


Abstract
CP-122721 and CP-141938 are potent and selective neurokinin-1 (NK1) receptor antagonists with very different brain disposition and potency in models of centrally mediated activity. These investigations sought to determine whether differences in potency were related to differences in P-glycoprotein (P-gp) transport at the blood-brain barrier. Both compounds stimulated ATPase activity of human recombinant MDR1 with similar kinetic parameters. Cell-associated drug concentrations of CP-141938 were 9.4-fold lower in KBV1 cells expressing P-gp compared with KB3.1 control cells. In Madin-Darby canine kidney (MDCK) cells expressing human MDR1, asymmetric transport of CP-141938 was 5-fold higher than in wild-type MDCK cells, whereas no asymmetry was observed with CP-122721. In agreement with these differences in cellular transport, the differences in brain/plasma ratio between mdr1a/b(−/−) and FVB mice 1 h following a 3 mg/kg s.c. dose were 3- and 50-fold for CP-122721 and CP-141938, respectively. The effect of inhibiting P-gp efflux on the effects of these agents was evaluated using GR73632-induced foot tapping in gerbils as a model to measure centrally mediated NK1antagonism. When gerbils were pretreated with the P-gp inhibitor MS-073 (50 mg/kg s.c.), there was no effect on the activity of CP-122721 (0.05 mg/kg), whereas the percent reversal for CP-141938 (10 mg/kg) increased from 60 to 100%. In gerbils, the brain/plasma ratio for CP-122721 was unaffected by MS-073 pretreatment, whereas the brain/plasma ratio for CP-141938 brain concentrations increased 13-fold. This suggested that P-gp efflux influences the brain disposition and pharmacologic activity of CP-141938, but not CP-122721. Complete response curves for CP-141938 were then determined with respect to dose, and drug concentration in the plasma and brain in the presence and absence of MS-073 pretreatment. The dose and plasma concentration-response curves of CP-141938 were shifted to the left in the presence of MS-073, yet brain concentrations associated with the response were unchanged. This suggested that once in the brain the interaction of CP-141938 with the NK1 receptor was not affected by P-gp transport. In conclusion, these studies show that brain disposition and centrally mediated in vivo activity of NK1 antagonists can be profoundly affected by P-gp transport and that such transport should be considered during the design of new agents.
Neurokinin-1 receptor (NK1) antagonists continue to be of interest for a number of disorders, including pain, chemotherapy-induced emesis, anxiety, and depression (Sakurada et al., 1997; Kramer et al., 1998; Rupniak and Kramer, 1999; Saria, 1999). The first nonpeptide NK1 receptor antagonist, CP-96345, a diphenyl-quinuclidine, was first described by McLean et al. (1991) and Snider et al. (1991). This lead was further modified through substitution of the “boat”-locked quinuclidine for the equally stable “chair” conformer of the piperidine (Desai et al., 1994). The obvious simplicity that this substitution offers is enhanced by a reduction in lipophilicity. Subsequent modification of the activity profile of CP-99994 through substitution at the para-position to the methoxy group has produced several highly potent NK1 antagonists, such as CP-122721 (McLean et al., 1996; Rosen et al., 1998). During the lead optimization phase of drug discovery, it is common to search for a position open to a wide variety of substituents to modify drug disposition properties while retaining receptor affinity. This strategy led to the synthesis of CP-141938, a sulfonamide analog of CP-122721 (see Fig.1). CP-141938 had similar receptor affinity for the substance P receptor in human IM-9 cells compared with CP-122721, with IC50 values of 0.11 and 0.19 nM, respectively (Rosen et al., 1998). However, the studies reported herein reveal that the in vivo potency of CP-141938 was substantially weaker in a model of centrally mediated NK1 antagonism.
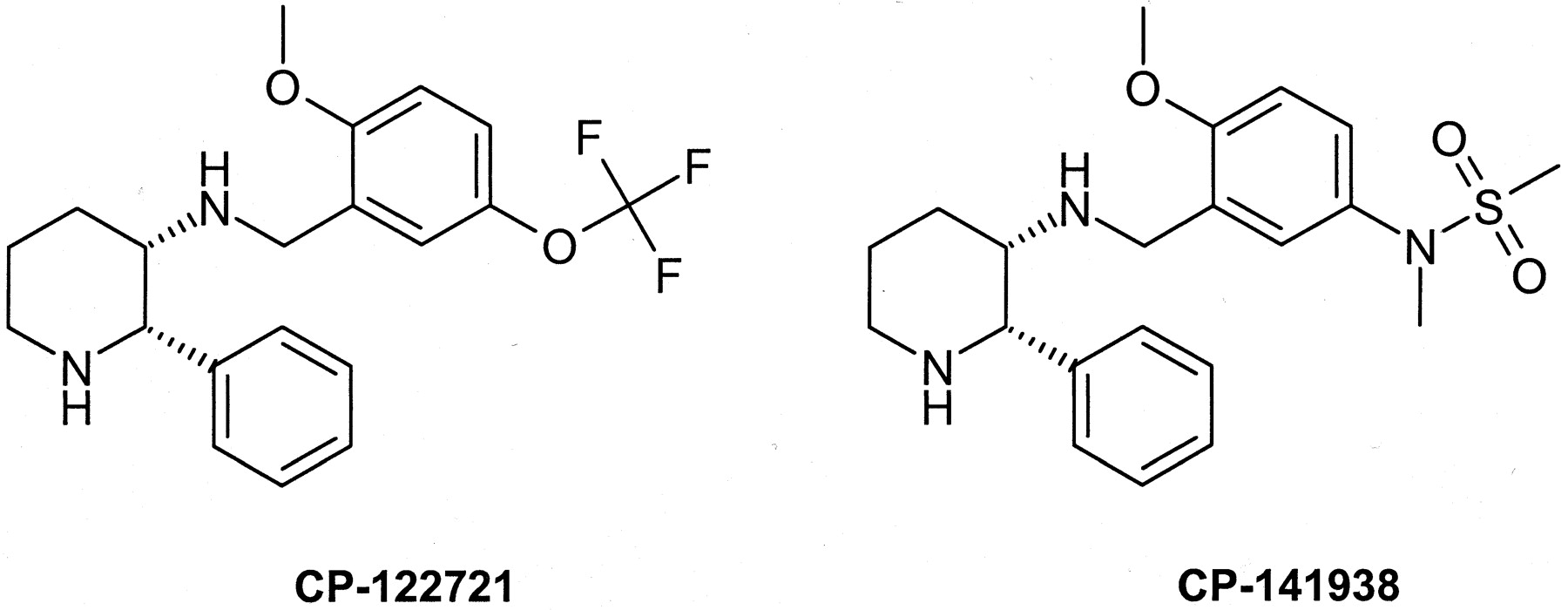
Structures of NK1 antagonists.
The divergence in the in vivo activity between CP-122721 and CP-141938 appears to stem from the exchange of the trifluoromethoxy substituent for the more polar sulfonamide. The sulfonamide functionality has previously been implicated in cases of poor brain penetration, and a recent example is sumatriptan, a sulfonamide-containing 5-HT1D agonist (Barf et al., 1996). The respective measured log DpH 7.4 oct values for CP-122721 and CP-141938 are 2.53 and 0.21, indicating that the sulfonamide group produces a substantial difference in octanol partitioning between these compounds. Recent studies have suggested that there are factors in addition to physical properties that influence the ability of drugs to gain access to the central nervous system. Advances in the understanding of the blood-brain barrier have strongly implicated a role for the MDR gene product, P-glycoprotein (P-gp), as an efflux transporter present on the luminal membrane of brain microvessel endothelial cells (Cordon-Cardo et al., 1989). P-glycoprotein has broad substrate specificity and has been shown to impair the brain penetrability of a number of drugs, including DPDPE, loperamide, ondansetron, and human immunodeficiency virus protease inhibitors (Schinkel et al., 1994, 1996; Chen and Pollack, 1998; Kim et al., 1998). Thus, in addition to physical properties, transporter activity must be considered when assessing the brain penetrability of new chemical entities. The purpose of the current investigation was to determine the mechanism of the poor in vivo activity of CP-141938 relative to CP-122721, with a focus on P-glycoprotein efflux.
Experimental Procedures
Materials.
CP-122721 (Rosen et al., 1998), CP-141938 (Howard et al., 1994), and MS-073 (Fukazawa et al., 1989) were prepared at the Groton Laboratories of Pfizer Global Research and Development, Pfizer, Inc. (Groton, CT). GR73632 was purchased from Peninsula Laboratories (Belmont, CA). Quinidine was purchased from Sigma (St. Louis, MO).
Animals.
Male FVB and mdr1a/b(−/−) mice 4 to 5 weeks of age (∼20 g) were obtained from Taconic Labs (Germantown, NY). Upon receipt, the mice were maintained at controlled temperature and humidity in a 12-h light/dark cycle while housed in polycarbonate cages with sawdust bedding with free access to food and water. Male Mongolian gerbils (40–60 g) were purchased from Charles River (Kingston, NY). Gerbils were housed as stated above for mice.
Gerbil Foot Tapping.
The gerbil foot tapping studies were conducted as described by Rupniak and Williams (1994), with minor modifications. Male Mongolian gerbils (40–60 g) were pretreated with MS-073 (50 mg/kg s.c.) 30 min prior to antagonist (5% dimethyl sulfoxide/5% Emulphor/90% saline vehicle). Thirty minutes after antagonist administration, gerbils were anesthetized using an isoflurane/O2 mixture, and a midline incision was made to expose the skull. The selective NK1agonist GR73632 (Hagan et al., 1991), δ-Ava[l-Pro9,N-MeLeu10]SP(7–11) (3 pmol/5 μl), or vehicle (phosphate-buffered saline) was administered directly into the lateral ventricles (i.c.v.) through vertical insertion of a 25-gauge needle to a depth of 4.5 mm below the skull. Scalp incision was then closed using Vet Bond adhesive (3M, Minneapolis, MN). Animals were placed in 1000 ml beakers while regaining consciousness. Once righting reflex occurred, animals were videotaped and later scored for foot tapping response (double-blinded scoring). Foot tapping response was scored every 30 s for a 12-min duration. A positive response was recorded if the animal tapped during the span of a 5-s interval. Data were then expressed as “percent time tapping” as compared with the agonist alone (GR73632) and the percent reversal of this response calculated. Control experiments were conducted to show that 50 mg/kg s.c. of MS-073 was the maximal tolerated dose without effect on the GR73632-induced foot tapping response in gerbils.
In Vitro Assessment of P-gp Interaction and Transport.
Drug stimulation of orthovanadate-insensitive ATPase activity was determined using human P-gp membranes obtained from the expression of MDR1 in baculovirus-infected insect cells (Gentest, Woburn, MA). The experiments were conducted according to the protocol supplied by Gentest as modified from Sarkadi et al. (1992). Specific modifications to the Gentest protocol included reducing the membrane protein concentration to 1 mg/ml. Test compounds were incubated in duplicate at concentrations of 200, 67, 22, 7.4, 2.5, and 0 μM, and a quinidine control was tested at the same concentrations. Standards were run in duplicate over a concentration range from 2 to 160 nM. The reaction endpoint is the appearance of inorganic phosphate, which was quantified spectrophotometrically at 620 nm according to the method of Druekes et al. (1995). The Michaelis constant [Km(app)] and the maximal rate of phosphate production (Vmax) from the drug-stimulated human P-gp ATPase assay were determined using nonlinear regression to fit the data to the equation v =Vmax · [S]/[S] +Km(app) (WinNonlin Professional version 2.1, model 101, Pharsight Corp., Cary, NC).
Cellular transport was assessed by comparing the amount of cell-associated drug following incubation with KBV1 and KB3.1 cells. The parental line KB3.1 and KBV1 cells have low and high MDR1 expression, respectively (Shalinsky et al., 1990). CP-122721 and CP-141938 were incubated at a concentration of 3 μM in 5-ml volume of Dulbecco's phosphate-buffered medium with ∼106cells/well for 3 h. At the end of the 37°C incubation, the medium was removed, the cells were washed with drug-free medium, and then scraped from the dish following precipitation with a 50% methanol/water solution. The recovered cellular suspension was assayed for drug concentration using the methods described below. The amount of cell-associated drug per 106 cells was calculated for each cell line, then the KB3.1 value was divided by the KBV1 value to calculate a cell accumulation ratio. A ratio greater than 1 suggests that the compound was actively transported by P-gp, resulting in reduced drug accumulation in the KBV1 cells. Due to the variability generally observed in KBV1 cells, determinations of transport in those cells were conducted in triplicate, whereas singlicate determinations were conducted with the KB3.1 cells.
Human P-gp-mediated transport in a second cellular system was studied by measuring the apical to basal and basal to apical permeability coefficients in a control MDCKII cell line and MDCKII-MDR1 cells expressing the human MDR1 gene (Bakos et al., 1998). The cell lines were provided by Dr. Piet Borst (The Netherlands Cancer Institute, Division of Molecular Biology, Amsterdam, The Netherlands). The cells were grown on 96-well HTS multiwell insert systems (Becton Dickinson, Sparks, MD). CP-122721, CP-141938, and the positive control quinidine were incubated at 2 μM on the apical or basal side of the insert for 2 h at 37°C. The medium was removed and assayed using a high-speed HPLC/MS/MS analysis as described by Brockman et al. (2000). CP-122721, CP-141938, and quinidine were detected in the positive ion mode by monitoring the M + H ion conversions: 381.5 → 159.5, 404.5 → 160.0, and 325.2 → 79.1, respectively. The permeability coefficient was calculated using the following formula:
Plasma and Brain Exposure in FVB and mdr1a/b(−/−) Mice.
Male FVB and mdr1a/b(−/−) mice (Taconic Labs) were dosed with NK1 antagonist, and the concentrations in plasma and brain were determined at 1 h postdose. The doses were prepared in 0.9% saline and delivered in a dosing volume of 10 ml/kg. Mice were sacrificed in a CO2 chamber, and whole blood was collected by cardiac puncture. Brain tissue was harvested and stored whole at −20°C until analysis. Plasma was prepared by centrifugation of the whole blood at 3000 rpm for 15 min. Plasma and brain samples were processed and analyzed as described below.
Analysis of CP-141938 in Plasma and Brain.
Whole brains were homogenized in 3 volumes (w/v) of water. Plasma and brain samples (0.05 ml) were placed into 1.2-ml microtubes in a 96-well block, and spiked with 10 μl of internal standard-1 (2.5 μg/ml), a compound of related structure to CP-141938 with a mol. wt. of 379. Samples were basified using 25 μl of 1.0 M NaOH. Following the addition of the organic solvent, methyl tert-butyl ether (1 ml), the samples were capped and mixed on an orbital shaker for 5 min. The organic and aqueous phases were separated by centrifugation at 3000 rpm for 5 min. The aqueous layer was frozen in a solid carbon dioxide/propanol bath, and the organic layer was transferred into a clean microtube using a Soken 96 channel pipetter. The organic phase was evaporated to dryness under N2 at 40°C. Sample residues were reconstituted in 50 μl of mobile phase (50% 25 mM ammonium formate, pH 3.5/30% acetonitrile/20% methanol) and analyzed by HPLC/MS/MS. A 10-μl aliquot of each sample was injected onto a Waters Symmetry C18 column (4.6 × 50 mm: 3.5-μm particle size) equilibrated in mobile phase at a flow rate of 0.5 ml/min. The effluent was analyzed by a triple quadrupole mass spectrometric detector (Sciex API 3000) fitted with a Turbo Ionspray interface operated in the positive ion mode. CP-141938 eluted at 1.8 min and was monitored as the M + H ion conversion, 404.1 → 160.0. The internal standard eluted at 2.0 min and was monitored as the M + H ion conversion, 380.2 → 204.1. The dynamic range of the plasma and brain assays were from 0.5 to 1000 ng/ml and from 10 to 4000 ng/g, respectively.
Analysis of CP-122721 and MS-073 in Plasma and Brain.
Whole brains were homogenized in 3 volumes (w/v) of water. For the analysis of MS-073 in gerbil brain homogenate, samples were diluted 1:10 with control homogenate. Plasma and brain samples (0.05 ml) were processed as stated for CP-141938, and spiked with 10 μl of internal standard-2 (2.5 μg/ml), a compound with a mol. wt. of 447.5 and structurally related to CP-122721 but unrelated to MS-073. Subsequent processing steps were identical to those described for CP-141938. Sample residues were reconstituted in 50 μl of mobile phase (30% 25 mM ammonium formate, pH 3.5/70% methanol) and analyzed by liquid chromatography/MS.
A 10-μl aliquot of each sample was injected onto a Waters Symmetry C18 column (4.6 × 50 mm: 3.5-μm particle size) equilibrated in mobile phase at a flow rate of 0.5 ml/min. The effluent was analyzed by a single quadrupole mass spectrometric detector (Sciex API 150) fitted with a Turbo Ionspray interface operated in the positive ion mode. CP-122721 eluted at 1.8 min and monitored as the M + H ion, 381.2. MS-073 eluted at 2.0 min and monitored as the M + H ion, 480.2. The internal standard was eluted at 4.1 min and monitored as the M + H ion, 448.1. The dynamic range of the MS-073 plasma and brain assays were from 100 to 1000 ng/ml and from 10 to 4000 ng/g, respectively. The dynamic range of the CP-122721 plasma and brain assays were from 0.5 to 1000 ng/ml and from 10 to 4000 ng/g, respectively.
Data and Statistical Analysis.
A Box-Cox transform indicated that the log-transform was the appropriate transformation of the data to perform statistical analyses for KBV cell transport and animal disposition studies. This was done to account for variance that increases with increasing mean. A two-sided t test was performed on the log-transformed data to generate p values and determine significant differences between groups (p< 0.05). Where multiple comparisons were indicated, the Bonferroni correction to the individual comparison significance level was used. Differences between MDCKII and MDCKII-MDR1 cell transport were determined by comparing the asymmetry ratios (basal to apicalPapp/apical to basalPapp) for each compound. Asymmetry ratio data was evaluated for statistical significance using analysis of variance on log-transformed data. This test determines if the fold increase between MDCKII and MDCKII-MDR1 cell asymmetry ratios is >1. Finally, the curves describing the relationship between pharmacologic effect in gerbil foot tapping and administered dose, plasma concentration, or brain concentration were fit to a simpleEmax model (model 101) using WinNonlin Professional version 2.1. The nonlinear regression was conducted using the result from each individual animal. Initial estimates were obtained through visual inspection of the data and with upper and lower bounds for Emax set at 90 and 100%, respectively. The resulting dose or concentration producing 50% inhibition (ID50 or IC50) was reported with the standard error of the estimate. The ID50 or IC50 values between treatment groups were compared using t statistic (two-sided), with the level of significance set at p < 0.05.
Results
Interaction with P-gp in the in Vitro Systems.
P-glycoprotein is an ATP-dependent transport protein that reduces cellular accumulation of drugs. When substrates or inhibitors interact with the intracellular binding site(s) of recombinant human P-gp in crude membrane preparations, ATPase activity is stimulated (Sarkadi et al., 1992). Although this increased ATPase activity suggests interaction with the protein, it does not prove that the substrate is transported. When CP-122721 and CP-141938 were tested in this model, both compounds stimulated ATPase activity with similar apparentKm values suggesting similar interaction with P-gp (Table 1). The maximal velocity for ATPase stimulation for CP-122721 was significantly greater than that determined for CP-141938 and quinidine. Quinidine was used as a positive control and produced an expected level of ATPase stimulation when evaluated with each compound.
Kinetic parameters of stimulation of human P-glycoprotein ATPase activity following interaction with two related NK1 antagonists
A second in vitro model of P-gp transport that was evaluated was compound accumulation in KB3.1 and KBV1 cells. KBV1 cells are grown under selection pressure by vinblastine, which induces expression of P-glycoprotein, whereas KB3.1 cells have been selected for low P-gp expression. Following incubation of the two cell lines with the putative substrates, a measurement of cell-associated drug was compared to assess the extent of drug efflux from the KBV1 cells. The result of this assay with CP-122721, CP-141938, and the positive control quinidine is shown in Table 2. CP-122721 appeared to have similar cell-associated drug in the KBV1 and KB3.1 cells. The cell accumulation ratios for CP-141938 and quinidine were significantly greater than CP-122721, indicating that these compounds are clearly effluxed from the KBV1 cells resulting in low cell-associated drug. This suggested that it is likely that CP-141938 is transported by P-gp, thus reducing intracellular accumulation in the KBV1 cells. CP-122721 either interacts with P-gp but is not transported or has such high passive permeability that at equilibrium the rate of cell entry is equal to the rate of P-gp efflux.
Cell-associated accumulation of NK1 antagonists in KB3.1 and KBV1 cells
MDCKII (control) and MDCKII-MDR1 cells with polarized expression of P-gp on the apical membrane were used to study the cell permeability and bidirectional drug transport of CP-122721, CP-141938, and the positive control quinidine. MDCKII-MDR1 cells express human P-glycoprotein on the apical membrane, and asymmetric transport of P-glycoprotein substrates has previously been shown (Bakos et al., 1998). When CP-122721 was incubated with MDCKII or MDCKII-MDR1 cells, the permeability coefficients were approximately equal in both directions resulting in no detectable asymmetric transport (Table3). The permeability coefficients determined for CP-141938 and quinidine were greater in the basal to apical direction in both cell lines. However, a marked increase was observed in the basal to apical permeability coefficient for CP-141938 and quinidine, with the MDCKII-MDR1 cells expressing human P-gp. The asymmetric transport observed with CP-141938 in control cells was presumably due to endogenous expression of canine P-glycoprotein. The P-glycoprotein substrate quinidine was included in these studies as a positive control, and the results were comparable with previous reports (Fromm et al., 1999). These data suggested that CP-141938 interacts with P-gp and is transported, whereas the interaction of CP-122721 with P-gp does not result in detectable efflux transport.
Permeability coefficients for bidirectional drug transport in MDCKII and MDCKII-MDR1 cells
Brain Disposition in mdr1a/1b Knockout Mice.
Assessment of the role of P-gp in brain penetration has been recently advanced by the development of the mdr1a/1b double knockout mouse model through the pioneering work of Schinkel et al. (1994). Several groups have demonstrated the enhanced brain penetration of P-gp substrates by comparing brain and blood or plasma concentrations in the knockout and wild type mice following drug administration (Schinkel et al., 1996;Chen and Pollack, 1998; Kim et al., 1998). CP-122721 and CP-141938 were administered at a dose of 3 mg/kg s.c. to the knockout and wild type mice and drug concentration in brain and plasma measured at 1 h post dose. The results presented in Table4 showed that CP-122721 brain concentrations exceeded plasma in the wild type mice, and a modest but statistically significant increase was observed in the brain concentrations and brain-to-plasma ratio in knockout mice. There was no significant difference in the plasma concentrations of CP-122721 between wild type and knockout mice. This suggested that in vivo in mice there is some modulation of brain penetration of CP-122721 by mouse mdr1a P-gp. In contrast, CP-141938 brain concentrations were only 10% of the plasma concentration in wild type mice. However, in the knockout mice, an enormous and statistically significant increase in brain concentration and brain/plasma ratio was observed. This resulted in a 50-fold difference in the brain/plasma ratio for CP-141938 between the knockout and wild type mice. These data suggested that P-gp efflux at the lumen of the brain microvesicular endothelium present a formidable barrier to the central nervous system penetration of CP-141938.
Comparison of brain and plasma concentrations of CP-122721 and CP-141938 in FVB wild-type and mdr1a/1b(−/−) knockout mice
Modulation of Pharmacodynamic Activity and Drug Disposition of CP-122721 and CP-141938 through Inhibition of P-gp.
To determine whether P-gp efflux modulated the pharmacodynamic activity of these NK1 antagonists, pharmacology experiments were performed in the presence and absence of a P-gp inhibitor, MS-073. MS-073 has previously been shown to be a potent P-gp inhibitor in vitro and in vivo (Sato et al., 1991). Pharmacology experiments were performed using the gerbil foot tapping model (Rupniak and Williams, 1994). In this model, gerbils given an i.c.v. injection of GR73632, a NK1 receptor agonist, exhibit a hind limb foot tapping response. This response can be inhibited by pretreatment with a centrally active NK1 antagonist. In a pilot experiment, gerbils were pretreated subcutaneously with CP-122721 or CP-141938 at their approximate ID50 doses 30 min prior to the i.c.v. administration of GR73632. A separate group received a 50 mg/kg subcutaneous dose of MS-073 30 min prior to the NK1 antagonists. As shown in Fig.2, CP-122721 produced a 47% reduction in the foot tapping response induced by the agonist. Pretreatment with MS-073 produced no significant increase in the antagonism of the foot tapping response produced by CP-122721. CP-141938 produced a 60% reduction in the foot tapping response when given alone. However, complete blockade of the foot tapping response was observed in the group pretreated with MS-073, then given the same dose of CP-141938.

Effects of the P-glycoprotein inhibitor MS-073 (50 mg/kg s.c., 30 min) on the antagonist response of CP-122721 and CP-141938 on GR73632-induced foot tapping in gerbils. Data shown as mean ± S.E.M. (n = 3–4 animals/group). ∗, significantly different from no MS-073 pretreatment (p < 0.05).
Plasma and brain concentrations of the NK1antagonists and MS-073 were determined in the gerbils used in the pharmacology experiments, and the results are shown in Table5. Brain and plasma concentrations of CP-122721 were not significantly altered by MS-073 pretreatment. The fact that the disposition of CP-122721 was essentially the same in the two groups was consistent with the foot tapping results. In contrast, CP-141938 brain concentrations were increased 17-fold in gerbils pretreated with MS-073, with no significant change in plasma concentration. The dramatic increase in brain concentration of CP-141938 by MS-073 pretreatment is consistent with the foot tapping results in which complete blockade was observed. The concentration of MS-073 in plasma and brain was not significantly different, when given with either NK1 antagonist. Thus, equal in vivo inhibition of P-gp by MS-073 would have been expected in the gerbils given CP-122721 and CP-141938.
Plasma and brain concentrations of CP-122721 and CP-141938 in gerbils with and without MS-073 pretreatment (50 mg/kg s.c., 30 min).
Effects of P-gp Inhibition on the Potency of CP-141938 in the Gerbil Foot Tapping Model.
In the pilot experiment, it was not possible to determine the extent to which MS-073 pretreatment could influence the potency of CP-141938. Because it appeared that the brain concentrations increased at least 10-fold with the MS-073 pretreatment, a dose-response study was conducted with a 10-fold lower dose range in the group given the P-gp inhibitor. As was done in the previous experiment, after the pharmacodynamic assessment was completed, the gerbil was sacrificed and blood and brain tissue were collected for measurement of plasma and brain CP-141938 concentration. The results of these studies are shown in Fig. 3, with the pharmacodynamic parameters presented in Table6. The calculated ID50 based on dose was 5.84 mg/kg for CP-141938, similar to that expected based on the response observed at 10 mg/kg in the pilot study (Fig. 3A). As expected, the ID50for CP-141938 in the group given the MS-073 pretreatment was dramatically lower at 0.19 mg/kg (Fig. 3A). When the pharmacodynamic response was based on plasma concentration, there was an 18-fold decrease in the plasma concentration necessary to produce the same antagonist response in the MS-073 pretreated animals compared with untreated gerbils (Fig. 3B). Finally, there was no significant difference in the brain concentration required to produce a 50% blockade of the NK1 agonist response in the presence or absence of MS-073 (Fig. 3C).

Pharmacodynamic response curves for CP-141938 antagonism of GR73632-induced foot tapping in gerbils based on administered dose (A), plasma concentration (B), or brain concentration (C): effect of P-glycoprotein inhibition. Symbols represent the mean [CP-141938 alone (▪) and CP-141938 with MS-073 pretreatment (●)], and the error bars represent the S.E.M. Lines are the predicted data based on the nonlinear regression analysis using a simple Emax model.
Pharmacodynamic parameters for CP-141938 antagonism of GR73632-induced foot tapping in gerbils: effect of P-glycoprotein inhibition
Discussion
The objectives of the present investigation were to assess the role of P-gp efflux on the disposition and pharmacodynamics of CP-122721 and CP-141938, two structurally related high-affinity NK1 receptor antagonists. CP-122721 stimulates ATPase activity in recombinant human MDR1 in crude membrane preparations with saturable kinetics. This suggested that CP-122721 interacts with the substrate binding site of P-gp, but does not provide definitive proof that the compound is transported. However, experiments that compared the bidirectional transport of CP-122721 in MDCKII and MDCKII-MDR1 were unable to detect basal to apical efflux mediated by human P-gp in an intact cellular system. Similarly, the cell-associated drug concentration of CP-122721 in KB3.1 and KBV1 cells was similar despite the expression of P-gp in the latter. These results suggested that CP-122721 is either not transported by P-gp or readily re-equilibrates with the intracellular compartment due to rapid diffusion across the cell membrane. This was confirmed in whole animals by comparing the brain/plasma ratio of CP-122721 in mdr1a/b(−/−) and wild type mice. These results showed that, in normal animals, CP-122721 has a high brain/plasma ratio and that there was only a 3-fold increase in the brain/plasma ratio in the knockout mice for CP-122721. One potential cause for the difference in the in vitro and in vivo results is species differences in transport between human and mouse P-gp (Yamazaki et al., 2001). Since there are no available models of centrally mediated NK1 blockade in mice, a study was performed in gerbils to determine whether P-gp inhibition could modulate the disposition and pharmacodynamic activity of CP-122721. At P-gp inhibitor concentrations that profoundly increased the brain concentration and activity of CP-141938 in gerbil foot tapping, only a slight change in brain concentration and activity of CP-122721 was observed. Thus, when the disposition and pharmacodynamic profiles of CP-122721 are considered, it is clear that that the combination of high receptor affinity, extensive brain penetration, and minimal P-gp efflux act in concert to produce a compound with a superb in vivo activity profile.
In both in vitro and in vivo systems, CP-141938 clearly interacts with and is transported by human and mouse P-gp. The efflux of CP-141938 by P-gp resulted in low brain concentration in mice and gerbils and weak in vivo pharmacodynamic activity in gerbils. When P-gp was inhibited, the brain/plasma ratio increased substantially, with a corresponding leftward shift in the dose- and plasma concentration-response curves for CP-141938 in the gerbil foot tapping model of NK1 antagonism. Interestingly, there was absolutely no difference in the brain concentration-response curves for CP-141938 in the presence or absence of MS-073 pretreatment (Fig. 3C and Table 6). This suggests that P-gp efflux acts as a direct functional barrier that impairs that ability of NK1 antagonists that are transported P-gp substrates to reach the receptor site.
Although there are numerous studies in the literature on the ability of P-gp to alter the central nervous system disposition of xenobiotics, there are few reports where the affect on pharmacodynamics has been studied simultaneously. One notable exception is the work of Chen and Pollack (1998, 1999), where they studied the effect of P-gp on the disposition and antinociceptive activity of the constrained peptide opioid analog DPDPE in mice. Chen and Pollack showed that the brain concentrations of DPDPE were higher and antinociceptive potency was enhanced in mdr1a(−/−) relative to the FVB background strain. Surprisingly, the brain concentration of DPDPE required for antinociceptive activity was considerably lower in mdr1a(−/−) compared with FVB mice. Using a sophisticated pharmacokinetic-pharmacodynamic modeling approach, the authors proposed a multicompartmental model of the brain where the δ-opiate receptor site was distinct from the blood-brain barrier for FVB mice and a simple one-compartment brain model in mdr1a(−/−), where no transporter was present. This is in contrast to the present work showing no difference in brain concentrations of CP-141938 required to antagonize NK1 receptors under control conditions or when P-gp was inhibited. This suggests that there may be regional differences in P-gp efflux within the brain that have varied effects on pharmacodynamic response, depending on the receptor and brain region affected by the pharmacologic agent. Alternatively, the disposition within the brain for P-gp substrates may be related to physicochemical properties of the compound and therefore may be drug specific.
In summary, the studies herein demonstrate that the in vivo brain disposition and pharmacologic potency of NK1antagonists can be profoundly affected if they are transported P-gp substrates. This emphasizes the need during drug discovery to understand the mechanism of poor brain penetration and low potency of compounds with high receptor affinity so that structural modifications can be considered to specifically address these issues. Toward that aim in vitro models of P-gp transport, mdr1a/b(−/−) mice and the use of P-gp inhibitors in pharmacology models comprise a triad of important tools that are available to better understand the relationships between efflux transport at the blood-brain barrier, brain disposition, and centrally mediated drug effects.
Acknowledgments
We gratefully acknowledge the contributions of Ralph Davidson and Dr. Adam Brockman (Exploratory Medicinal Sciences, Pfizer, Inc.), and Simone Paillet (Cancer Discovery Biology, Pfizer, Inc.) for the MDCK and KBV cell transport experiments, respectively, as well as Dr. David Raunig for statistical analyses.
Footnotes
- Abbreviations:
- NK1
- neurokinin-1 receptor
- P-gp
- P-glycoprotein
- mdr
- multidrug resistance
- DPDPE
- d-Pen2,d-Pen5-enkephalin
- HPLC
- high-performance liquid chromatography
- MS
- mass spectroscopy
- Papp
- apparent permeability coefficient
- Received February 19, 2001.
- Accepted May 9, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}