


Abstract
Administration of the bacterial endotoxin lipopolysaccharide (LPS) causes induction of cytochrome P-450 (CYP) 4A mRNAs in rat liver and kidney. Because induction of the CYP4A subfamily by chemicals requires peroxisome proliferator-activated receptor-α (PPARα), we determined whether CYP4A induction by LPS also requires PPARα by comparing the responses of PPARα-null (−/−) and wild-type (+/+) mice. Renal expression of CYP4A10, CYP4A14, and acyl-CoA oxidase was induced by LPS treatment in (+/+) mice, and these effects were absent in the (−/−) mice. In contrast, hepatic expression of CYP4A10 was down-regulated in the (+/+) animals, and no significant induction of acyl-CoA oxidase or CYP4A14 was detected in liver. Expression of the peroxisomal bifunctional enzyme was not significantly affected by LPS treatment. These results indicate that PPARα is activated in mouse kidney after LPS treatment and that this leads to modulation of some PPARα-regulated genes. However, the species and tissue specificity of these effects suggest that inflammatory pathways may modulate the induction via PPARα. Mice pair fed with LPS-treated mice showed no induction of renal CYP4A10 or CYP4A14, indicating that renal CYP4A induction during endotoxemia is not due to hypophagia. Down-regulation of CYP2A5, CYP2C29, and CYP3A11 by LPS was attenuated or blocked in the (−/−) mice, suggesting a role for PPARα in CYP down-regulation as well. Finally, we found that clofibrate caused an acute induction of two hepatic acute-phase mRNAs that was only partially dependent on PPARα.
Inflammatory agents such as bacterial endotoxin (lipopolysaccharide; LPS) can affect both the mRNA and protein levels of various isoforms of the cytochrome P-450 (CYP) family (Stanley et al., 1988). In LPS-treated rats, hepatic CYP4A subfamily mRNAs are induced, whereas mRNA and protein levels of CYP2E1, CYP2C11, and CYP3A2 are decreased (Sewer et al., 1996b). In the kidneys of rats treated with either LPS or particulate irritants, expression of both CYP4A and CYP2E1 mRNAs is induced (Sewer et al., 1996a). The CYP4A family is also induced by peroxisome proliferators, a structurally diverse class of compounds that cause several effects in the liver including hepatomegaly, increased peroxisomal β-oxidation, proliferation of parenchymal peroxisomes, and hepatocarcinogenesis (Gonzalez et al., 1998). These effects of peroxisome proliferators are mediated by the peroxisome proliferator-activated receptor-α (PPARα) (Gonzalez et al., 1998). PPARα is a member of the nuclear hormone receptor superfamily, which requires another dimerization partner, retinoid X receptor (RXR), for gene activation (Issemann et al., 1993). Target genes of PPARα include those encoding peroxisomal and mitochondrial β-oxidation enzymes, CYP4A subfamily enzymes, and fatty acid binding proteins (Aoyama et al., 1998; Gonzalez et al., 1998). Recently, PPARα-null (−/−) mice have been developed, and these animals are refractory to the effects of peroxisome proliferators, including induction of CYP4A or peroxisomal and mitochondrial fatty acid β-oxidation enzymes (Lee et al., 1995;Aoyama et al., 1998).
PPARα has been implicated in inflammatory pathways. Mediators of inflammation such as arachidonate (Issemann et al., 1993) and hydroxyeicosatetraenoic acids (Yu et al., 1995) are activators of PPARs. Leukotriene B4-induced inflammation is prolonged in (−/−) mice (Devchand et al., 1996), and peroxisome proliferators can suppress the expression of negative acute-phase genes (Motojima et al., 1997; Corton et al., 1998). PPARα ligands inhibit induction of cyclooxygenase 2 and prostaglandin production in human aortic smooth muscle cells and reduce the plasma concentrations of interleukin-6 and acute-phase proteins in hyperlipidemic patients (Staels et al., 1998). Conversely, peroxisome proliferators induce cyclooxygenase 2 expression in immortalized mouse liver cells (Ledwith et al., 1997).
The objective of this study was to investigate whether the induction of the CYP4A subfamily mRNA and protein by LPS treatment was mediated by PPARα. This was done by comparing the responses to LPS in wild-type and PPAR (−/−) mice. Our results indicate that endotoxin causes a tissue-specific induction of CYP4A10 and CYP4A14 in mouse kidney and that this effect is dependent on a functional PPARα. Another PPAR-responsive gene, the β-oxidation enzyme acyl-CoA oxidase (ACO), was induced by LPS, providing further evidence for activation of PPARα during inflammation.
Materials and Methods
Animals and Treatments.
All procedures were approved by the Institutional Animal Care and Use Committee of Emory University. In the experiments to determine the role of PPARα in CYP regulation, groups of four female 9- to 11-week-old wild-type (−/−) or (+/+) mice (F5 Sv/129) homozygous for a disruption in the ligand-binding domain of the PPARα gene were used (Lee et al., 1995). Animals were housed in group cages containing three to five animals per cage and provided food and water ad libitum unless otherwise indicated. Animals were given a single i.p. injection of either 1 mg/kg of chromatographically purified Escherichia coli LPS, serotype 0127:B8 (Sigma Chemical Co., St. Louis, MO) dissolved in sterile 0.9% saline, 40 mg/kg of clofibrate (Sigma) dissolved in corn oil (Wesson), or 0.9% saline in a volume of 0.1 ml. Animals were sacrificed 24 h after injection by CO2 asphyxiation before collection of organs.
In the experiments to examine the effects of hypophagia, female C57/BL6 mice (9–11 weeks old; Charles River Laboratories, Wilmington, MA) were divided into three groups of nine mice, each cage containing three mice. Mice in the first and second groups were injected with 1 mg/kg of LPS or sterile saline, respectively, and their food intakes were recorded every 6 h. Mice in the third group were injected with saline the following day and fed every 6 h with an amount equal to the mean intake of the LPS-treated groups for the corresponding period. Mice from all three groups were injected at 8:00 AM and sacrificed either 12 or 24 h after injection.
Microsome and RNA Preparation.
Livers and kidneys were collected from mice, and total RNA was prepared from fresh tissues according to the method of Chomczynski and Sacchi (1987). Samples were stored at −80°C until analysis. RNA concentrations were determined by absorbance at 260 nm.
Northern Blots.
RNA was fractionated on a 1% agarose gel electrophoresis in the presence of 5% formaldehyde and transferred to nylon MagnaGraph transfer membrane filters (Micron Separations, Inc., Westboro, MA). Blots were prehybridized, hybridized, and washed according to previously described procedures (Sewer et al., 1996b). To control for RNA loading and transfer artifacts, Northern blots were normalized to the content of glyceraldehyde 3-phosphate dehydrogenase (GAP) mRNA with a cDNA probe (Morgan et al., 1994). The amount of probe bound to the filter was quantitated with a Molecular Dynamics (Sunnyvale, CA) 445si PhosphorImager and ImageQuant software.
cDNA and Oligonucleotide Probes.
The cDNA probes for rat CYP4A1, ACO, and bifunctional enzyme (BIEN) were described previously (Lee et al., 1995). The nomenclature system for the CYPs analyzed in this article has been published previously (Nelson et al., 1996). Fibrinogen β cDNA was donated by Dr. Gerald Fuller (University of Alabama at Birmingham, Birmingham, AL). Oligonucleotide probes for mouse CYP genes were designed by selecting candidate probes with minimum homology to closely related genes via multiple sequence alignment. Lack of identity to other sequences was confirmed by searching the GenEMBL rodent database with the FastA program (University of Wisconsin GCG package).
cDNA probes were labeled with the Megaprime random nonamer labeling kit (Amersham, Arlington Heights, IL) and [α-32P]dCTP. All blots probed with cDNA were hybridized at 42°C and washed at 55°C. Oligonucleotide probes were labeled with T4 polynucleotide kinase and [γ-32P]ATP. Hybridization and washing conditions for the CYP4A oligonucleotide probes were as described previously (Sewer et al., 1996b). The general conditions for washing and hybridization of blots with the other oligonucleotide probes were as described before (Morgan et al., 1994). All hybridizations and final washes were done at 45°C, except for CYP2A5, which was hybridized at 52°C and washed at 55°C, and CYP4A14, which was hybridized at 45°C and washed at room temperature. The oligonucleotide sequences used and their Genbank accession numbers are: CYP4A10—acaatg- cctgggacaggtgagtagagcctt nt 1146–1175 of X69296; CYP4A14—tctgctcacagatattactggtggatagag nt 1229–1258 of Y11640; CYP2C29—ggccaggccctctccagcacaaatccgttt nt 1301–1330 of D17674; CYP3A11—tgtccgatgttcttagacactgcctttctg nt 1631–1660 of X60452; CYP2A5—cttggtccaccagagcttccttgactgcct nt 351–380 of M19319; and α1-acid glycoprotein (AGP)—cggagttcagagagctgagttcatgcctgg nt 648–677 of M27008.
Statistical Analysis.
Statistical analyses were performed with the Quick Statistica package (StatSoft, Inc., Tulsa, OK). One-way ANOVA and the Student-Newman-Keuls test were used to determine differences among treatment groups. In cases where the variances of the experimental groups were not equivalent, the Mann–Whitney Utest was used instead. The null hypothesis was rejected atP < .05.
Results
Effects of Endotoxin on CYP4A Expression in the Mouse.
We previously found that LPS treatment of Fischer 344 rats induced CYP4A2 and CYP4A3 mRNAs in both liver and kidney and CYP4A1 in liver (Sewer et al., 1996a). The effect of LPS on expression of CYP4A mRNAs in mice has not been reported. Three CYP4a genes have been reported in mice: CYP4A10 and CYP4A14 mRNAs are expressed in the livers and kidneys of male and female mice (Heng et al., 1997), whereas CYP4A12 mRNA is male specific (Bell et al., 1993). Therefore, for this study in female mice, we measured the expression of CYP4A10 and CYP4A14 mRNAs. All of the relative mRNA measurements in this article are presented relative to GAP. Essentially identical results were obtained when the signals were normalized to the content of 18S rRNA (not shown), validating the use of GAP as a control.
In contrast to the induction of CYP4A mRNAs in the rat, we found that CYP4A10 mRNA was significantly reduced (to 20% of control levels) in (+/+) mouse liver by treatment with LPS (Fig.1). In (−/−) mice, the level of CYP4A10 mRNA in the control group was reduced (P < .05) to below the limit of accurate measurement, such that it was impossible to observe whether there was any further decrease on treatment with LPS. As a control, we examined the hepatic expression of CYP4A10 mRNA 24 h after a single injection of clofibrate. This treatment caused a PPARα-dependent induction of CYP4A10 mRNA in (+/+) mice (Fig. 1), in agreement with previous findings via chronic clofibrate treatment (Lee et al., 1995).

Effects of endotoxin on CYP4A10 mRNA expression. Northern blots of RNA from liver and kidney of mice treated with saline (control), LPS, or clofibrate (Clof) were probed with an oligonucleotide complementary to CYP4A10. All values are normalized to the GAP mRNA signal and are expressed as a percentage of the control group mean. The third sample of the LPS-treated (+/+) group in liver gave a very low signal on several blots with different probes and was omitted from analysis of this and all other blots. Numbers represent the mean ± S.E. of each group. *P < .05, significantly different from respective control group.
In contrast to the findings in the liver, we found that CYP4A10 mRNA was significantly induced in the mouse kidney to 324 and 291% of control levels by treatment with LPS or clofibrate, respectively (Fig.1). In (−/−) mice, basal CYP4A10 expression was lower than in (+/+) mice (P < .05), and there were no detectable changes due to treatment with LPS or clofibrate (Fig. 1).
The effect of LPS on expression of CYP4A14 in the livers and kidneys of (+/+) and (−/−) mice was similar to those observed with CYP4A10 (Fig.2). However, the basal expression of CYP4A14 in the livers of both (+/+) and (−/−) mice was below the level of detection, so that it was not possible to detect any further decrease in liver after LPS treatment. CYP4A14 expression in (+/+) mouse kidney was induced by LPS or clofibrate treatment (Fig. 2). No effect of either LPS or clofibrate was observed in the kidneys of (−/−) mice. Similar results were obtained when the liver and kidney RNA Northern blots shown in Fig. 1 were probed with the rat CYP4A1 cDNA (data not shown).
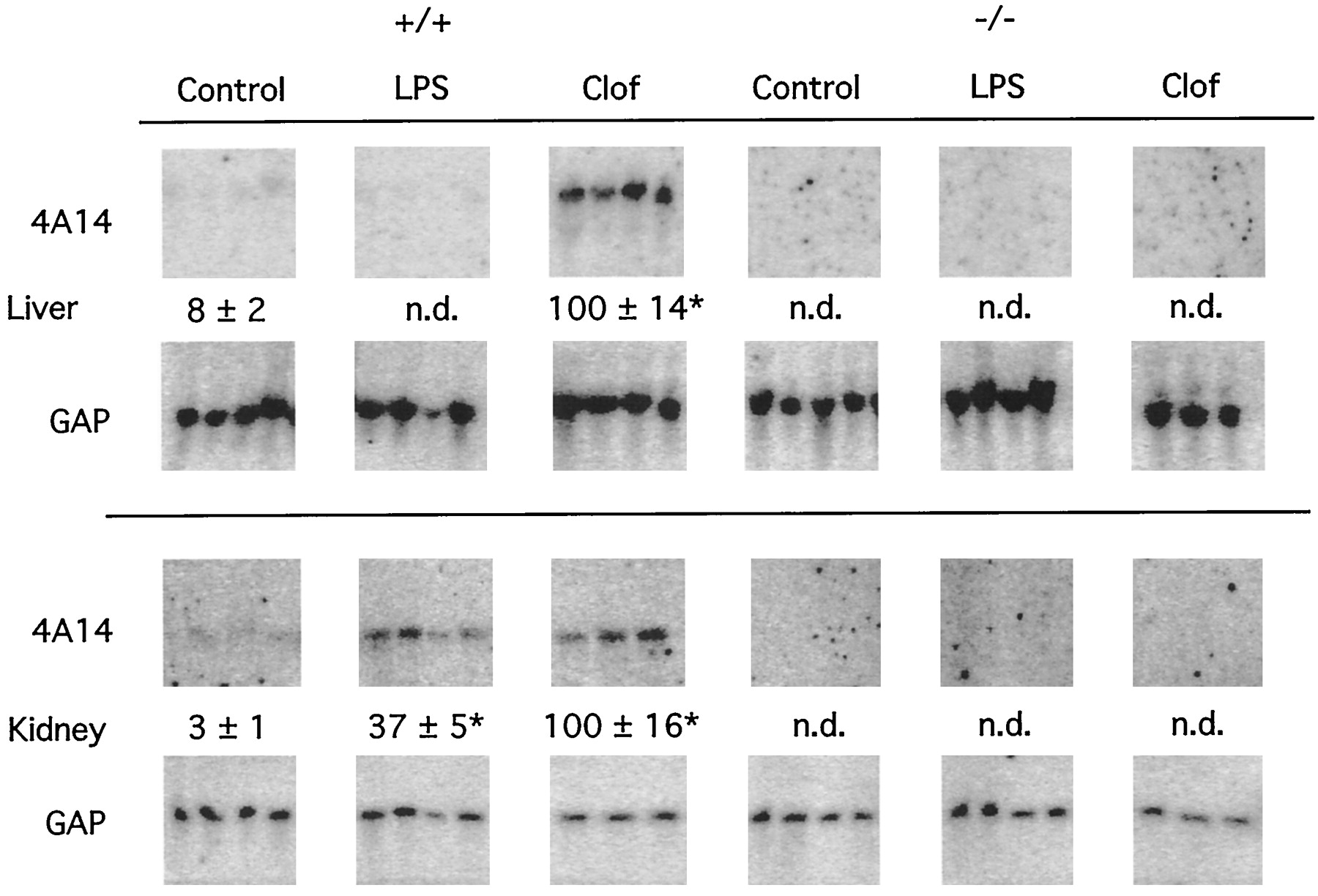
Effects of endotoxin on CYP4A14 mRNA expression. Northern blots of RNA from liver and kidney of mice treated with saline (control), LPS, or clofibrate were probed with an oligonucleotide complementary to CYP4A14. All values are normalized to the GAP mRNA signal and are expressed as a percentage of the mean of the clofibrate-treated group. Numbers represent the mean ± S.E. of each group. n.d., values below detection limit, less than three times background value. *P < .05, significantly different from respective control group.
Induction of Peroxisomal β-Oxidation Genes by Endotoxin Treatment.
During treatment with clofibrate and other peroxisome proliferators, hepatocytes undergo several prototypical changes in gene expression, including induction of the mRNAs for the peroxisomal β-oxidation enzymes ACO, BIEN, and 3-ketoacyl coenzyme A thiolase. Induction of the genes encoding these enzymes by peroxisome proliferators is abolished in the PPARα (−/−) mouse (Lee et al., 1995). Because we found that the induction of CYP4A10 by LPS in mouse kidney is also regulated by PPARα, we investigated the effects of LPS treatment on the expression of genes encoding β-oxidation enzymes.
In the kidneys of (+/+) mice, ACO was induced to 293 and 142% of control levels by LPS or a single dose of clofibrate, respectively, and these effects were not found in (−/−) mice (Fig.3). There was no significant effect of LPS on ACO expression in the liver. BIEN expression in liver and kidney was up-regulated about 13- and 3-fold, respectively, by a single dose of clofibrate in (+/+) mice but was not significantly affected by LPS (Fig. 3). The levels of both ACO and BIEN mRNAs in the livers and kidneys of control (−/−) animals tended to be lower than in (+/+) animals, but this was only significant (P < .05) for BIEN expression in the liver. Treatment with either clofibrate or LPS had no effect in (−/−) mice (Fig. 3)

Effects of endotoxin on mRNA expression of ACO and peroxisomal bifunctional enzyme. Northern blots of RNA from liver and kidney of mice treated with saline (control), LPS, or clofibrate were probed with cDNA probes for ACO or BIEN. All values were normalized to the GAP mRNA signal and are expressed as a percentage of the control group mean. Numbers represent the mean ± S.E. of each group. *P < .05, significantly different from respective control group.
Down-Regulation of CYPs.
Because PPARα is activated during LPS-induced inflammation, it was of interest to determine whether PPARα could also be involved in down-regulation of CYP expression during inflammation. Therefore, we examined the hepatic expression of CYP2A5, CYP2C29, and CYP3A11. Basal levels of CYP2A5 and CYP3A11 mRNAs were lower in the (−/−) than in the (+/+) group (Fig.4). Treatment of (+/+) mice with LPS resulted in decreases in CYP2A5, CYP2C29, and CYP3A11 mRNAs to less than 2, 20, and 15% of control values, respectively (Fig. 4). Clofibrate treatment also caused lower levels of these mRNAs, to a lesser degree. The down-regulation of CYP2A5 and CYP2C29 by LPS or clofibrate was attenuated but not abolished in (−/−) mice. Neither LPS nor clofibrate had an effect on CYP3A11 expression in the (−/−) mice (Fig. 4).

Involvement of PPARα in the down-regulation of CYPs by endotoxin. Northern blots of RNA from liver of mice treated with saline (control), LPS, or clofibrate were probed with an oligonucleotide complementary to CYP2A5, CYP2C29, or CYP3A11. All values are normalized to the GAP mRNA signal and are expressed as a percentage of the control group mean. Numbers represent the mean ± S.E. of each group. *P < .05, significantly different from respective control group.
Induction of Acute-Phase Genes by Clofibrate.
To determine the efficacy of the LPS treatment, we examined its effect on the acute-phase genes fibrinogen and AGP in the liver (Fig.5). These are prototypic acute-phase proteins, whose expression in inflammation is thought to be regulated mainly by interleukin-6 (fibrinogen) and interleukins-1 and -6 (AGP) (Baumann and Gauldie, 1990). In (+/+) mice, hepatic expression of both mRNAs was induced by LPS treatment as expected. However, fibrinogen mRNA was also significantly induced by clofibrate treatment. The response of fibrinogen and AGP mRNAs to LPS and clofibrate was similar in (+/+) and (−/−) mice (Fig. 5).

Effects of endotoxin and clofibrate on mRNA expression of acute-phase genes. Northern blots of RNA from liver of mice treated with saline (control), LPS, or clofibrate were probed with an oligonucleotide complementary to AGP or a cDNA complementary to fibrinogen. All values are normalized to the GAP mRNA signal and are expressed as a percentage of the control group mean. Numbers represent the mean ± S.E. of each group. *P < .05, significantly different from respective control group.
Role of LPS-Induced Hypophagia in Induction of CYP4A by Endotoxin.
Endotoxin treatment causes hypophagia in mice (Kozak et al., 1994), and fasting is known to induce CYP4A expression in rodents (Imaoka et al., 1990). We found that 24 h of fasting caused a significant induction of CYP4A10 mRNA in mouse liver and kidney, without affecting CYP2C29, CYP3A11, or CYP2A5 (in kidney, the induction was similar to that caused by endotoxin; data not shown). However, fasting is not a good model for endotoxin-induced hypophagia, because LPS-treated mice eat about 20% as much as control animals (Kozak et al., 1994). To discover whether the induction of CYP4A mRNA in mouse kidney could be caused by endotoxin-induced hypophagia, we measured the food intake of LPS-treated mice and pair fed a saline-injected group with the LPS-treated animals. As shown in Table1, saline-injected mice consumed about 25% of their total daily intake in the daytime (8:00 AM to 8:00 PM). LPS-injected mice consumed about 30% that of the control mice over the entire 24-h period, and only 24% of their food intake was in the first 12 h. In contrast to LPS treatment, pair feeding did not induce CYP4A10 or CYP4A14 in the kidney and even produced a slight decrease in CYP4A14 expression (Table 2). Similarly, hypophagia was not responsible for the down-regulation of CYP mRNAs by LPS in the liver, because the pair feeding had no effect on CYP3A11 and induced CYP4A10, CYP4A14, CYP2C29, and CYP2A5 mRNAs (Table 2). The data in Table 2 are from animals sacrificed 24 h after initiation of treatment. Qualitatively similar effects of LPS and pair feeding were seen for all parameters when analyzed at 12 h (data not shown).
Food intake in saline- and LPS-treated mice
Effect of LPS-induced hypophagia on liver and kidney CYP mRNA expression
Discussion
The aims of this study were to determine whether inflammation causes an induction of CYP4A mRNA expression in the mouse and whether the effect was dependent on PPARα. We found that CYP4A10 and CYP4A14 expression are induced in mouse kidney after LPS treatment and that these effects are PPARα dependent. The most reasonable explanation of this result would be that the transcriptional activity of PPARα is activated in mouse kidney during the response to LPS, and this interpretation is supported by the finding that another PPARα-regulated gene, ACO, is also induced by LPS treatment. Whereas a significant effect of LPS treatment on BIEN expression was not detected, the results with this enzyme are also consistent with the hypothesis. Because, as noted, arachidonic acid, prostaglandins, leukotrienes, and hydroxyeicosatetraenoic acid are potent activators of PPARs (Yu et al., 1995), it is reasonable to hypothesize that one or more of these compounds, or perhaps another unidentified endogenous compound that is generated during an inflammatory response, contribute to the observed effects.
Because endotoxin is a lipopolysaccharide, the lipid moiety of LPS rather than the inflammation associated with LPS administration could be responsible for activation of PPARα-dependent gene expression. Endotoxin is deacylated in the liver, with the subsequent production of 3-hydroxytetradecaenoic, hexadecaenoic, and dodecaenoic acids (Fox et al., 1996). These fatty acids are potential ligands for PPARα (Issemann et al., 1993). However, it is unlikely that the lipid moiety of LPS underlies PPARα activation, because the single dose of endotoxin used here is two orders of magnitude lower than the daily doses of fatty acids that have been reported to induce CYP4As in rats in a chronic treatment protocol (Göttlicher et al., 1993).
Fasting induces CYP4A activity and protein levels in rat liver (Orellana et al., 1992) and kidney (Imaoka et al., 1990). Moreover,Kroetz et al. (1998) recently showed that induction of the CYP4A subfamily as a result of fasting is regulated through PPARα. LPS treatment of mice reduces food intake and stimulates weight loss in a dose-dependent manner (Kozak et al., 1994), but the weight loss is mainly because of factors other than hypophagia (Kozak et al., 1994). Our results show that the induction of renal CYP4A in the mouse and the down-regulation of several hepatic CYP mRNAs are not caused by hypophagia. Thus, PPAR-dependent induction of CYP4A may be caused by other metabolic effects of endotoxemia. For instance, LPS injection stimulates hepatic fatty acid synthesis within hours of injection and causes later increases in serum triglycerides and cholesterol that appear to be mediated by tumor necrosis factor (Memon et al., 1993).
In contrast to the kidney, we found that LPS treatment resulted in lower levels of CYP4A10 mRNA in the liver and did not induce CYP4A14 mRNA. The tissue specificity of this effect could be explained by the activation of Kupffer cells, which are present in liver but not in kidney, by LPS. The resulting high local concentrations of cytokines and interferons in the vicinity of the hepatocyte (Geller et al., 1994) may oppose any inductive effect caused by PPAR activators. In support of this theory, Knickle et al. (1992) found that treatment of rats with an interferon inducer attenuated the induction of CYP4A mRNA by clofibrate. Cytokines can also suppress induction of CYP4A in cultured fetal rat hepatocytes, and this is accompanied by down-regulation of PPARα expression (Parmentier et al., 1997). Another plausible mechanism is that other signaling pathways, activated by cytokines, may interact with PPARα. For instance, cytokine-activated transcription factors such as nuclear factor-κB, activator protein-1, or signal transducer and activator of transcription-3 could interact directly with PPAR to modulate its activity. PPARα cotransfection attenuated the transcriptional activation of an nuclear factor-κB-responsive promoter in a ligand-dependent manner (Staels et al., 1998): if these transcription factors were mutually antagonistic, it could explain the down-regulation of CYP4A10 by LPS in liver.
The possible reasons for the species differences in the responses of hepatic CYP4A mRNAs to LPS are less obvious. CYP4A10 and CYP4A14 are induced in mouse kidney but not in liver, in contrast to the F344 rat, in which CYP4A mRNAs are induced in both tissues (Sewer et al., 1996a). It is likely that species differences in induction of CYP4A by LPS treatment are due to genetically controlled factors affecting the general response to LPS or the generation of PPAR ligands during inflammation.
Constitutive expression of genes encoding peroxisomal enzymes (e.g., ACO and BIEN), microsomal CYP4A enzymes, and basal levels of peroxisomes are similar between male (−/−) and (+/+) mice on a mixed genetic background (C57BL/6N × Sv/129) (Lee et al., 1995). These results are consistent with another study that used purebred male Sv/129 mice (Aoyama et al., 1998). In contrast, our study shows that the levels of CYP4A10, ACO, and BIEN mRNAs in purebred female Sv/129 (−/−) mice are significantly lower than in (+/+) mice, suggesting that in female mice, a low level of constitutive activation by PPARα occurs. The apparent discrepancy could be due to inherent differences between sexes, because there are significant differences in the phenotype between male and female (−/−) and (+/+) mice (Costet et al., 1998; Djouadi et al., 1998).
Another novel finding of this study is that expression of the mRNA encoding the hepatic acute-phase gene, fibrinogen, is induced by the peroxisome proliferator clofibrate. In contrast, Corton et al. (1998)recently reported that expression of fibrinogen mRNA was suppressed by peroxisome proliferators. This disparity could be attributed to several possibilities. In this study, the acute effect of clofibrate (24 h) was examined, whereas Corton et al. (1998) used chronic treatment (1–13 weeks) with either diethylhexyl phthalate or WY-14,643. PPAR-dependent gene expression can be influenced by protein-protein interactions with coactivators and/or corepressors (Dowell et al., 1997). Thus, differences in the levels of coactivators and/or corepressors could explain why acute activation of PPARα by clofibrate causes increases in fibrinogen mRNA, whereas chronic exposure results in decreases in this mRNA. Clearly, further work is needed to identify the mechanisms underlying this difference. Peroxisome proliferators are known to activate Kupffer cells, resulting in stimulation of tumor necrosis factor-α production in the liver (Rose et al., 1998). This suggests that the release of inflammatory cytokines by Kupffer cells after clofibrate treatment could contribute to the increase in acute-phase proteins. Whether the PPARα is required for Kupffer cell activation by peroxisome proliferators has not been reported. In this study, the acute induction of fibrinogen mRNA by clofibrate occurred through a PPARα-independent mechanism. In contrast, subsequent chronic down-regulation clearly requires the receptor (Corton et al., 1998).
The down-regulation of multiple CYPs that occurs during inflammation can be mimicked by the treatment with inflammatory cytokines in vivo and in vitro (hepatocyte cultures; Morgan, 1997). This study examined the effect of LPS and peroxisome proliferator treatment on CYP mRNA expression in (−/−) mice. LPS and clofibrate administration caused decreases in mRNAs encoding CYP2A5, CYP2C29, and CYP3A11, effects that were attenuated or blocked in the PPARα (−/−) mice. The results with clofibrate are consistent with the observations that peroxisome proliferator-induced down-regulation of CYPs (Corton et al., 1998) and other genes, including apolipoproteins A-I and C-III (Motojima et al., 1997; Peters et al., 1997), requires the PPARα. However, the results with LPS suggest that the PPARα is also implicated in the down-regulation of CYPs during inflammation. Because the effects of LPS on CYP2C29 and CYP2A5 expression were not completely absent in the PPARα (−/−) mice, it is likely that both PPARα-dependent and PPARα-independent pathways are involved in the down-regulation of these CYPs.
Our study confirms the hypothesis that PPARα was involved in the induction of CYP4A during endotoxemia. More surprising was the finding that PPARα appears to have a role in CYP down-regulation in this model as well. This work adds to the growing body of information on the relationship between peroxisome proliferation and inflammation. We show here that not only is inflammation associated with PPAR activation, but peroxisome proliferators activate acute-phase gene expression. The exact role of PPAR in pathology and homeostasis of the inflammatory response remains to be determined.
Acknowledgments
The excellent technical assistance of Qi Chen is gratefully acknowledged.
Footnotes
-
Send reprint requests to: Dr. Edward T. Morgan, Department of Pharmacology, Emory University, Atlanta, GA 30322. E-mail: etmorga{at}bimcore.emory.edu
-
↵1 This work was supported by Grant GM-46897 from the National Institutes of Health (to E.T.M.). A preliminary report was presented at the 10th International Symposium on Cytochrome P-450, Biochemistry, Biophysics, and Molecular Biology, San Francisco, California, August 1997.
- Abbreviations:
- LPS
- lipopolysaccharide
- ACO
- acyl-CoA oxidase
- AGP
- α1-acid glycoprotein
- BIEN
- peroxisomal bifunctional enzyme enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase
- CYP
- cytochrome P-450
- GAP
- glyceraldehyde 3-phosphate dehydrogenase
- PPAR
- peroxisome proliferator-activated receptor
- RXR
- retinoid X receptor
- Received December 18, 1998.
- Accepted April 27, 1999.
- U.S. Government





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}