-amidinophenyl)methylsulfonyl fluoride
- PVDF
- polyvinylidene difluoride
- HPLC
- high-performance liquid chromatography
- EDTA
- ethylenediamine-N,N,N′,N′-tetraacetic acid
- UGT
- UDP-glucuronyltransferase
- EROD
- 7-ethoxyresorufin-O-deethylase
- GST
- glutathioneS-transferase
- PBS-T
- phosphate-buffered saline (pH 7.5) containing 0.1% (v/v) Tween 20
- AUC
- area under the plasma concentration-time curve from time 0 to infinity
- t1/2
- half-life of elimination
- CL
- total body clearance
- Cmax
- maximum concentration
- Received November 29, 2000.
- Accepted February 5, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}